

Abstract
Background
Nance-Horan syndrome (NHS) is a rare X-linked disorder typified by dense congenital central cataracts, microcornea, anteverted and simplex pinnae, brachymetacarpalia, and numerous dental anomalies due in most cases to a mutation in the NHS gene.
Material and Methods
We present a case of clinical manifestation and ocular pathology in a patient with NHS. This article also reviews and discusses the relevant literature.
Results
Classic and novel ocular pathological findings of a young male with NHS are described, including congenital cataracts, infantile glaucoma, scleral staphyloma, and severe retinal cystoid degeneration.
Conclusions
We report a new pathological finding of severe retinal cystoid degeneration in this NHS patient and confirm abnormal development of the anterior chamber angle structure. These findings, coupled with our analysis of the available NHS literature, provide new understanding of the histopathological basis of ocular abnormalities and vision loss in NHS.
Keywords: Nance-Horan syndrome, eye pathology, cataract, glaucoma, retinal cystoid retinopathy
Introduction
Nance-Horan syndrome (NHS), also known as cataracts-oto-dental syndrome,1 X-linked cataract-dental syndrome,2 or X-linked congenital cataracts and microcornea,3 was first described in detail by Walter E. Nance4 in the United States and Margaret B. Horan5 in Australia. In 1974, Nance reported a six-generation family, which included 7 males presenting with congenital cataracts, microcornea (averaging 9.0 mm horizontally), anteverted pinnae, and dental anomalies, and 9 female carriers with very similar but less severe cataracts and dental anomalies.
Despite cataract surgery during infancy, all of the affected males exhibited severely reduced visual acuity and infantile nystagmus.4 In the same year, Horan and Billson reported a three-generation Australian family, including three affected males described with opacification of the nucleus of the fetal lens, posterior Y-sutures, and zonular variations, as well as dental anomalies; affected heterozygote females had punctate lens opacities surrounding the Y-suture.5
Nance and Horan were, in fact, not the first to describe this disease. Before 1974, there were several reports of male patients with congenital cataracts and microcornea, female carriers with posterior Y-sutural opacities and dental abnormalities, and an X-linked inheritance pattern. Early reports varied in the thoroughness with which features of the condition are documented.
In 1937, Walsh et al.6 presented a pedigree of hereditary cataract with microcornea and marked reduction of vision. Seven affected males showed manifest lens opacities, which were “of sufficient size that they appeared to fill or almost fill the pupillary space, microcornea and nystagmus.” Five females were found to have mild lens opacities with normal vision.
Fraccaro and colleagues,7 in 1967, described a northern Italian family of four generations in which nine males were affected with congenital total nuclear dense and homogeneous opacities requiring surgical intervention before age 2. Although visual acuity was not mentioned, nystagmus was present in at least one male and esotropia in at least four males in the family. Krill8 described in 1969 a family of X-linked cataract in which one of the affected males also exhibited microcornea and mental deficiency. The female carriers had posterior Y-sutural opacities without any effect on vision.
NHS is a rare X-linked hereditary disorder, mapping to chromosome Xp22.31–Xp22.13, which is characterized by severe congenital cataracts, microcornea, anteverted and simplex pinnae, brachymetacarpalia, and numerous dental and craniofacial anomalies.9–11 Affected males have severe bilateral cataracts which lead to profound vision loss and usually require surgery at an early age. Microcornea and microphthalmia have also been reported in some families.12,13
Dental abnormalities include screwdriver-shaped incisors, supernumerary maxillary incisors, and diastema (wide space of teeth). Although this constellation of ocular and craniofacial symptoms constitutes the major clinical manifestation of NHS, 30% of cases also display mental retardation. Heterozygous females manifest less pronounced forms of these same features.
To our knowledge, no histological studies of whole eyes afflicted with NHS have been reported to date. This article describes both typical and novel ocular pathological findings of a young male with NHS, including congenital cataracts, developmental glaucoma, scleral staphyloma, and retinal cystoid degeneration, and discusses the relevant literature.
Case Report
A male patient, who was born in 1977, had a medical history significant for bilateral congenital cataracts, infantile glaucoma, bilateral microcornea, dental abnormalities with screwdriver-shaped incisors and diastema, and mild facial dysmorphism. He also noticed his middle finger of the left hand slightly pointed towards the left. Cataract surgery and placement of intraocular lenses were performed during early childhood. Multiple glaucoma surgeries were also performed in both eyes.
When examined in November 2007, at age 30, the patient exhibited marked bilateral infantile nystagmus, microcornea, and left buphthalmos. His visual acuity was limited to count fingers in the right eye and no light perception in the left eye at that time. Two staphylomas had developed on the temporal side of sclera in the left eye. Due to severe, unremitting pain from glaucoma, which was unresponsive to medical and surgical management, the blind left eye was ultimately enucleated in December 2007.
The patient's family history was positive for NHS and has been described previously by Lewis and colleagues (Family XL-56), although the details of the patient's clinical presentation were not included in the previous reports.3,14,15 His older brother, great uncles, uncles, and male cousin, all on his maternal side, were diagnosed with NHS and presented typical manifestations of the disease.
Ocular Pathological Findings
Pathological examination of the enucleated left eye disclosed an elongated globe 26 mm in length. The cornea was small, measuring 6.5 mm horizontally, with irregular epithelia and loss of Bowman's layer. Peripherally, there was scarring, neovascularization, and T-cell infiltration in the anterior one-third corneal stroma (Figure 1). Descemet's membrane was mildly thickened and irregular. Endothelial cells were attenuated. There was a small clump of retrocorneal pigmentation in the periphery.
