

The collagen family of proteins
Collagens, the major extracellular matrix components in most vertebrate tissues, comprise a superfamily of proteins [1]. A total of 29 genetically distinct collagens have been described so far in the vertebrate tissues and designated by Roman numerals I-XXIX in order of their discovery [2, 3]. The collagen molecules consist of three subunit polypeptides, so-called α-chains, and while some collagens are homotrimers, others can be heterotrimers containing two or even three genetically distinct subunit polypeptides. Consequently, there are well over 40 genes in vertebrate tissues that encode the subunits polypeptides of different, genetically distinct collagen molecules [1, 2].
A characteristic structural feature of all collagens is the presence of a protein domain in triple-helical conformation which provides stability to these molecules to serve as structural building blocks providing integrity to connective tissues. The triple-helical conformation resists non-specific proteolysis, such as digestion with pepsin. The folding of the individual α-chains into the triple-helical conformation is predicated upon the characteristic primary sequence, consisting of repeating Gly-X-Y triplet sequences. In some collagens, such as in type I collagen, the most abundant collagen in the skin and bones, the central collagenous domain of individual α-chains contains an uninterrupted Gly-X-Y repeat segment spanning approximately 1000 amino acids. In some collagens, such as in type IV (the basement membrane collagen) and type VII (the anchoring fibril collagen), the Gly-X-Y repeat sequence contains imperfections which interrupt the triple-helical conformation [1]. These interruptions then provide flexibility to the rod-like collagen molecules and also provide sites susceptible to non-specific proteolytic cleavage of the primary sequence.
On the basis of their fiber architecture in tissues, the genetically distinct collagens have been divided into different subgroups [1]. Collagens types I, II, III, V, and X align into large extracellular fibrils and are designated as fibril-forming collagens. Type IV collagen is arranged in an interlacing network within the basement membranes, while type VI collagen forms distinct microfibrils, and type VII collagen forms anchoring fibrils. FACIT collagens (fibril-associated collagens with interrupted triple helices) [4] include types IX, XII, XIV, XIX, XX, and XXI. Several of the latter types of collagens associate with larger collagen fibers and serve as molecular bridges, stabilizing the organization of the extracellular matrix.
The major collagens in human skin are types I and III which account for approximately 80% and 10% of the total bulk of collagen, respectively (Table 1). These two collagens associate to form broad extracellular fibers characteristic for human dermis. Type V collagen is present in most connective tissues, including dermis, where it represents less than 5% of the total collagen. In dermis, type V collagen is located on the surface of the large collagen fibers, formed by type I and III collagens, and type V collagen regulates the lateral growth of these fibers. Another major collagen in the skin is type IV collagen, present within the dermal-epidermal junction as well as in the vascular basement membranes.
TABLE 1.
Genetic heterogeneity of collagen in human skin
| Collagen Type | Chain Composition | Supramolecular Assembly | Tissue Distribution c |
|---|---|---|---|
| I | [α1(I)]2α2(I) | Fibrillar | Dermis, bone, tendons |
| III | [α1(III)]3 | Fibrillar | Fetal dermis, blood vessels, GI tract |
| IV | [α1(IV)]2α2(IV) a | Basement membrane | Ubiquitous |
| V | [α1(V)]2α2(V) a | Fibrillar | Ubiquitous |
| VI | α1(VI)α2(VI)α3(VI) a | Microfibrils | Ubiquitous |
| VII | [α1(VII)]3 | Anchoring fibrils | Anchoring fibrils (see table 2) |
| VIII | [α1(VIII)]3 | Network forming | Endothelia |
| XIII | [α1(XIII)]3 | Transmembrane | Ubiquitous, including epidermis |
| XIV | [α1(XIV)]3 | FACIT b | Skin, cornea |
| XV | [α1(XV)]3 | Basement membrane | Ubiquitous |
| XVII | [α1(XVII)]3 | Transmembrane | Hemidesmosomes in skin, cornea, mucous membrane |
| XXIX | unknown | unknown | Epidermis |
Additional α-chains have been identified
Fibril-associated collagens with interrupted triple helices
Distribution in the skin and other major tissues is indicated; lesser amounts may be present in other tissues
In addition to these major collagens, human skin contains several minor collagens which demonstrate spatially restricted location, yet they play a critical role in providing integral stability to the skin (Table 1). One of them is type VII collagen, the major, if not the exclusive, component of anchoring fibrils [5]. Another one is type XVII collagen, a transmembrane collagen in type II topography [6]. Type XVII collagen resides in hemidesmosomes complexed with α6β4 integrin, plectin, and laminin-332 (laminin -5) [7]. Finally, type XXIX collagen has been recently reported to be a putative epidermal collagen with the highest level of expression in suprabasal layers [3]. This collagen has been suggested to play a role in atopic dermatitis but its characterization is currently incomplete.
The biology of type VII collagen
Type VII collagen was initially described as an extended, unusually long molecule, hence the original designation as long-chain (LC) collagen [8]. Rotary shadowing electron microscopy of type VII collagen molecules synthesized and secreted by human keratinocytes in culture revealed a long, 424 nm, triple-helical domain and flanking non-collagenous sequences (Fig. 1a). The amino-terminal domain was particularly noticeable with individual α-chains contributing an extended arm. At the same time, visualization of type VII collagen isolated by limited pepsin proteolysis of amniotic membranes revealed a 780-nm dimer of two identical molecules in anti-parallel orientation, with a 60-nm overlap stabilized by disulfide bonds (Fig. 1b, c) [5]. Further proteolytic digestion with pepsin revealed that type VII collagen molecules consists of a central collagenous, triple-helical segment flanked by the non-collagenous NC-1 and NC-2 domains. Subsequent cloning of the human type VII collagen and the corresponding cDNA indicated that the initially synthesized type VII collagen subunit polypeptide, the pro-α1(VII) chain is a complex modular protein consisting of a central, 1,530-amino acid triple-helical domain (Fig. 2a) [9, 10]. Unlike interstitial collagens, however, the repeating Gly-X-Y sequence is interrupted by 19 imperfections due to insertions or deletions of amino acids in the Gly-X-Y repeat sequence. Most notably, in the middle of the triple-helical domain, there is a 39-amino acid non- collagenous “hinge” region which is susceptible to proteolytic digestion with pepsin. The amino-terminal NC-1 domain of type VII collagen [NC-1(VII)], approximately 145 kDa in size, consists of sub-modules with homology to known adhesive proteins, including segments with homology to cartilage matrix protein (CMP), nine consecutive fibronectin type III-like (FN-III) domains, a segment with homology to the A domain of von Willebrand factor, and a short cysteine and proline-rich region [11]. The carboxy-terminal non-collageneous domain, NC-2, is relatively small, ~30kDa, and it contains a segment with homology to Kunitz protease inhibitor molecule (Fig. 2a) [10, 12].
