

Abstract
We developed a procedure to synthesize pinacolyl boronate containing stilbene derivatives and used this procedure to synthesize boron-containing combretastatin analogues. The key step involves the Wittig reaction of the ylide 4-(4,4,5,5-tetramethyl-1,3,2-dioxaboratophenyl)-methyl triphenylphosphonium bromide 11 with 3,4,5-trimethoxy benzaldehyde in the presence of tBuONa in DMF, providing 88% yield. We are now in a position to evaluate the biological activity of these derivatives as modulators of TGF-beta signaling pathways.
Keywords: Antimitotic agent, Combretastatin, Stilbene, Pinacolatoborane, TGF-beta
For an ongoing chemical biology project involving the study of the TGF-beta (Transforming Growth Factor-beta) pathways using developing zebrafish embryos, we synthesized a small library of 2-substituted 2H-chromene derivatives, and after screening, we identified a lead molecule BT7 capable of modulating a specific relevant pathway, namely p-SAPK/JNK, that is known to be down-stream of TGF-beta and can mediate Smad-independent signaling.1,2 To increase the activity and potency of our lead molecule, and for structure/activity relationship studies, we envisioned developing a boron-based stilbene-containing small molecule library (Fig. 1) based on a hypothesis that (a) introducing chromene isosteres like stilbene may increase potency and (b) introducing a boron atom into a biologically active framework, might allow interaction with a target protein not only through hydrogen bonds but also through covalent bonds, and this interaction would produce potent biological activity, and (c) from a literature search we also found that combretastatin A modulates TGF-beta signaling pathway. Therefore, we considered if different CA4 analogues could provide additional insight into TGF-beta signaling pathways to identify new gene targets or co-factors. In this context we undertook a project to develop synthetic methodology to synthesize boron-containing stilbene derivatives, and use those methods to synthesize CA4 analogues.
Figure 1.

Structure of BT7.
Stilbene (1,2-diphenylethene) represents a prototype of a 1,2-disubstituted olefin. Stilbene itself does not occur in nature, but substituted stilbenes (Fig. 2) such as pinosilvin 2, resveratrol 3,3,4 diethylstilbestrol 4,5 4,4-diaminostilbene-2,2-disulfonic acid,6 nitro- substituted stilbene boronate pinacol esters,7 arotinoids8, combretastatin 5, 69 (Fig. 2) are among the many synthetic and natural stilbene derivatives that are biologically active compounds.
Figure 2.

Biologically important stilbene derivatives.
The use of boron atoms in pharmaceutical drug design possesses a high potential for discovery of new biological activity.10–13 Among various boron compounds synthesized, much attention has been paid to boronic acid containing peptides such as Velcade and DPP-IV inhibitors.12 In these boropeptides, a carboxylic acid has been replaced by a boronic acid group.
Combretastatin A-4 (CA-4) has been found to be a potent inhibitor of tubulin polymerization through binding to the colchicine binding site. It is also a cytotoxic agent toward a wide variety of human cancer cell lines and also modulates TGF-beta signaling pathways.14 From a structure/activity relationship point of view, CA-4 belongs to the class of natural compounds related to biphenyls and contains, as a key structural feature, the cis-stilbene motif.
Due to the strong biological significance of stilbene-containing compounds, a general applicable synthetic protocol to synthesize these highly demanding molecules with boronic acid groups would be useful. The protocol should tolerate a large number of functional groups in different positions on the aromatic ring. The most familiar and general strategy for synthesis of boron-containing stilbenes 8 is based on disconnection A (Fig. 3) and involves the Wittig reaction of the various substituted benzyl phosphonium ylide 9 with the pinacol ester of boronate aldehyde 10.15
Figure 3.