FIG. 1.
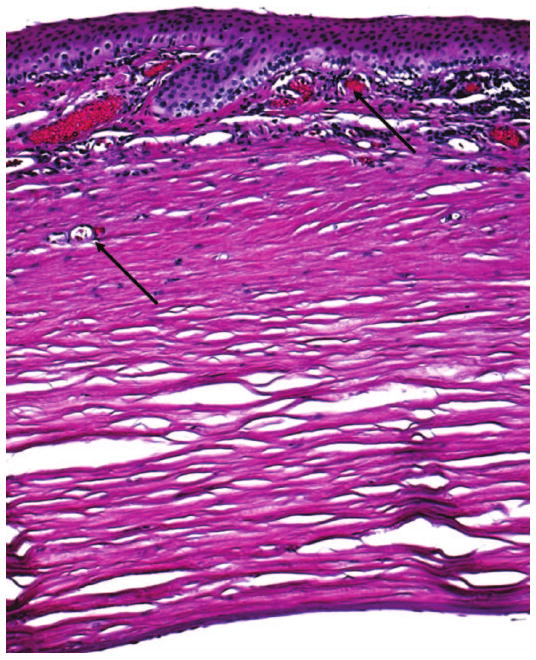
Photomicrograph showing corneal scarring, neovascularization (arrows), and lymphocytic infiltration from the basal epithelium to the anterior one-third of the stroma. Descemet's membrane is mildly thickened and irregular. Endothelial cells are attenuated. (hematoxylin & eosin, original magnification, ×100)
The anterior chamber angle appeared poorly developed with iris roots directly adherent to the posterior trabecular meshwork without fibrosis (Figure 2). The thin iris contained many small sclerotic vessels on the surface, suggesting rubeosis iridis. There was a small residue, dumbbell-shaped, cataractous lens consist of cortical fragmentation and globules. The ciliary body was also poorly developed with hypotrophic pars plicata and ciliary muscles.
FIG. 2.
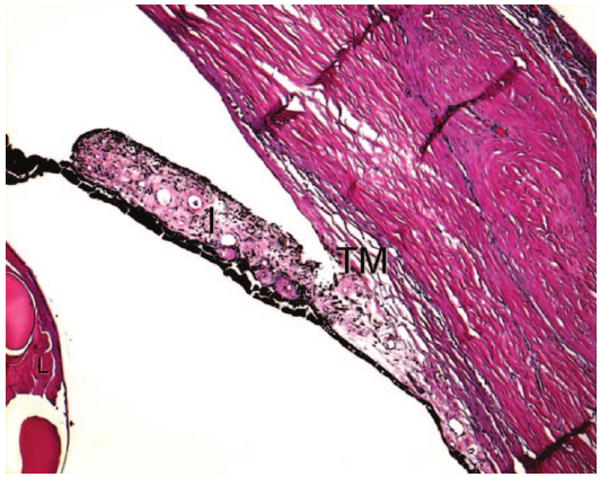
Photomicrograph showing poorly developed anterior chamber angle with iris roots (I) directly adherent to the posterior trabecular meshwork (TM). The iris is thin and contains many small sclerotic vessels on the surface. There are residues of the cataractous lens (L) with cortical fragmentation and globule formation. (hematoxylin & eosin, original magnification, ×50)
Most of the retina showed loss of photoreceptors and ganglion cells (Figure 3A). Remarkable cystoid degeneration was noted mainly in the plexiform layers from the far periphery extending near the equator, and areas of relatively preserved retinal tissues were also present (Figure 3B–D). The retinal pigment epithelium (RPE) exhibited focal hypotrophy, atrophy, hypertrophy, and migration.
FIG. 3.
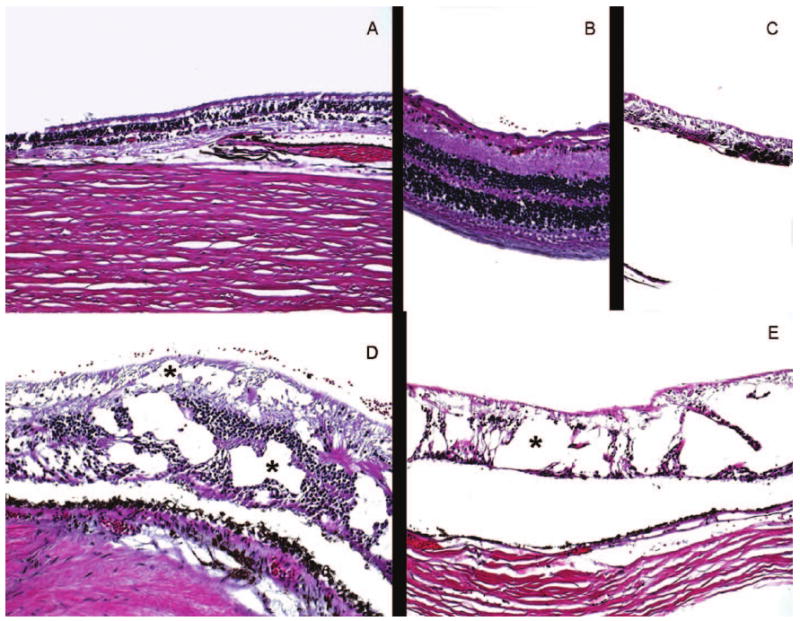
Photomicrographs of the retina showing A: areas with loss of photoreceptors and ganglion cells as well as retinal pigment epithelium (RPE); B: small area with relatively intact retina; C: a foci with retinal atrophy and RPE hypertrophy and migration; D,E: foci with remarkable cystoid degeneration (asterisks) in different retinal layers. (hematoxylin & eosin, original magnifications: A and C, ×25; B and E, ×50; D, ×100)
In the peripapillary region, there was a thin layer of glial elements (glial fibrillary acidic protein, GFAP-positive) (Figure 4); focal absence of neuroretina, RPE, and choroid; and an outward bulging of the sclera that formed staphylomas. There was focal infiltration of inflammatory cells in the choroid and absence of the choriocapillaris in most areas. Loss of neuronal tissue was observed from the head of the optic nerve to the lamina cribrosa (Figure 4). The optic nerve was severely atrophied and gliotic.