Figure 1.

Structural features of newly synthesized type VII collagen. (A) Rotary shadowing image of a type VII collagen molecule synthesized and secreted by human keratinocytes in culture. Note the central collagenous domain, flanked by non-collagenous NC-1 and NC-2 sequences. (B) Identification of NC-1 domains at both ends of a type VII collagen dimer molecule, as visualized by a monoclonal anti-type VII collagen antibody in rotary shadowing image. Note that the dimer has an overlapping region of the carboxy-terminal ends of the two molecules, as schematically illustrated in C. (Adapted from Sakai LY, Keene DR, Morris NP, et al. Type VII collagen is a major structural component of anchoring fibrils. J Cell Biol 1986; 103: 1577-86. and Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol 1993; 101: 252–5., with permission).
Figure 2.
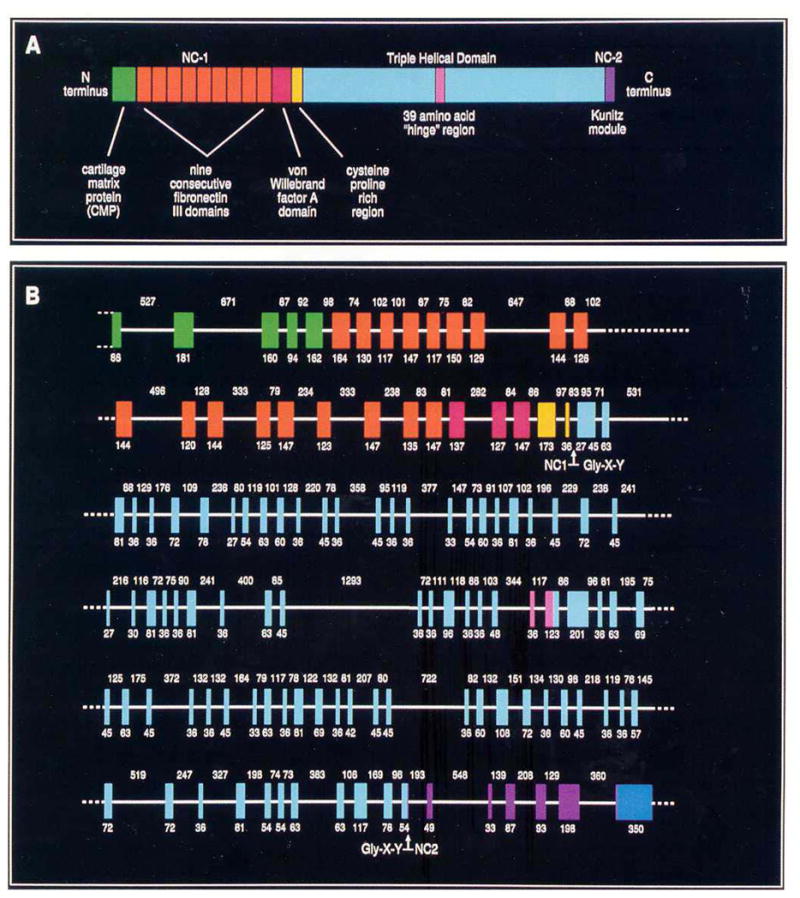
Domain organization of type VII collagen polypeptides and the intron-exon organization of the corresponding gene, COL7A1. (A) The amino acid sequence of the pro-α1(VII) polypeptide, as deduced from cDNA sequences, indicates that type VII collagen consists of triple-helical central domain containing a 39-amino acid non-collagenous “hinge” region. The triple-helical domain is flanked by amino-terminal (NC-1) and carboxy-terminal (NC-2) non-collagenous domains. The NC-1 domain consists of submodules with homology to known adhesive proteins. The NC-2 domain contains a segment with homology to the Kunitz protease inhibitor molecule. (B) The intron-exon organization of COL7A1 reveals that the gene consists of 118 distinct exons (vertical colored blocks) separated by relatively small introns (horizontal white lines). The sizes (in base pairs) of the exons and introns are indicated below and above the gene structure, respectively. The exons encoding distinct protein domains within the type VII collagen polypeptides, as shown in Panel A, are color-matched. (Adapted from Christiano AM, Uitto J. Impact of molecular genetic diagnosis on dystrophic epidermolysis bullosa. Current Op Dermatol 1996; 3: 225–32., with permission).
Cloning of the human type VII collagen gene, COL7A1, revealed a complex structure consisting of a total of 118 separate exons (Fig. 2b) [9]. The gene is, however, relatively compact, and most of the intervening sequences (introns) are relatively small; consequently, the size of the entire human COL7A1 gene is only ~32 kb, encoding a messenger RNA of ~8.9 kb. COL7A1 has been mapped to the short-arm of human chromosome 3, region 3p21.1 [13]. At the time of the report of its structural organization, the type VII collagen gene was noted to be composed of more exons than any previously characterized gene [9]. At the same time, most of the COL7A1 introns are small, including a 71 nucleotide intron that was the smallest intron yet reported in a collagen gene. The type VII collagen gene structure and the encoded primary sequence of the protein are well conserved, and for example, the mouse gene shows 84.7 percent homology at the nucleotide and 90.4 percent identity at the protein level, attesting to the importance of type VII collagen as a structural protein [14].
