

Agelastatin A and its congeners are a structurally intriguing class of bromopyrrole-based natural products comprised of a densely functionalized cyclopentane core adorned with four contiguous nitrogen substituent groups (Figure 1).[1] Agelastatin A and B were first isolated in 1993 from the Coral Sea marine sponge Agelas dendromorpha.[2] Subsequently, agelastatin C and D were identified in extracts from the Australian sponge Cymbastela sp.[3] The unique structural features of these compounds together with their powerful cytotoxic activities against certain human cancer cell lines have fueled efforts aimed at their de novo synthesis.[4,5] To date, seven completed syntheses of agelastatin A have appeared, each presenting a decidedly different strategy for assembly of the natural product.[6,7] For our purpose, structures such as agelastatin A serve to inspire the development of new catalytic methods for oxidative C–N bond formation. In this report, we detail an 11-step synthesis of this natural product made possible with the advent of a highly selective and efficient intramolecular olefin aziridination method.[8,9] The unique heterocyclic intermediate generated in this sequence is easily manipulated through two selective nucleophilic ring-opening reactions to afford the substituted cyclopentane core of the target. The finished work offers a flexible and highly efficient preparation of (–)-agelastatin A, easily amenable to analogue design.[10]
Figure 1.
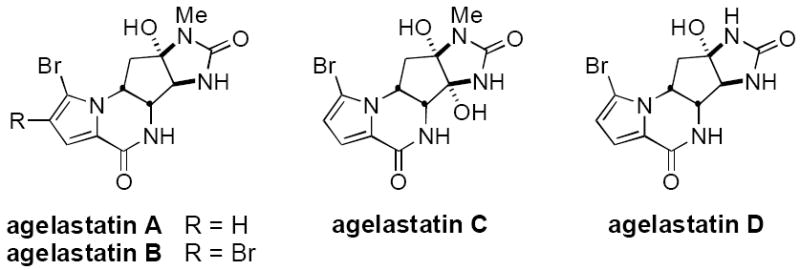
The agelastatin family of natural products.
Recent work from our lab and others has demonstrated that homoallyl and bis-homoallyl sulfamate esters react under oxidative conditions to furnish unique bicyclic aziridine derivatives (Figure 2).[8,11,12] This process generally affords high levels of diastereocontrol with both cyclic and acyclic starting materials. The products can be smoothly converted to polyfunctionalized amine derivatives through sequential, regioselective ring opening. For the purpose of assembling (–)-agelastatin A, an attractive plan emerged that would capitalize on such a sequence of steps to establish the trans-substituted vicinal diamine unit embedded at C4 and C8 (Figure 3). Prior to initiating these investigations, we had little sense if a substrate such as 3 would undergo chemoselective oxidation to generate the unusual tricyclic structure 2 and whether such a product would be isolable. Selectivity in the subsequent aziridine displacement reaction presented an additional concern. This plan, however, could be quickly assessed due to the ready availability of sulfamate 3.
Figure 2.
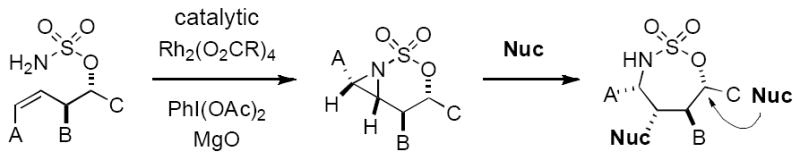
Rh-catalyzed aziridination: a versatile method for assembling polyfunctionalized amines.
Figure 3.
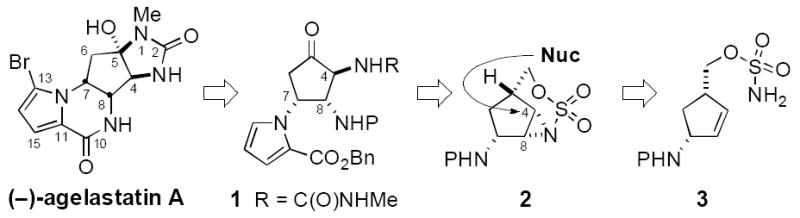
Retrosynthetic analysis of (–)-agelastatin A.
Optically enriched lactam 4 is prepared on industrial scale and may be obtained in either antipode at a relatively inexpensive cost (Figure 4).[13] In two high yielding transformations, this material can be converted to alcohol 5, also an item of commerce. Sulfamoylation of 5 following a standard protocol that involves in situ generation of ClSO2NH2 is then easily accomplished.[14]
Figure 4.
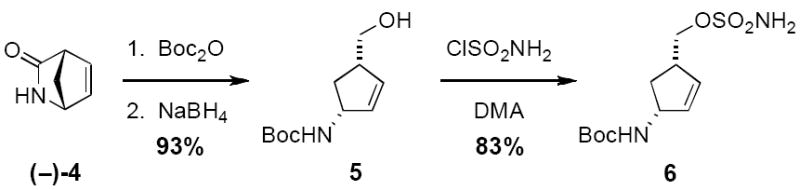
Homoallylic sulfamate synthesis from commercial lactam.