FIG. 4.
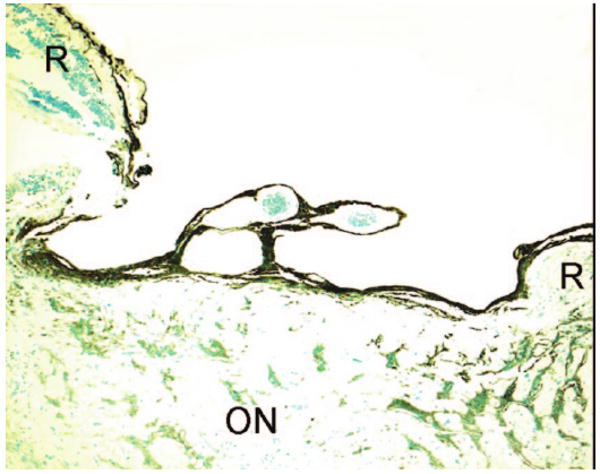
Photomicrograph showing severely atrophied and gliotic optic nerve and a deep cupping. (R: retina; ON: optic nerve; avidin-biotin-immunoperoxidase with primary antibody of GFAP, original magnification, ×50)
Discussions and Literature Review
NHS is a rare genetic X-linked disorder, characterized by dental and ocular anomalies, and mental retardation more pronounced in males than in heterozygous females. The incidence has not been fully evaluated, but the disease is probably underdiagnosed. Fewer than 20 families have been reported in the literature.
Systemic Features of NHS
Typical systemic features described in the literature are listed in Table 1. The diagnosis of NHS in males is made early, generally in the first year of life, and most often at birth, particularly in cases where there is a known family history. It is characterized by the association in male patients of congenital cataracts with microcornea, dental anomalies, and facial and finger dysmorphism.15 Dental anomalies in NHS are typical, specific, and often marked, either by their type or by their aggregation in the same individual. Thus, recognition of dental anomalies in patients may play an important role in the diagnosis of NHS, especially in cases where other dysmorphological features are not well manifested.
TABLE 1.
Summary of clinical manifestations of NHS
| Author(s) | Family No. |
Affected males |
Congenital cataract |
Nystag- mus |
Micro- cornea |
Glau- coma |
Dental anomalies |
Brachy- metacarpalia |
Dysmorphic facial features |
Mental retardation |
|---|---|---|---|---|---|---|---|---|---|---|
| Nance4 | − | 7 | + | + | + | NS | + | + | + | − |
| Horan5 | − | 3 | + | + | + | NS | + | NS | NS | + |
| van Dorp16 | P8598 | 7 | + | + | + | NS | + | NS | NS | + |
| Seow17 | − | 1 | + | NS | NS | NS | + | NS | + | NS |
| Zhu et al.28 Levin et al.34 | − | 7 | + | NS | + | NS | + | NS | + | − |
| Huang,14 Lewis3,15 | XL-11 | 7 | + | + | + | + | + | + | + | − |
| Huang,14 Lewis3,15 | XL-56 | 7 | + | + | + | + | + | + | + | + |
| Huang,14 Lewis3,15 | XL-51 | 3 | + | + | + | + | + | + | + | + |
| Krill,8 Huang,14 Lewis3,15 | XL-116 | 2 | + | + | + | + | + | + | + | − |
| Huang,14 Lewis3,15 | XL-39 | 3 | + | + | + | + | + | + | + | − |
| Florijn,13 Pinckers24 | P21540 | 2 | + | + | − | NS | + | NS | + | − |
| Florijn13 | P8598 | 3 | + | + | + | NS | + | NS | + | − |
| Florijn13 | P20079 | 1 | + | + | − | NS | − | NS | − | − |
| Florijn13 | P24486 | 1 | + | − | + | − | + | NS | + | + |
| Reches22 | − | 3 | + | + | − | − | + | + | + | − |
| Van Esch35 | − | 1 | + | #* | + | #* | #* | #* | + | #* |
| Ramprasad36 | − | 8 | + | + | + | − | + | − | + | − |
| Toutain A37 | 1 | 7 | + | NS | NS | NS | + | NS | + | + |
| Toutain A37 | 2 | 7 | + | NS | NS | NS | + | NS | + | + |
| Toutain A37 | 3 | 2 | + | NS | NS | NS | + | NS | + | + |
| Toutain A37 | 4 | 1 | + | NS | NS | NS | + | NS | + | + |
Too young to draw conclusions.
NS = not stated.
XL and P are from the references; They represent the family and patient.
The spectrum of dental disease includes maxillary and mandibular diastema of both the central and the lateral incisors, notched or serrated incisal edges, narrow gingival and incisal margins creating a “screwdriver” shape, conical distortion of other teeth, “mulberry-like” molars, and mesiodens.6,9,16–18 Mesiodens, which are peg-shaped, supernumerary teeth, most frequently medial and in the upper jaw, are reported in 65% of cases.19 Besides the shape and alignment abnormalities, the teeth are “soft,” with yellow-grey discoloration and caries. Both the deciduous and permanent dentitions are involved.6,9,16–18 Many male patients with dental abnormalities undergo partial or full-mouth dental extractions in the third, fourth, and fifth decades of life.15 In heterozygous females, dental anomalies are present but are generally less severe and less varied.