Type VII collagen gene expression displays a restricted, tissue-specific pattern. Specifically, type VII collagen has been localized by immunomapping to a select number of epthelia, including human skin, and the presence of type VII collagen correlated with the presence of ultrastructurally detected anchoring fibrils (Table 2) [5]. The expression of the type VII collagen gene can be modulated by a number of cytokines, and in particular, transforming growth factor-β is a powerful upregulator of COL7A1 in fibroblasts and keratinocytes, the regulation taking place primarily at the transcriptional level [15, 16].
TABLE 2.
Correlation of the presence of type VII collagen as detected by immunofluorescence with anchoring fibrils detected ultrastructurally
| Immunofluorescence | Anchoring fibrils | |
|---|---|---|
| Skin | + | + |
| Chorioamnion | + | + |
| Placenta | - | - |
| Skeletal muscle | - | - |
| Cornea (Bowman’s membrane) | + | + |
| Oral mucosa | + | + |
| Cervix | + | + |
| Oesophagus | + | + |
| Anal canal | + | + |
| Kidney cortex | - | - |
| Lung alvoli | - | - |
| Liver sinusoids | - | - |
| Stomach (fundus) | - | - |
| Large intestine | - | - |
| Elastic cartilage | - | - |
Modified from reference [5]
Type VII collagen is a major component of the anchoring fibrils
Anchoring fibrils are specialized attachment complexes at epithelium/mesenchyme interface in a number of tissues (Table 2). In human skin, anchoring fibrils extend from the lower portion of the dermal-epidermal basement membrane to the underlying upper papillary dermis (Fig. 3). Initially, it was suggested that anchoring fibrils attach at one end to the lamina densa of the cutaneous basement membrane and at the other end to basement membrane-like structures, so-called anchoring plaques, which were thought to reside within the upper papillary dermis [8]. Subsequent immunoelectron microscopic analyses indicated, however, that most, if not all, anchoring fibrils attach at both ends to the lamina densa, allowing entrapment of interstitial collagen fibers into the U-shaped structures (Fig. 3) [17]. In retrospect, the appearance of “anchoring plaques” in the upper papillary dermis may be an artifact resulting from preparation of skin samples for electron microscopy.
Figure 3.

Schematic presentation of the synthesis of pro-α1(VII) collagen polypeptides and their assembly into anchoring fibrils under physiological conditions (left side of the figure) and perturbations in these processes leading to dystrophic epidermolysis bullosa (right side). Within the intracellular (IC) space of cells, such as keratinocytes and fibroblasts, pro-α1(VII) polypeptides are synthesized on ribosomes (I). Three polypeptides associate through their carboxy-terminal ends, and their collagenous domains fold into a characteristic triple-helical conformation (II and III). After secretion into the extracellular (EC) space, triple-helical typeVII collagen molecules form anti-parallel dimers (IV), and after proteolytic removal of a part of the carboxy-terminal end, the dimer assembly is stabilized by intermolecular disulfide bonds (V). Subsequently, a large number of dimer molecules laterally assemble in register to form cross- striated, centro-symmetric anchoring fibrils (VI). The amino-terminal non-collagenous globular domains (NC-1) attach to the extracellular macromolecules of the lamina densa, stabilizing the association of the lower part of the dermo-epidermal basement membrane to the underlying dermis. Mutations in the COL7A1 gene can result in premature termination codons (PTCs), manifesting with the severe Hallopeau-Siemens (HS)-type recessive dystrophic epidermolysis bullosa (RDEB) when present in both alleles. Recessive missense mutations can interfere with chain assembly (a), triple helix formation (b), or stability of the triple-helix (c), resulting in mild (mitis, M) non-HS-RDEB. Glycine substitutions in the collagenous domain destabilize the triple helix and can result through dominant-negative interference in dominantly inherited DEB (DDEB). (Adapted from Varki R, Sadowski S, Uitto J, et al. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype/genotype correlations in the dystrophic subtypes. J Med Genet 2007; 44: 181–92., with permission).
Type VII collagen is synthesized both by epidermal keratinocytes and dermal fibroblasts in culture (Fig. 4) [18]. Upon synthesis of complete pro-α1(VII) polypeptides, three polypeptides associate through their carboxy-terminal ends to a trimer molecule which in its collagenous portion folds into the triple-helical formation (Fig. 3). The triple-helical molecules are then secreted to the extracellular milieu where two type VII collagen molecules align into an nti-parallel dimer with the amino-terminal domains present at both ends of the molecule (Figs. 1b, c and 3) [5]. This dimer assembly is accompanied by proteolytic removal of a portion of the carboxy-terminal end of both type VII collagen molecules and stabilization by inter-molecular disulfide bond formation (Fig. 3) [19]. Subsequently, a large number of these anti-parallel dimers aggregate laterally to form anchoring fibrils that can be recognized by transmission electron microscopy of the skin through their characteristic, centro-symmetric banding patterns.
Figure 4.