Retrosynthetic analysis.
Although Wittig and Horner–Wadsworth–Emmons reactions have been carried out on aldehyde derivatives of boronate esters16,17 the problem with this approach (A) for the synthesis of a library of boron-containing stilbene derivatives is the need to utilize various substituted benzyl phosphonium ylides and boron-containing aldehydes. Boron-containing aldehydes are very prone to self-dimerization and oxidation and are unstable in air, so this method is limited in scope. To overcome this problem, we undertook the disconnection B approach (Fig. 3), envisaging the use of the pinacolylboronate phosphonium ylide 11, which has not previously been explored at all. However, organotrifluoroborato phosphonium ylides are used to synthesize alkene derivatives by Wittig reactions.18
To synthesize our model compound 1, we first synthesized compound 11. 4-(4,4,5,5-tetramethyl-1,3,2 dioxaboratophenyl)- methyl triphenylphosphonium bromide 11 was synthesized from 2-[4′-(bromomethyl)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 13 in the presence of 1.01 equiv of triphenylphosphine in acetonitrile at reflux conditions. The minor excess PPh3 was removed from the product by trituration with ether 2–3 times and the product is stable under normal atmospheric conditions (Scheme 1).
Scheme 1.

Synthesis of boronate ylide 11.
Compound 13, was synthesized by refluxing 14, NBS (N-bromosuccinimide) and AIBN (azobisisobutyronitrile) in carbon tetrachloride for six hours to provide the intermediate 13 (85% yield).19
In this study, we found that 4-(4,4,5,5-tetramethyl-1,3,2-dioxaboratophenyl)- methyl triphenylphosphonium bromide 11, when treated with 2-hydroxy-3,5-dichloro benzaldehyde as a model substrate in the presence of 3 equiv of sodium tert-butoxide in DMF as solvent at room temperature produced the 2,4-dichloro-6-{2-[4- (4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-phenyl]-vinyl}-phenol 1 in 86% yield with a mixture of E and Z isomers (E:Z = 42:58) (Scheme 2).
Scheme 2.

Synthesis of model compound 1.
After successfully synthesizing our target compound 1, next we examined the scope of the Wittig reaction for the synthesis of pinacolylboronate- substituted stilbenes using various aryl aldehydes. 20 The results are summarized in Table 1. The reaction proved tolerant of both electron-withdrawing (NO2; Table 1, entry 4) and electron-donating groups (B(OH)2, di-t-butyl; Table 1, entries 2 and 3) on the phenyl ring of the aldehyde. For the Wittig reaction of aryl aldehydes containing a heteroatom such as nitrogen (Table 1, entry 5) gave the product 1f with good yield with high E selectivity.
Table 1.
Wittig reaction of boronate ylide (11) with various aldehydesa
| Entry | Aldehyde | Product | Yieldb (%) |
|---|---|---|---|
| 1 | 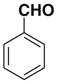 |
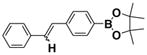 1b E:Z= 74:26 |
86 |
| 2 | 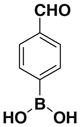 |
1c E:Z= 64:36 |
72 |
| 3 | 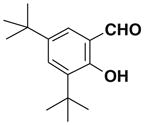 |
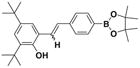 1dE:Z= 75:25 |
71 |
| 4 | 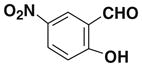 |
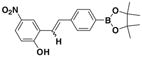 1e E:Z= 55:45 |
76 |
| 5 | 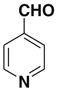 |
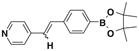 1f E:Z= 100:0 |
78 |
All reactions were performed using 1 equiv of aldehyde and 3 equiv of tBuONa in DMF for 2–12 h.
Isolated yield (mixture of E and Z) refers to aldehyde and the E/Z ratios were determined by 1H NMR.
Next we focussed on target compound CA4 analogues 7a and 7b, bearing a boronic acid system that replaces the OMe group of the natural CA-4 in ring B (Fig. 2). To synthesize 7a and 7b, the ylide 11 was reacted with aldehydes 12a and 12b. Wittig reaction between the aldehyde 12a and ylide 11 in the presence of tBuONa, afforded the corresponding products 7a and 7b with 88% and 82% yields, respectively. The products were a mixture of E and Z isomers (Scheme 3). As Rf value is very close to each other, it was very difficult to separate them, but from TLC we found Rf 0.69 is considerably more fluorogenic than Rf 0.76 (1:1 ethyl acetate/hexane; solvent system).
Scheme 3.