Distinctive facial features in males with NHS include prominent ears with anteverted pinnae, a long face with a narrow mandible, and a narrow nasal bridge.15 Though the incidence varies, patients may also present with broad and/or short fingers, largely due to brachymetacarpalia, or shortened metacarpals. Brachymetacarpalia has been described in all three males in the original report by Nance et al.4 In the report by Lewis and colleagues, all the 5 families (XL-51 in Nance's report included) showed the consistent appearance of shortened metacarpals IV and V in both affected males and carriers, affecting 95% of both affected males and carrier females.18
Mild mental retardation has been reported in 20–30% of affected individuals.13,15,20,21 The etiology of NHS-related mental retardation is likely complex, given that in some cases, some affected individuals exhibit mental retardation whereas other affected individuals within the same family are of normal intelligence.13,15 The phenotype of female carriers is generally less severe, and rarely includes intellectual impairment. There are, however, some female cases with unusually strong expression of the NHS phenotype, including mental retardation.15 Heterozygote detection and prenatal diagnosis are possible by linkage analysis with polymorphic markers.22
Our patient presents with dental anomalies and the facial and the third metacarpal features described above, but without intellectual anomalies.
Ocular Manifestations of NHS
The ocular manifestations of NHS in affected males consist of bilateral, severe congenital cataracts present from an early age in 100% of cases, typically involving the fetal nucleus and posterior Y-suture with variable zonular extensions into the posterior cortex, associated with microcornea (96% cases) or even microphthalmia.14,15 In the majority of cases (93%), NHS leads to severe visual impairment leading to nystagmus (93%). Strabismus occurs in 43%. Many patients exhibit glaucoma (51%).15
Pathological processes, which might arise as complications of cataract extraction, possible, even necessitate enucleation.23 Overall visual prognosis remains poor, despite cataract extraction and glaucoma surgeries.10 Our patient exhibits these typical clinical ocular findings of NHS including congenital cataract, microcornea, glaucoma, and loss of visual acuity.
In heterozygous females, clinical ocular features may be identical to those of affected males, but they are attenuated and are often subclinical.3 Ocular signs are observed in more than 90% of female cases. They consist of bilateral, but often asymmetrical, lens opacities, especially posteriorly.15 Extensive or progressive opacities are observed in only 18% of the cases.7,16 Microcornea is rare (6%) and microphthalmia has never been reported in females.12,13,15,20
Surgery is required in only a minority of cases with progressive cataractous lesions at an advanced age or with extensive congenital cataract. Postoperative complications do not usually occur and visual prognosis is good overall, generally with normal vision (>75% cases) or mildly decreased vision at an advanced age.23 Significant impairment in visual acuity is reported in only 3% of cases, and strabismus is noted in only 2% of cases.12,13,15,20
Congenital Cataracts
Much like other X-linked conditions such as Lenz syndrome and Lowe syndrome, NHS is characterized by congenital cataracts.4,5 Bilateral dense stellate and nuclear cataracts are often noted at birth in males affected with NHS. Lens abnormalities in affected males usually require cataract extraction, although the results are generally poor.9,17,18 Our patient underwent cataract extraction at an early age. Complications of cataract extraction such as glaucoma and/or retinal detachment are treated medically or surgically depending on the type and severity.
In female carriers, the cataracts are characteristically fine, punctate opacities aligning with or outlining the posterior Y-suture, sometimes with a dense, solid organization.12,13,15,20 Secondary and even tertiary sutural bifurcations are opacified in some older women. Older women also tend to have both posterior and anterior Y-opacities, often with peripheral cortical dots, with no distinguishable radial or anteroposterior anatomic organization.15
Cornea
The normal adult cornea measures 10.0–11.5 mm horizontally (median: 10.5 mm). Most males with NHS have microcornea with horizontal corneal measurements of 6 to 10 mm.9–11 Microcornea is reported in almost all affected males, except in those eyes where complicated and prolonged glaucoma in the early years of life would explain symmetric or asymmetric enlargement. Our patient exhibited marked microcornea measuring 6.5 mm. Corneal scarring, neovascularization, and inflammation in this patient are likely secondary to poor intraocular pressure control and multiple ocular operations.
Although some authors have mentioned the association of NHS with “microphthalmia,”13,14,16 none of the available literature indicates whether quantitative evaluations (for example, by ultrasonic scan or MRI) were conducted to determine the precise axial dimensions of the globes of NHS patients. Cycloplegic refractions and A-scan echobiometry performed in a study by Pinckers and colleagues demonstrated that both phakic and aphakic globes in patients with NHS have dimensions appropriate for the chronologic age of the subjects.24 Normal head circumference, normal midfacial dimensions, and the absence of blepharophimosis or orbital hypoplasia, which normally accompany true microphthalmia—also provide indirect evidence for normal globe growth in NHS patients. In our patient, the length of the globe is 26 mm, which is longer than normal, most likely due to his longstanding glaucoma and staphyloma.
Glaucoma
Glaucoma is reported in approximately 50% of male NHS patients.15 In general, glaucoma is always severe and unremitting, despite both medical and surgical therapy. When present, it develops early in life, manifesting with photophobia, blepharospasm, corneal edema, and epiphora accompanied by enlargement and stretching of the limbal tissues and cornea.