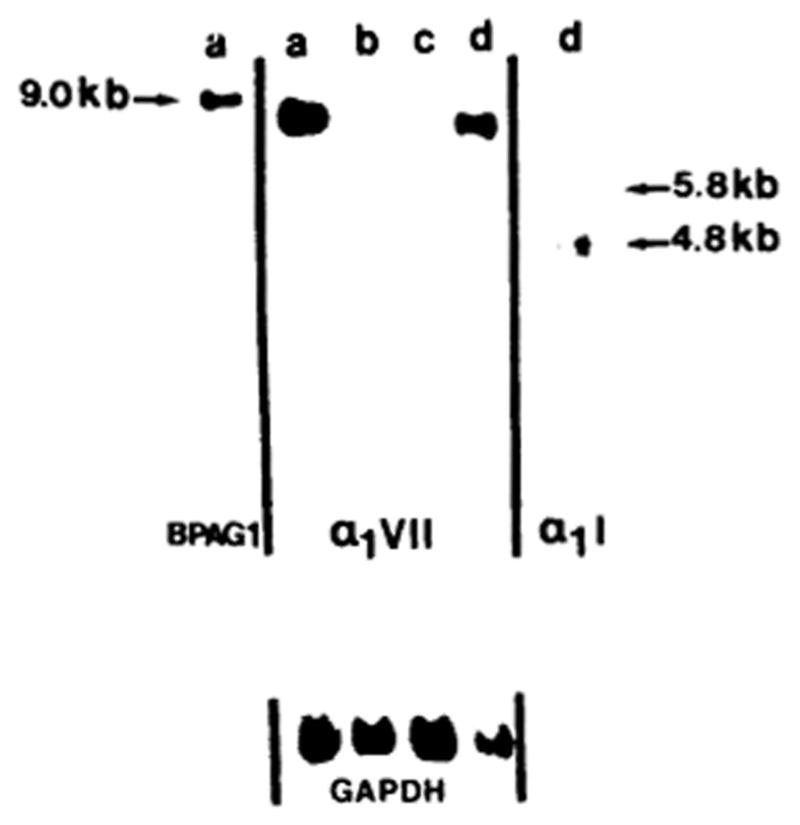
Demonstration of type VII collagen gene expression in epidermal keratinocytes (lane a), Ras oncogene transformed human epidermal keratinocytes (RHK) (lane b), human papilloma virus-transformed epidermal keratinocytes (HPK) (lane c), and skin fibroblasts (lane d). Total mRNA was isolated from the cultured cells and subjected to Northern hybridization with a human type VII collagen cDNA probe (middle panel). Note the expression of human type VII collagen mRNA of approximately 8.9 kb in epidermal keratinocytes and dermal fibroblasts (a, d). For reference, hybridizations were performed with the 230-kDa bullous pemphigoid antigen (BPAG1) and type I collagen (α1I) cDNAs. Rehybridization of the same filter got the GAPDH “housekeeping” gene served as an internal control for RNA loading. (Adapted from Ryynänen J, Sollberg S, Parente MG, et al. Type VII collagen gene expression by cultured human cells and in fetal skin. Abundant mRNA and protein levels in epidermal keratinocytes. J Clin Invest 1992; 89: 163–8., with permission).
Type VII collagen is a major component of the anchoring fibrils which provide stability to the dermal-epidermal adhesion on the dermal site at the lamina lucida/papillary dermis interface [5]. This stability has been attributed to the affinity of the NC-1(VII) domain to bind the principal components of the cutaneous basement membrane, laminin-332 (laminin-5), laminin-311 (laminin-6), and type IV collagen [20, 21]. Kinetic assays of such associations have demonstrated that the binding of the NC-1(VII) domain to laminin-332 and collagen IV are of high affinity, and the NC-1 domain utilizes the same region to bind both of these macromolecules (Fig. 5a, b). In contrast, the NC-1(VII)-mediated binding to type I collagen is relatively weak (Kd value of ~10−6 M) [21]. Thus, the high affinity binding of type VII collagen, articularly at the NC-1(VII) domains, appears to facilitate stabilization of the structure of the basement membrane zone, and type VII collagen interactions with the interstitial collagen fibers in the dermis, consisting primarily of type I, III, and V collagens, may be due to physical entrapment of these fiber structures (Fig. 5b, c) [21, 22].
Figure 5.
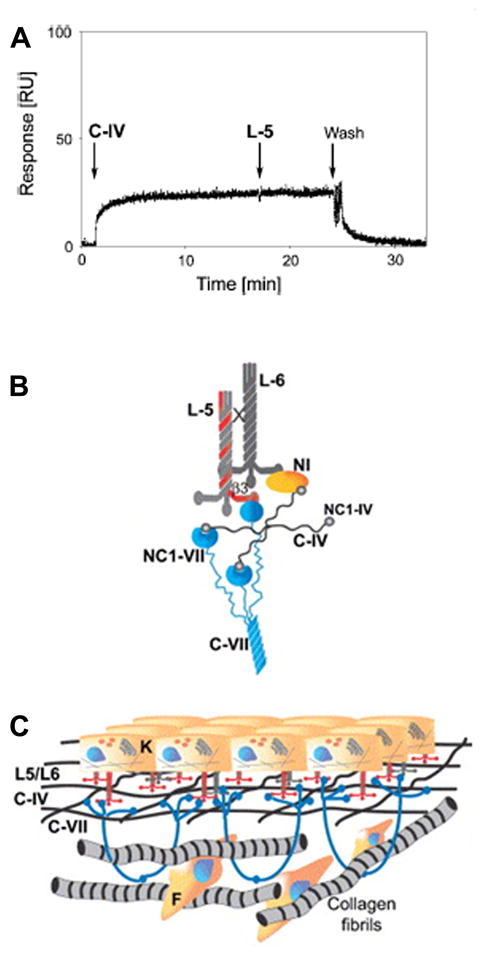
Demonstration of the binding of type VII collagen to type IV collagen, and schematic representation of the dermal-epidermal junction with type VII collagen binding to basement membrane macromolecules. (A) Kinetic biosensor analysis demonstrates that recombinant NC-1 domain of human type VII collagen binds to type IV collagen (C-IV). After a wash, the NC-1/C-IV complex dissociates indicating reversible nature of the binding. Addition of laminin-332 (laminin-5, L-5) did not result in additional binding, suggesting that the L-5 binding site corresponds to or is located close to that for binding of type IV collagen. (B) Binding of the NC-1-VII collagen domains to C-IV, L-5, and laminin-6 (L-6). (C) The anchoring fibrils entrap type VII collagen fibers, stabilizing the association of lamina densa of the dermo-epidermal basement membrane to underlying dermis. (Adapted from Brittingham R, Uitto J, Fertala A. High-affinity binding of the NC-1 domain of collagen VII to laminin 5 and collagen IV. Biochem Biophys Res Commun 2006; 343: 692–9., with permission).