Synthesis of boronate containing combretastatin analogues 7a–b.
Moreover, we have also developed a one-pot Wittig reaction for synthesizing the compound 7a. In this case, 4-(4,4,5,5-tetramethyl- 1,3,2-dioxaboratophenyl)methyltriphenylphosphonium bromide 11, the intermediate generated by reaction of 13 with triphenylphosphine, was reacted directly with 3,4,5-trimethoxy benzaldehyde 12a in the presence of tBuONa as a base, and the desired boron-containing combretastatin analogue 7a was isolated in 74% overall yield as a one-pot Wittig reaction (Scheme 4).
Scheme 4.

One-pot synthesis of 7a.
To compare our approach B with the approach A in (Fig. 3), we synthesized the 7a starting from 16 using aldehyde 17 via the Wittig ylide 15 (Scheme 5), and found our approach gives higher yields with high stereoselectivity.
Scheme 5.

Synthesis of 7a using approach A.
In summary, we developed a procedure to synthesize pinacolylboronate containing stilbene derivatives and used this procedure to synthesize boron-containing combretastatin analogues. Experiments are currently underway to separate the E and Z isomers and test the biological activity of these derivatives and to determine their utility as modulators of TGF-beta signaling pathways.
Supplementary Material
Acknowledgments
The author B.C.D. is thankful to AECOM for start up funding. T.E. is supported by grants from the NIH (HL56182 and HL64282). S.M.M. is thankful to K. Hema for initiating this project. The instrumentation in the AECOM Structural NMR Resource is supported by the Albert Einstein College of Medicine and in part by Grants from the NSF (DBI9601607 and DBI0331934), the NIH (RR017998) and the HHMI Research Resources for Biomedical Sciences.
Footnotes
Supplementary data (experimental procedures and copies of 1H, 13C NMR) associated with this article can be found, in the online version, at doi:10.1016/j.tetlet.2009.04.003.
References and notes
- 1.Liu F, Evans T, Das BC. Tetrahedron Lett. 2008;49:1578. doi: 10.1016/j.tetlet.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torregroza I, Evans T, Das BC. Chem Biol Drug Des. 2009;73:339. doi: 10.1111/j.1747-0285.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hart JH. Annu Rev Phytopathol. 1981;19:437. [Google Scholar]
- 4.Farina A, Ferranti C, Marra C. Nat Prod Res. 2006;20:247. doi: 10.1080/14786410500059532. [DOI] [PubMed] [Google Scholar]
- 5.a The Merck index. 9. Merk & Co; 1976. p. 414. [Google Scholar]; (b) Dodds EC, Goldberg L, Lawson W, Robinson R. Nature. 1938;141:247. [Google Scholar]; (c) Huggins C, Hodges CV. Cancer Res. 1941;1:293. [Google Scholar]
- 6.Kirk RE, Othmer DF. (Kirk-Othmer) Encyclopedia of Chemical Technology. 2. Vol. 19. Wiley-Interscience; 1969. p. 1.p. 13. [Google Scholar]
- 7.Oehlke A, Auer AA, Jahre I, Walfort B, Rffer T, Zoufal P, Lang H, Spange S. J Org Chem. 2007;72:4328. doi: 10.1021/jo070084v. [DOI] [PubMed] [Google Scholar]
- 8.Tolomeo M, Simoni D. Curr Med Chem: Anti-Cancer Agents. 2002;2:387–401. doi: 10.2174/1568011024606361. [DOI] [PubMed] [Google Scholar]