Glaucoma is a complication of cataract surgery in infants, and thus cataract extraction might contribute to the high rate of glaucoma in patients with NHS. In Lewis' report,15 nearly half (10/21) of males with NHS developed glaucoma within several months to several decades of cataract surgery. Two adult males developed glaucoma only in the eye that was operated previously (1 and 10 years post-cataract extraction), which suggests that lens surgery is an important factor in glaucoma development in NHS patients. Still, pediatric patients with NHS exhibit glaucoma at a much higher rate than non-NHS patients with a history of childhood cataract surgery (15–45%).25 The mechanism underlying this increased risk of glaucoma in NHS patients is unclear, although Lewis et al. suggested that it is “inextricably linked to the primary genetic trait.”15
The pathological findings in this case suggest that this increased risk of glaucoma may be mediated through abnormal development of the aqueous humor drainage system in NHS patients. Our patient suffered from bilateral glaucoma, with total loss of vision in his left eye. Pathological examination revealed an abnormal anterior insertion of the iris on the trabecular meshwork, combined with a poorly developed scleral spur, as if there had been incomplete separation of the structures. The trabecular tissue appeared as a compact mass of trabecular sheets in the posterior portion, and the iris root was not well separated from the trabecular meshwork. Schlemm's canal was not identified. The ciliary body was markedly hypotrophic and flattened.
All these abnormalities in the absence of identifiable scar tissue suggest that a developmental abnormality in the trabecular meshwork was present which might have impaired aqueous humor drainage and led ultimately to infantile glaucoma. Thus, in this patient, both developmental glaucoma and secondary glaucoma due to cataract surgery elevated intraocular pressure. The high intraocular pressure, in turn, led to typical glaucomatous complications, such as a deep optic nerve head cupping, optic nerve atrophy, and staphyloma formation.
Glaucoma, unlike most other causes of optic nerve atrophy, usually produces significant optic nerve cupping, which was noted on clinical examination in this patient. Staphylomas can also develop under the influence of long-standing, continually increased intraocular pressure, especially in young persons. These ectasias most commonly are seen in the equatorial region around the openings of the vortex veins, which is consistent with the ocular findings in this patient.
Retinal Cystoid Degeneration
Based upon our review of the literature, no NHS-related retinal pathology has been documented previously. In our patient, we found severe cystoid degeneration, particularly in the peripheral retina. The cystic cavities are present in both outer and inner plexiform layers. They are delineated by the inner and outer nuclear layers, and they are fortified by the middle and external limiting membranes and by the interbridging of neural fibers and Müller cells coursing between the inner and outer retinal layers. The mechanisms underlying these degenerative retinal changes in NHS are not yet clear. We suggest that it might be related to NHS protein-mediated dysfunction of Müller cells' adhesion in the retina, which will be discussed below.
Loss of visual acuity in NHS patients was initially attributed to the cataract itself. However, males with NHS often have poor vision even despite early surgical intervention, regardless of nystagmus at time of surgery early in life, the surgical technique, or the method of postoperative visual rehabilitation.15
The mechanisms underlying the reduced visual acuity are not clear. We suggest that retinal degeneration might be one of multiple reasons for vision loss. Mathys et al.26 recently posited that the mechanisms underlying the visual impairment in NHS might include not only deprivation amblyopia and secondary glaucoma, but also dysfunction of the retina. He presented ophthalmologic findings in a boy with NHS. Both of his eyes were aphakic and with normal intraocular pressure. Flash visual evoked potentials (VEP) obtained at the age of 10 weeks and at 5 months showed a response, while the electroretinogram performed under anesthesia at the age of 10 months showed a reduction in the overall retinal function with a more pronounced deficit of the rods (rod-cone dystrophy). Our pathological findings in this case are consistent with the report by Mathys et al., suggesting that retinal pathology might play a role in NHS-related vision loss.
Genetics of NHS
Progress towards mapping and identification of the NHS locus has been facilitated by the development of the Xcat mouse, a proposed murine model for the disease.27 Affected mice develop congenital cataracts that are inherited as an X-linked trait, leading to total lens opacity both in hemizygous males and homozygous females, while heterozygous females display a variable phenotype from totally opaque to clear lenses.
Recently, a large insertion mutation in the first intron of Nhs/Nhs1 gene was identified as the cause of congenital cataract in the Xcat mouse, which confirms the latter as an animal model of Nance-Horan syndrome.12 Two variants of the Nhs gene, Nhs1 and Nhs_v1, are expressed in the developing mouse embryo as a result of transcription initiation at two distinct sites and alternative splicing of exon 2.
In humans, the NHS gene is localized to Xp22.31–p22.13 b.2,3,28–31 Its locus has been refined to a critical region of 1.3 Mb between short tandem repeat markers DXS1195 and DXS999.30 Fourteen protein truncation (nonsense) mutations have been identified in several domains of the NHS gene (Table 2). Some families, such as the family of the patient reported here, with typical NHS symptoms, actually possess none of these mutations, suggesting that yet undiscovered mutations in intronic or regulatory NHS gene sequences or some unknown non-genetic mechanisms may exist.14
TABLE 2.