The pathologic consequences of type VII collagen gene mutations
Considering the complexity of type VII collagen gene and protein structures, and the critical importance of its distinct domains in macromolecular interactions, one would have initially redicted that mutations in the COL7A1 gene can have clinical consequences in terms of integrity of the skin. The dystrophic forms of epidermolysis bullosa (DEB) emerged as candidate diseases for type VII collagen mutations when immunofluorescent staining of skin of patients with the most severe recessive dystrophic EB (RDEB) demonstrated lack of type VII collagen epitopes [23]. At the same time, anchoring fibrils were shown by ultrastructural analysis to be morphologically altered, reduced in number, or completely absent in patients with different forms of DEB [24, 25]. The cloning of human type VII collagen gene and cDNAs then provided the opportunity to assess the hypothesis that type VII collagen serves as the candidate gene/protein system for this group of blistering disorders.
Initial cloning of the COL7A1 gene provided the tools to perform genetic linkage analyses in families with DEB and to explore the possibility that the inheritance of the disease in these families is linked to a specific genetic markers which allows tracking of the segregation of COL7A1 alleles through the family pedigrees. One of the early markers was an intragenic PvuII restriction fragment length polymorphism (RFLP) which was shown to reflect a single base-pair substitution within the type VII collagen gene in exon 21. This polymorphism occurs within the third nucleotide of a redundant proline codon, and, consequently, does not change the amino acid within the encoded polypeptide. The first genetic linkage analysis was performed with this marker in a large dominantly inherited DEB family (Fig. 6). In all cases within this family, there was complete co-segregation of the inheritance of one COL7A1 allele and the DDEB phenotype [26]. The subsequent examination of 14 families with DDEB resulted in the combined LOD score of Ẑ = 41.42 at θ= 0, establishing a robust linkage between the type VII collagen gene and the disease locus causing skin fragility in DEB [27]. Similar genetic linkage studies were subsequently performed in families with RDEB, particularly with the most severe, Hallopeau- Siemens type [28, 29]. Again, an unequivocal genetic linkage between COL7A1 and the disease locus in RDEB was established. These early linkage studies were consistent with the notion that the majority of, if not all, cases with the dystrophic forms of EB are the result of mutations in the COL7A1 gene.
Figure 6.

Demonstration of genetic linkage between the dominant dystrophic epidermolysis bullosa phenotype and a type VII collagen allele in a family with 20 affected and 22 unaffected living individuals in four generations. The type VII collagen allele was tracked by inheritance of a PvuII polymorphic marker. Note that the disease allele tracks with the B-allele of COL7A1. (Adapted from Ryynänen M, Knowlton RG, Parente MG, et al. Human type VII collagen: genetic linkage of the gene (COL7A1) on chromosome 3 to dominant dystrophic epidermolysis bullosa. Am J Hum Genet 1991; 49: 797–803., with permission).
Subsequent development of streamlined mutation detection strategies [30, 31] has allowed examination of a large number of cases with dystrophic forms of EB with respect to type VII collagen mutations. In fact, well over 300 distinct mutations in the COL7A1 gene have now been disclosed [32, 33]. The types of mutations range from premature termination codon (PTC)-causing mutations as a result of nonsense mutations, small insertions or deletions or splice junction mutations resulting in frame shift of translation, to more subtle missense mutations. In fact, genotype/phenotype correlations in general terms have been established [34]: In recessively inherited forms of DEB, presence of PTC-causing mutations in both alleles results in complete absence of type VII collagen, manifesting with severe mutilating scarring and blistering (Fig. 7). Combinations of a PTC-causing mutation with a more subtle missense mutation can result in milder autosomal recessive form of DEB. Most of the dominantly inherited cases of DEB result from glycine substitution mutations in the collagenous domain replacing one of the glycines in the Gly-X-Y repeat triplet sequence (Fig. 7). Collectively, the precise degree of severity of DEB reflects the combinations of mutations in COL7A1 and their consequences at the mRNA and protein levels, combined with the effects of modifier genes on the individuals’ genetic background and the exposure to environmental trauma [34]. (For details on phenotypic presentations and genetic basis of different forms of DEB, see Chapter --, Leena Bruckner-Tuderman; in this issue.)
Figure 7.

Schematic presentation of molecular mechanisms leading to a spectrum of phenotypic severity and different types of inheritance in families with dystrophic forms of EB. (Adapted from Christiano AM, Uitto J. Molecular diagnosis of inherited skin diseases: the paradigm of dystrophic epidermolysis bullosa. Adv Dermatol 1996; 11: 199–214., with permission).
Circulating autoantibodies to type VII collagen in patients with EB Acquisita (EBA)
In addition to inherited forms of EB, the acquired form of epidermolysis bullosa (EBA) involves pathology in type VII collagen. Specifically, circulating autoantibodies in patients with EBA recognize epitopes in type VII collagen molecules, and molecular cloning of the type VII collagen cDNAs again provided the tools to identify the most predominant immunoepitopes within the amino-terminal NC-1 domain of type VII collagen (Fig. 8) [35, 36]. The antigenic properties of the NC-1(VII) domain are further highlighted by the fact that monoclonal antibodies, such as H3A and L3D, which are in clinical use to map type VII collagen in the skin of patients with inherited forms of EB, also identify epitopes in this portion of the protein (Fig. 8) [36]. In addition to circulating autoantibodies recognizing type VII collagen epitopes in EBA, bullous lesions in some patients with systemic lupus erythematosus have also been associated with anti-type VII collagen antibodies [36, 37].