- 9.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. J Med Chem. 2006;49:3033. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 10.Groziak MP. In: Progress in Heterocyclic Chemistry. Gribble GC, Gilchrist TL, editors. Vol. 12. Pergamon; Oxford: 2000. pp. 1–21. [Google Scholar]
- 11.(a) Morin C. Tetrahedron. 1994;50:12521–12569. [Google Scholar]; (b) Yang W, Gao X, Wang B. Med Res Rev. 2003;23:346. doi: 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]
- 12.(a) Matteson DS. Tetrahedron. 1989;45:1859. [Google Scholar]; (b) Matteson DS. Chem Rev. 1989;89:1535. [Google Scholar]; (c) Tian ZQ, Brown BB, Mack DP, Hutton CA, Bartlett PA. J Org Chem. 1997;62:514. doi: 10.1021/jo9615007. [DOI] [PubMed] [Google Scholar]; (d) Fevig JM, Abelman MM, Brittelli DR, Kettner CA, Knabb RM, Weber PC. Bioorg Med Chem Lett. 1996;6:295. [Google Scholar]; (e) Skordalakes E, Eligendy S, Goodwin CA, Green D, Scully MF, Kakkar VV, Freyssinet JM, Dodson G, Deadman JJ. Biochemistry. 1998;37:14420. doi: 10.1021/bi980225a. [DOI] [PubMed] [Google Scholar]; (f) Leung D, Abbenante G, Fairlie DP. J Med Chem. 2000;43:305. doi: 10.1021/jm990412m. [DOI] [PubMed] [Google Scholar]
- 13.Koehler KA, Lienhard GE. Biochemistry. 1971;10:2477. doi: 10.1021/bi00789a008. [DOI] [PubMed] [Google Scholar]
- 14.Thorpe PE, Chaplin DJ, Blakey DC. Cancer Res. 2003;63:1144. [PubMed] [Google Scholar]
- 15.(a) Athar M, Ho Back J, Tang X, Ho Kim K, Kopelovich L, Bickers DR, Kim AL. Toxicol Appl Pharmacol. 2007;224:274. doi: 10.1016/j.taap.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolas Di C, Lakowicz JR. J Photochem Photobiol A: Chem. 2001;143:39. doi: 10.1016/S1010-6030(01)00471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wittig: Lautens M, Mancuso J. J Org Chem. 2004;69:3478. doi: 10.1021/jo049874k.Lautens M, Marquardt T. J Org Chem. 2004;69:4607. doi: 10.1021/jo049722p.Kobayashi Y, Tokoro Y, Watatani K. Tetrahedron Lett. 1998;39:7537.Kobayashi Y, Tokoro Y, Watatani K. Eur J Org Chem. 2000:3825.
- 17.Horner–Wittig–Emmons: Schmidt U, Leitenberger V, Griesser H, Schmidt J, Meyer R. Synthesis. 1992:1248.Park KC, Yoshino K, Tomiyasu H. Synthesis. 1999:2041.Busnel O, Carreaux F, Carboni B, Pethe S, Goff SVL, Mansuy D, Boucher JL. Bioorg Med Chem. 2005;13:2373. doi: 10.1016/j.bmc.2005.01.053.Gopalarathnam A, Nelson SG. Org Lett. 2006;8:7. doi: 10.1021/ol051861l.
- 18.Molander GA, Figueroa R. J Org Chem. 2006;71:6135. doi: 10.1021/jo060863w. [DOI] [PubMed] [Google Scholar]
- 19.Zhang C, Zhemg G, Fang L, Li Y. Synlett. 2006;3:475. [Google Scholar]
- 20.General procedure for the synthesis of stilbenes (Table 1): A flask was equipped with a magnetic stirring bar, a septum inlet, charged with 4-(4,4,5,5- tetramethyl-1,3,2-dioxaboratophenyl)methyltriphenylphosphonium bromide (11) (1 mmol), dry DMF (5 mL), and tBuONa (3 mmol) under nitrogen. The mixture was stirred at room temperature for 5–10 min. To this solution was added aldehyde (1 mmol) and the resulting mixture was then stirred at room temperature for 4–6 h. The reaction mixture was treated with water (20 mL) and was neutralized with 1 M HCl and the product was extracted with ethyl acetate (3 × 10 mL) washed with brine and dried over Na2SO4. The product was isolated by chromatography over silica gel.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.