Summary of mutation in the NHS gene
| Authors | Ref. | Family No. | Sequence change | Exon/Intron | Predicted protein change |
|---|---|---|---|---|---|
| Huang et al. | 14 | XL-116 | c.4129 C>T | Exon 6 | p.Q1358X |
| Huang et al. | 14 | XL-39 | c.3624 C>A | Exon 6 | p.C1208X |
| Huang et al. | 14 | XL-11 | c.1108 C>T | Exon 5 | p.Q370X |
| Huang et al. | 14 | XL 51 | No mutation | — | — |
| Huang et al. | 14 | XL 56 | No mutation | — | — |
| Florijn et al. | 13 | P21540 | c.1117 C>T | Exon 5 | p. R378X |
| Florijn et al. | 13 | P8598 | c.853-2 A>G | IVS3-2 | Splice site change |
| Florijn et al. | 13 | P20079 | c.2601-2602 insG | Exon 6 | p.K868E fsX5 |
| Florijn et al. | 13 | P24486 | c.2635 C>T | Exon 6 | p.R879X |
| Ramprasad et al. | 36 | 1 | c.115C>T | Exon 1 | p.Q39X |
| Brooks et al. | 32 | 1 | c.3738-3739 delTG | Exon 6 | p.C1246-A1247fsX15 |
| Brooks et al.* | 32 | 2 | c.400delC | Exon 1 | p.R134fsX61 |
| Brooks et al. | 32 | 3 | c.2687 delA | Exon 6 | p.Q896fsX10 |
| Brooks et al. | 32 | CRX | No mutation | — | — |
| Burdon et al. | 20 | 1 | c.2387insC | Exon6 | p.S797fsX35 |
| Burdon et al. | 20 | 2 | c.3459delC | Exon 6 | p.L1154fsX28 |
| Burdon et al. | 20 | 3 | c.1117C>T | Exon 5 | p.R378X |
| Burdon et al. | 20 | 4 | c.718insG | Exon3 | p.E240fsX36 |
| Burdon et al. | 20 | 4 | c.IVS2-3C>G | Intron2 | |
| Burdon et al. | 20 | 5 | c.400delC | Exon 1 | p.R134fsX61 |
| Burdon et al.* | 20 | 6 | No mutation | — | — |
| Reches et al. | 22 | — | c.3908del11bp | Exon 6 | Ile1302 |
| Van EH et al. | 35 | — | 2.8 Mb microdeletion at Xp22.2-Xp22.13 | Whole gene | deletion |
Related family.
XL and P are from the references; They represent the family and patient.
Functional Studies of NHS
The NHS gene is conserved in human and other vertebrates such as mouse, rat, and zebra-fish.20 It is expressed in the developing brain, eye, and teeth consistent with developmental defects found in these organs in individuals with NHS, supporting a possible regulatory role for the NHS gene.20,26 NHS protein has also been detected in human fetal brain, thymus, lung, and kidney32; however, the significance of expression in these tissues is presently unclear. There are multiple NHS isoforms and their localizations have distinct cellular compartments, which suggest that the pleiotropic features of NHS might be attributed to the complex regulation and multiple functions of the isoforms.
Disruption of NHS expression or translation leads to cataract formation in humans and in mice, suggesting that it is a pivotal gene in ocular lens development. Both Huang and Sharma detected Nhs protein in animal lens.10,12 Isoform NHS-A appears to be the most relevant form for NHS.
Using in situ hybridization, Sharma et al. showed continuous expression of NHS-A in the developing mammalian lens and subsequent restriction of NHS-A expression to the lens epithelium in pre- and postnatal lens.10 While Sharma and colleagues reported that Nhs protein was absent from lens fibers in the postnatal lens, Huang et al. have employed immunohistochemistry to identify Nhs in the developing lens fibers.12 These discrepancies in Nhs protein expression might be attributable to the different antibodies used by the investigators for Nhs detection.
Interestingly, Figure 8 in the article by Sharma and colleagues, which exhibits in situ hybridization for Nhs transcripts in the developing lens, also demonstrates its expression in the retina, especially in the periphery (Figures E11.5, E14.5, E16.5 in Sharma et al.).10 The authors also demonstrated that Nhs-A colocalizes with the tight junction protein zona occludens-1 (ZO-1) in the apical aspect of cell membrane in epithelial cells, including lens epithelial cells.10
The localization of NHS-A protein in the peripheral retina and to ZO-1 in tight junctions of epithelial cells suggests a putative mechanism by which NHS-A deficiency in NHS might lead to cystoid retinal degeneration, which we described above as a potential cause of vision loss in our and perhaps other patients with NHS.
Studies have demonstrated the presence of ZO-1 along the outer limiting membrane of the retina, especially at the adherens junctions between the Müller cells and at the outer segments of photoreceptor cells.33 The abundance of ZO-1 in the outer limiting membranes, which are formed by the Müller cell junctions, may therefore be important in maintaining retinal architecture, probably by stabilizing the arrangement of retinal neural cells. Since NHS-A is found in the retina and localizes with ZO-1, it is possible that NHS-A mutations lead to impaired ZO-1-mediated cell-to-cell adherence in Müller cells of the retina, leading therefore to cystoid degeneration such as that seen in our patient. In addition, in accordance with the peripheral retinal expression of NHS-A illustrated by Sharma et al.,10 the cystoid degeneration in our patient is mainly localized to the peripheral retina.
Conclusions
To our knowledge, this is the first description of pathological findings of an eye with NHS. Although the eye has had multiple operations and may represent at an end-stage of the disease, it demonstrates certain findings unique to NHS. In addition to the documented glaucoma secondary to cataract surgery, and glaucoma-associated optic nerve atrophy, we identify anatomical aberrations in the anterior chamber angle, trabecular meshwork, Schlemm's canal, and iris root, which could potentially contribute to the development of infantile glaucoma in some patients with NHS.