Figure 8.
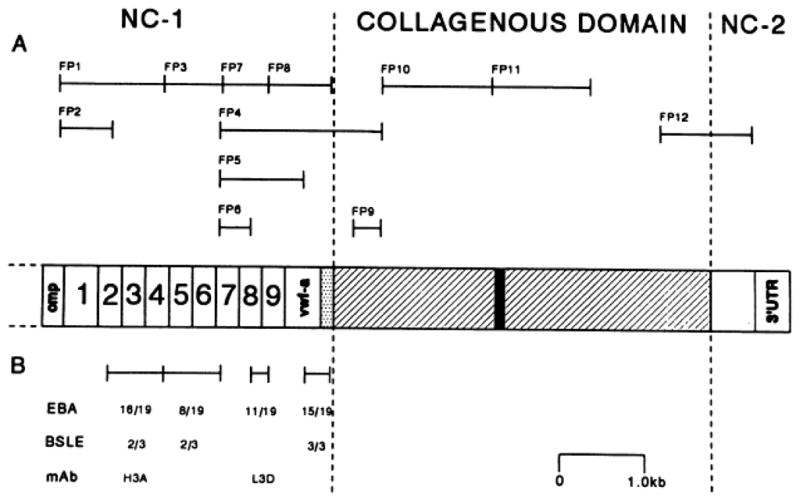
Mapping of antigenic epitopes in type VII collagen recognized by autoantibodies in patients with acquired EB (EBA) and bullous systemic lupus erythematosus (BSLE). (A) the positions of fusion proteins that correspond to the type VII collagen sequences are indicated by brackets. (B) The number of sera positive among the 19 EBA and 3 BSLE patients is indicated by the regions tested using recombinant fusion-proteins. Also, note the areas recognized by two monoclonal antibodies, H3A and L3D. (Adapted from Lapière JC, Woodley DT, Parente MG, et al. Epitope mapping of type VII collagen: identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J Clin Invest 1993; 92: 1831–9., with permission).
The role of type VII collagen in epidermal squamous cell carcinoma
Type VII collagen has also been suggested to be required for human epidermal tumorigenesis. This suggestion relates to the observation that with extended life span of the affected individuals with RDEB, an increasing number of life-threatening complications related to development of squamous cell carcinomas (SCCs) has emerged [38]. The RDEB-associated SCCs usually manifest early in life, and they are distinguished by a particularly aggressive clinical course. These tumors have a very high rate of metastatic spread often leading to early demise of the affected individual. The association of type VII collagen expression and development of SCCs in RDEB derived from observations on tumorigenic conversion of keratinocytes cultured from these patients and xenotransplanted to immunodeficient mice [39]. Those keratinocytes expressing NC-1(VII) domain developed cancer while those keratinocytes which did not express the same domain did not develop SCCs. A molecular mechanism potentially involving “anchorless” NC-1 activation of α6β4 integrin-mediated signal transduction as a result of terminally truncated NC-1/laminin-332 interactions has also been proposed (Fig. 9) [40].
Figure 9.
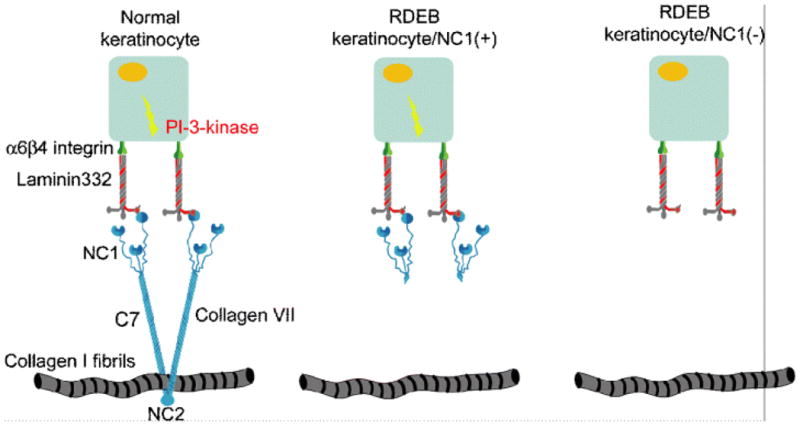
Schematic representation of “anchorless” activation of α6β4 integrin-mediated signal transduction by NC-1(VII) in RDEB keratinocytes. In normal skin, type VII collagen is firmly anchored to the basement membrane zone through interactions with other components of the extracellular matrix. Thus, activation of α6β 4 integrin signaling is restricted to the appropriate tissue compartment within the epidermis (left panel). In contrast, expression of N-terminally truncated type VII collagen lacking the collagenous and the carboxy-terminal domains but depicting the presence of NC-1 may enable α6β4 integrin dependent signal transduction in RDEB keratinocytes which are not firmly anchored in the BMZ, potentially supporting inappropriate cell survival during invasion and metastasis (middle panel). In the case of complete absence of NC-1 expression, activation of α6β4 integrin-dependent signal transduction will not occur (right panel) (Adapted from Rodeck U, Fertala A, Uitto J. Anchorless keratinocyte survival. An emerging pathogenic mechanism for squamous cell carcinoma in recessive dystrophic epidermolysis bullosa. Exp Dermatol 2007; 16: 465–7., with permission).
It should be noted that the notion that NC-1(VII) expression is required for SCC development in RDEB has been challenged by isolation of keratinocytes from RDEB patients with SCC yet with complete absence of type VII collagen [41]. It should also be noted that NC-1-dependent tumor formation has been described only in keratinocytes that were immortalized by co-expression of Ha-RasV12 and mutant IκBα to inhibit cellular NF-κB activity [39]. Nevertheless, the importance of the suggestion for the role of type VII collagen in SCC development in patients with RDEB is emphasized by the fact that the lack of information of the pathomechanistic features of SCC has precluded rational development of targeted therapies for this complication of DEB. Meanwhile, significant progress has been made towards development of molecular therapies to ameliorate, and eventually to provide cure for this, currently intractable, disease with significant morbidity and mortality [42, 43].
Acknowledgments
GianPaolo Guercio assisted in preparation of this manuscript. The authors’ original research summarized in this article was supported by DHHS, NIH/NIAMS grants P01 AR38923 and R01 AR54876, and by the Dermatology Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Uitto J, Chu ML, Gallo R, et al. Collagen, elastic fibers, and the extracellular matrix of the dermis. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, editors. Fitzpatrick’s Dermatology in General Medicine. 7. New York: McGraw-Hill; 2008. pp. 517–42. [Google Scholar]
- 2.Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Söderhäll C, Marenholz I, Kerscher T, et al. Variants in a novel epidermal collagen gene (COL29A1) are associated with atopic dermatitis. PLoS Biol. 2007;5:e242. doi: 10.1371/journal.pbio.0050242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olsen BR. New insights into the function of collagens from genetic analysis. Curr Opin Cell Biol. 1995;7:720–7. doi: 10.1016/0955-0674(95)80115-4. [DOI] [PubMed] [Google Scholar]
- 5.Sakai LY, Keene DR, Morris NP, et al. Type VII collagen is a major structural component of anchoring fibrils. J Cell Biol. 1986;103:1577–86. doi: 10.1083/jcb.103.4.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franzke CW, Tasanen K, Schäcke H, et al. Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J. 2002;21:5026–35. doi: 10.1093/emboj/cdf532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Pereda JM, Lillo MP, Sonnenberg A. Structural basis of the interaction between integrin alpha6beta4 and plectin at the hemidesmosomes. EMBO J. 2009;28:1180–90. doi: 10.1038/emboj.2009.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol. 1993;101:252–5. doi: 10.1111/1523-1747.ep12365129. [DOI] [PubMed] [Google Scholar]
- 9.Christiano AM, Hoffman GG, Chung-Honet LC, et al. Structural organization of the human type VII collagen gene (COL7A1), comprised of more exons than any previously characterized gene. Genomics. 1994;21:169–79. doi: 10.1006/geno.1994.1239. [DOI] [PubMed] [Google Scholar]
- 10.Christiano AM, Greenspan DS, Lee S, et al. Cloning of human type VII collagen. complete primary sequence of the α1(VII) chain and identification of intragenic polymorphisms. J Biol Chem. 1994;269:20256–62. [PubMed] [Google Scholar]
- 11.Christiano AM, Rosenbaum LM, Chung-Honet LC, et al. The large non-collagenous domain (NC-1) of type VII collagen is amino-terminal and chimeric. Homology to cartilage matrix protein, the type III domains of fibronectin and the A domains of von Willebrand factor. Hum Mol Genet. 1992;1:475–81. doi: 10.1093/hmg/1.7.475. [DOI] [PubMed] [Google Scholar]
- 12.Greenspan DS. The carboxyl-terminal half of type VII collagen, including the non-collagenous NC-2 domain and intron/exon organization of the corresponding region of the COL7A1 gene. Hum Mol Genet. 1993;2:273–8. doi: 10.1093/hmg/2.3.273. [DOI] [PubMed] [Google Scholar]
- 13.Parente MG, Chung LC, Ryynänen J, et al. Human type VII collagen: cDNA cloning and chromosomal mapping of the gene. Proc Natl Acad Sci USA. 1991;88:6931–5. doi: 10.1073/pnas.88.16.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kivirikko S, Li K, Christiano AM, et al. Cloning of mouse type VII collagen reveals evolutionary conservation of functional protein domains and genomic organization. J Invest Dermatol. 1996;106:1300–6. doi: 10.1111/1523-1747.ep12349019. [DOI] [PubMed] [Google Scholar]
- 15.Ryynänen J, Sollberg S, Olsen DR, et al. Transforming growth factor-beta up-regulates type VII collagen gene expression in normal and transformed epidermal keratinocytes in culture. Biochem Biophys Res Commun. 1991;180:673–80. doi: 10.1016/s0006-291x(05)81118-0. [DOI] [PubMed] [Google Scholar]
- 16.Vindevoghel L, Kon A, Lechleider RJ, et al. Smad-dependent transcriptional activation of human type VII collagen gene (COL7A1) promoter by transforming growth factor-β. J Biol Chem. 1998;273:13053–7. doi: 10.1074/jbc.273.21.13053. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu H, Ishiko A, Masunaga T, et al. Most anchoring fibrils in human skin originate and terminate in the lamina densa. Lab Invest. 1997;76:753–63. [PubMed] [Google Scholar]
- 18.Ryynänen J, Sollberg S, Parente MG, et al. Type VII collagen gene expression by cultured human cells and in fetal skin. Abundant mRNA and protein levels in epidermal keratinocytes. J Clin Invest. 1992;89:163–8. doi: 10.1172/JCI115557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colombo M, Brittingham RJ, Klement JF, et al. Procollagen VII self-assembly depends on site-dpecific interactions and is promoted by cleavage of the NC-2 domain with procollagen C-proteinase. Biochemistry. 2003;42:11434–42. doi: 10.1021/bi034925d. [DOI] [PubMed] [Google Scholar]