We also report severe extensive retinal cystoid degeneration, which might be related to NHS protein expression in the cell-to-cell junctions between Müller cells and neuroretinal cells. The presence of several variants of the NHS gene and their localization to different parts of the cell indicates their ability to perform diverse functions, possibly through interaction with a wide range of protein molecules. Further studies are needed to investigate the NHS genes, NHS proteins, and their functional roles in NHS pathogenesis.
Acknowledgments
The study was supported by the NEI intramural research program. Sincere appreciation is extended to the families described here for their willing and continuing cooperation in these investigations. Dr. Richard A. Lewis reviewed the manuscript.
Footnotes
Declaration Of Interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
This article is not subject to United States copyright laws.
Publisher's Disclaimer: Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article may be used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
Contributor Information
Xiaoyan Ding, Immunopathology Section, Laboratory of Immunology, National Eye Institute, Bethesda, Maryland, USA; Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China.
Mrinali Patel, Immunopathology Section, Laboratory of Immunology, National Eye Institute, Bethesda, Maryland, USA; Howard Hughes Medical Institute, Chevy Chase, Maryland, USA.
Alexandra A. Herzlich, Immunopathology Section, Laboratory of Immunology, National Eye Institute, Bethesda, Maryland, USA
Pamela C. Sieving, National Institutes of Health Library, National Institutes of Health, Bethesda, Maryland, USA
Chi-Chao Chan, Immunopathology Section, Laboratory of Immunology, National Eye Institute, Bethesda, Maryland, USA.
References
- 1.Sonoda T. Cataracts-oto-dental defects (Nance-Horan syndrome) Ryoikibetsu Shokogun Shirizu. 2001;33:344–345. [PubMed] [Google Scholar]
- 2.Stambolian D, Lewis RA, Buetow K, Bond A, Nussbaum R. Nance-Horan syndrome: Localization within the region Xp21.1-Xp22.3 by linkage analysis. Am J Hum Genet. 1990;47:13–19. [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis RA, Nussbaum RL, Stambolian D. Mapping X-linked ophthalmic diseases. IV. Provisional assignment of the locus for X-linked congenital cataracts and microcornea (the Nance-Horan syndrome) to Xp22.2-p22.3. Ophthalmology. 1990;97:110–120. doi: 10.1016/s0161-6420(90)32644-1. [DOI] [PubMed] [Google Scholar]
- 4.Nance WE, Warburg M, Bixler D, Helveston EM. Congenital X-lined cataract, dental anomalies and brachymetacarpalia. Birth Defects Orig Art Ser. 1974;10(4):285–291. [PubMed] [Google Scholar]
- 5.Horan MB, Billson FA. X-linked cataract and Hutchinsonian teeth. Aust Paediatr J. 1974;10:98–102. [Google Scholar]
- 6.Walsh FB, Wegman ME. Pedigree of hereditary cataract, illustrating sex-limited type. Bull Johns Hopkins Hosp. 1937;61:125–135. [Google Scholar]
- 7.Fraccaro M, Morone G, Manfredini U, Sanger R. X-linked cataract. Ann Hum Genet. 1967;31:45–50. [PubMed] [Google Scholar]
- 8.Krill AE, Woodbury G, Bowman JE. X-chromosomal-linked sutural cataracts. Am J Ophthalmol. 1969;68:867–872. doi: 10.1016/0002-9394(69)94582-6. [DOI] [PubMed] [Google Scholar]
- 9.Bixler D, Higgins M, Hartsfield J., Jr The Nance-Horan syndrome: A rare X-linked ocular-dental trait with expression in heterozygous females. Clin Genet. 1984;26:30–35. doi: 10.1111/j.1399-0004.1984.tb00783.x. [DOI] [PubMed] [Google Scholar]
- 10.Sharma S, Ang SL, Shaw M, et al. Nance-Horan syndrome protein, NHS, associates with epithelial cell junctions. Hum Mol Genet. 2006;15:1972–1983. doi: 10.1093/hmg/ddl120. [DOI] [PubMed] [Google Scholar]
- 11.Toutain A, Ronce N, Dessay B, et al. Nance-Horan syndrome: Linkage analysis in 4 families refines localization in Xp22.31-p22.13 region. Hum Genet. 1997;99:256–261. doi: 10.1007/s004390050349. [DOI] [PubMed] [Google Scholar]
- 12.Huang KM, Wu J, Duncan MK, et al. Xcat, a novel mouse model for Nance-Horan syndrome inhibits expression of the cytoplasmic-targeted Nhs1 isoform. Hum Mol Genet. 2006;15:319–327. doi: 10.1093/hmg/ddi449. [DOI] [PubMed] [Google Scholar]
- 13.Florijn RJ, Loves W, Maillette de Buy Wenniger-Prick LJ, et al. New mutations in the NHS gene in Nance-Horan syndrome families from the Netherlands. Eur J Hum Genet. 2006;14:986–990. doi: 10.1038/sj.ejhg.5201671. [DOI] [PubMed] [Google Scholar]
- 14.Huang KM, Wu J, Brooks SP, Hardcastle AJ, Lewis RA, Stambolian D. Identification of three novel NHS mutations in families with Nance-Horan syndrome. Mol Vis. 2007;13:470–474. [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis RA. Mapping the gene for X-linked cataracts and microcornea with facial, dental, and skeletal features to Xp22: An appraisal of the Nance-Horan syndrome. Trans Am Ophthalmol Soc. 1989;87:658–728. [PMC free article] [PubMed] [Google Scholar]
- 16.van Dorp DB, Delleman JW. A family with X-chromosomal recessive congenital cataract, microphthalmia, a peculiar form of the ear and dental anomalies. J Pediatr Ophthalmol Strabismus. 1979;16:166–171. doi: 10.3928/0191-3913-19790501-08. [DOI] [PubMed] [Google Scholar]
- 17.Seow WK, Brown JP, Romaniuk K. The Nance-Horan syndrome of dental anomalies, congenital cataracts, microphthalmia, and anteverted pinna: Case report. Pediatr Dent. 1985;7:307–211. [PubMed] [Google Scholar]
- 18.Walpole IR, Hockey A, Nicoll A. The Nance-Horan syndrome. J Med Genet. 1990;27:632–634. doi: 10.1136/jmg.27.10.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hibbert S. A previously unreported association between Nance-Horan syndrome and spontaneous dental abscesses. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:207–211. doi: 10.1016/j.tripleo.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Burdon KP, McKay JD, Sale MM, et al. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am J Hum Genet. 2003;73:1120–1130. doi: 10.1086/379381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang KM, Geunes-Boyer S, Wu S, Dutra A, Favor J, Stambolian D. Organization and annotation of the Xcat critical region: Elimination of seven positional candidate genes. Genomics. 2004;83:893–901. doi: 10.1016/j.ygeno.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Reches A, Yaron Y, Burdon K, et al. Prenatal detection of congenital bilateral cataract leading to the diagnosis of Nance-Horan syndrome in the extended family. Prenat Diagn. 2007;27:662–664. doi: 10.1002/pd.1734. [DOI] [PubMed] [Google Scholar]
- 23.Toutain A. Nance-Horan syndrome. Orphanet Encyclopedia. [May 5, 2008];2003 http://www.orpha.net/data/patho/GB/uk-Nance-Horan.pdf.