- 20.Chen M, Marinkovich MP, Veis A, et al. Interactions of the amino-terminal noncollagenous (NC-1) domain of type VII collagen with extracellular matrix components. A potential role in epidermal-dermal adherence in human skin. J Biol Chem. 1997;272:14516–22. doi: 10.1074/jbc.272.23.14516. [DOI] [PubMed] [Google Scholar]
- 21.Brittingham R, Uitto J, Fertala A. High-affinity binding of the NC-1 domain of collagen VII to laminin 5 and collagen IV. Biochem Biophys Res Commun. 2006;343:692–9. doi: 10.1016/j.bbrc.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 22.Villone D, Fritsch A, Koch M, et al. Supramolecular interactions in the dermo-epidermal junction zone: anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J Biol Chem. 2008;283:24506–13. doi: 10.1074/jbc.M802415200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruckner-Tuderman L, Mitsuhashi Y, Schnyder UW, et al. Anchoring fibrils and type VII collagen are absent from skin in severe recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 1989;93:3–9. doi: 10.1111/1523-1747.ep12277331. [DOI] [PubMed] [Google Scholar]
- 24.Tidman MJ, Eady RA. Evaluation of anchoring fibrils and other components of the dermal-epidermal junction in dystrophic epidermolysis bullosa by a quantitative ultrastructural technique. J Invest Dermatol. 1985;84:374–7. doi: 10.1111/1523-1747.ep12265460. [DOI] [PubMed] [Google Scholar]
- 25.McGrath JA, Ishida-Yamamoto A, O’Grady A, et al. Structural variations in anchoring fibrils in dystrophic epidermolysis bullosa: correlation with type VII collagen expression. J Invest Dermatol. 1993;100:366–72. doi: 10.1111/1523-1747.ep12471830. [DOI] [PubMed] [Google Scholar]
- 26.Ryynänen M, Knowlton RG, Parente MG, et al. Human type VII collagen: genetic linkage of the gene (COL7A1) on chromosome 3 to dominant dystrophic epidermolysis bullosa. Am J Hum Genet. 1991;49:797–803. [PMC free article] [PubMed] [Google Scholar]
- 27.Uitto J, Christiano AM. Molecular basis for the dystrophic forms of epidermolysis bullosa: mutations in the type VII collagen gene. Arch Dermatol Res. 1994;287:16–22. doi: 10.1007/BF00370713. [DOI] [PubMed] [Google Scholar]
- 28.Hovnanian A, Duquesnoy P, Blanchet-Bardon C, et al. Genetic linkage of recessive dystrophic epidermolysis bullosa to the type VII collagen gene. J Clin Invest. 1992;90:1032–6. doi: 10.1172/JCI115916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunnill MG, Richards AJ, Milana G, et al. Genetic linkage to the type VII collagen gene (COL7A1) in 26 families with generalised recessive dystrophic epidermolysis bullosa and anchoring fibril abnormalities. J Med Genet. 1994;31:745–8. doi: 10.1136/jmg.31.10.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pulkkinen L, Uitto J. Mutation analysis and molecular genetics of epidermolysis bullosa. Matrix Biol. 1999;18:29–42. doi: 10.1016/s0945-053x(98)00005-5. [DOI] [PubMed] [Google Scholar]
- 31.Christiano AM, Hoffman GG, Zhang X, et al. Strategy for identification of sequence variants in COL7A1 and a novel 2-bp deletion mutation in recessive dystrophic epidermolysis bullosa. Hum Mutat. 1997;10:408–14. doi: 10.1002/(SICI)1098-1004(1997)10:5<408::AID-HUMU12>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Varki R, Sadowski S, Uitto J, et al. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype/genotype correlations in the dystrophic subtypes. J Med Genet. 2007;44:181–92. doi: 10.1136/jmg.2006.045302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553–68. doi: 10.1111/j.1600-0625.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- 34.Uitto J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin Dermatol. 2005;23:33–40. doi: 10.1016/j.clindermatol.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 35.Woodley DT, Briggaman RA, O’Keefe EJ, et al. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007–13. doi: 10.1056/NEJM198404193101602. [DOI] [PubMed] [Google Scholar]
- 36.Lapière JC, Woodley DT, Parente MG, et al. Epitope mapping of type VII collagen: identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J Clin Invest. 1993;92:1831–9. doi: 10.1172/JCI116774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol. 1999;135:569–73. doi: 10.1001/archderm.135.5.569. [DOI] [PubMed] [Google Scholar]
- 38.Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Ortiz-Urda S, Garcia J, Green CL, et al. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science. 2005;307:1773–6. doi: 10.1126/science.1106209. [DOI] [PubMed] [Google Scholar]
- 40.Rodeck U, Fertala A, Uitto J. Anchorless keratinocyte survival. An emerging pathogenic mechanism for squamous cell carcinoma in recessive dystrophic epidermolysis bullosa. Exp Dermatol. 2007;16:465–7. doi: 10.1111/j.1600-0625.2007.00563.x. [DOI] [PubMed] [Google Scholar]
- 41.Pourreyron C, Cox G, Mao X, et al. Patients with recessive dystrophic epidermolysis bullosa develop squamous-cell carcinoma regardless of type VII collagen expression. J Invest Dermatol. 2007;127:2438–44. doi: 10.1038/sj.jid.5700878. [DOI] [PubMed] [Google Scholar]
- 42.Uitto J. Progress in heritable skin diseases: Translational implications of mutation analysis and pospects of molecular therapies. Acta Derm Venereol. 2009;89:228–35. doi: 10.2340/00015555-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamai K, Yasufumi K, Uitto J. Molecular therapies for heritable blistering diseases. Trends Mol Med. 2009;15:285–92. doi: 10.1016/j.molmed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 44.Christiano AM, Uitto J. Impact of molecular genetic diagnosis on dystrophic epidermolysis bullosa. Current Op Dermatol. 1996;3:225–32. [Google Scholar]
- 45.Christiano AM, Uitto J. Molecular diagnosis of inherited skin diseases: the paradigm of dystrophic epidermolysis bullosa. Adv Dermatol. 1996;11:199–214. [PubMed] [Google Scholar]