- 24.Pinckers A, Van AAI, Bosch TP, Verbeek AM, Hardus P. X-linked cataract. Ophthalmic Paediatr Genet. 1982;1:169–172. [Google Scholar]
- 25.Kirwan C, O'Keefe M. Paediatric aphakic glaucoma. Acta Ophthalmol Scand. 2006;84:734–739. doi: 10.1111/j.1600-0420.2006.00733.x. [DOI] [PubMed] [Google Scholar]
- 26.Mathys R, Deconinck H, Keymolen K, Jansen A, Van Esch H. Severe visual impairment and retinal changes in a boy with a deletion of the gene for Nance-Horan syndrome. Bull Soc Belge Ophtalmol. 2007;305:49–53. [PubMed] [Google Scholar]
- 27.Favor J, Pretsch W. Genetic localization and phenotypic expression of X-linked cataract (Xcat) in Mus musculus. Genet Res. 1990;56:157–162. doi: 10.1017/s0016672300035242. [DOI] [PubMed] [Google Scholar]
- 28.Zhu D, Alcorn DM, Antonarakis SE, et al. Assignment of the Nance-Horan syndrome to the distal short arm of the X chromosome. Hum Genet. 1990;86:54–58. doi: 10.1007/BF00205172. [DOI] [PubMed] [Google Scholar]
- 29.Bergen AAB, ten Brink J, Schuurman EJM, Bleeker-Wagemakers EM. Nance-Horan syndrome: Linkage analysis in a family from The Netherlands. Genomics. 1994;21:238–240. doi: 10.1006/geno.1994.1248. [DOI] [PubMed] [Google Scholar]
- 30.Toutain A, Dessay B, Ronce N, et al. Refinement of the NHS locus on chromosome Xp22.13 and analysis of five candidate genes. Eur J Hum Genet. 2002;10:516–20. doi: 10.1038/sj.ejhg.5200846. [DOI] [PubMed] [Google Scholar]
- 31.Brooks SP, Ebenezer ND, Poopalasundaram S, et al. Refinement of the X-linked cataract locus (CXN) and gene analysis for CXN and Nance-Horan syndrome (NHS) Ophthalmic Genet. 2004;25:121–131. doi: 10.1080/13816810490514360. [DOI] [PubMed] [Google Scholar]
- 32.Brooks SP, Ebenezer ND, Poopalasundaram S, Lehmann OJ, Moore AT, Hardcastle AJ. Identification of the gene for Nance-Horan syndrome (NHS) J Med Genet. 2004;41:768–771. doi: 10.1136/jmg.2004.022517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saitou M, Ando-Akatsuka Y, Itoh M, et al. Mammalian occludin in epithelial cells: Its expression and subcellular distribution. Eur J Cell Biol. 1997;73:222–231. [PubMed] [Google Scholar]
- 34.Levin LS, Cortin P. X-linked cataract, microcornea, and dental anomalies. Birth Defects Origl Art Ser. 1976;13(3C):241. [Google Scholar]
- 35.Van Esch H, Jansen A, Bauters M, Froyen G, Fryns JP. Encephalopathy and bilateral cataract in a boy with an interstitial deletion of Xp22 comprising the CDKL5 and NHS genes. Am J Med Genet A. 2007;143:364–369. doi: 10.1002/ajmg.a.31572. [DOI] [PubMed] [Google Scholar]
- 36.Ramprasad VL, Thool A, Murugan S, et al. Truncating mutation in the NHS gene: Phenotypic heterogeneity of Nance-Horan syndrome in an Asian Indian family. Invest Ophthalmol Vis Sci. 2005;46:17–23. doi: 10.1167/iovs.04-0477. [DOI] [PubMed] [Google Scholar]
- 37.Toutain A, Ayrault AD, Moraine C. Mental retardation in Nance-Horan syndrome: Clinical and neuropsychological assessment in four families. Am J Med Genet. 1997;71:305–314. doi: 10.1002/(sici)1096-8628(19970822)71:3<305::aid-ajmg11>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]