Exposure of sulfamate 6 to a dimeric Rh(II) catalyst, 1.1 equiv of PhI(OAc)2, and MgO, affords aziridine 7 as a single diastereomer in 95% yield (Figure 5). Less than 1% of the 5-membered ring product of allylic C–H insertion is obtained in this transformation. By capitalizing on our recently developed Rh2(esp)2 catalyst, loadings as low as 0.06 mol% (>1500 turnovers) can be used, thus enabling the reaction to be easily and inexpensively scaled.[15] The novel tricylic structure is quite stable and can be isolated in pure form following chromatography on silica gel. When treated with NaN3 in aqueous isopropanol, regioselective attack at C4 (agelastatin numbering) proceeds at ambient temperature to yield predominantly the bridging [1,2,3]-oxathiazepane-2,2-dioxide 8 (C4/C8 regioselectivity = 9:1).[16,17] This versatile intermediate incorporates three of the four stereogenic carbamine centers found in the natural product, all differentially masked. Accordingly, this aziridination/ring opening reaction sequence should offer ready access to several derivative forms of agelastatin.
Figure 5.
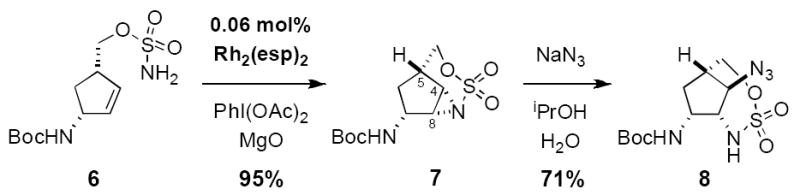
Catalytic aziridination and regioselective ring-opening affords the desired oxathiazepane heterocycle 8. Rh2(esp)2 = Rh2(α,α,α’,α’-1,3-benzenediproprionate)2.
To forward the synthetic plan, a series of maneuvers was needed that would ultimately enable a single carbon excision and introduction of the C5 ketone (see Figure 3). Six- and seven-membered ring cyclic sulfamates possess intrinsic reactivity as electrophilies, which can be modulated as a result of N-functionalization.[14b,18] Taking advantage of this property, oxathiazepane 8 was first treated with diethyl pyrocarbonate to furnish the N-acylated species 9; subsequent introduction of NaSePh (prepared in a separate reaction vessel) displaces the oxathiazepane C–O bond to afford in a single operation selenide 10 (Figure 6). Access to this product in just 4 steps from 5 underscores the effectiveness of our aziridination process for the rapid assembly of stereochemically complex, orthogonally protected polyamine intermediates.
Figure 6.
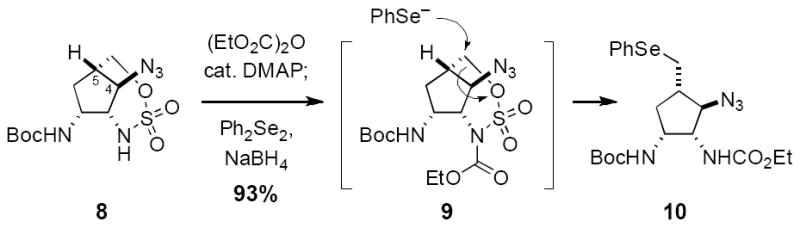
Oxathiazepane 8 activation and ring opening.
Oxidation of selenide 10 and elimination of the transient selenoxide was intended to furnish the C5 exo-methylene product 11 (Figure 7). Such a reaction does occur, however, the resulting allylic azide undergoes facile [3,3]-sigmatropic rearrangement to afford cyclopentene 12.[19] As it was not possible to prevent this isomerization process, a decision was made to postpone exo-methylene introduction until the latter steps of the synthesis. Accordingly, we opted to fashion first the requisite pyrrole unit from 10 (Figure 8). Removal of the Boc-group with CF3CO2H precedes an efficient Paal-Knorr condensation, which employs tricarbonyl 13 and mild acid catalysis to forge the heterocycle.[20,21] The desired pyrrole 14 is generated in 85% yield over this two-step sequence.
Figure 7.
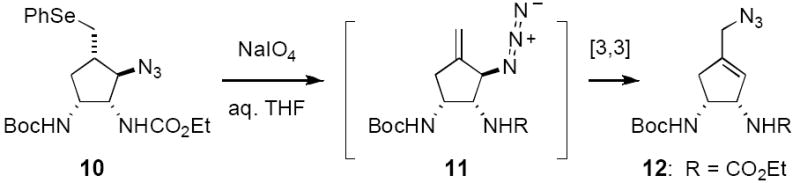
Rearrangement of allylic azide 11 necessitates strategic modification.
Figure 8.
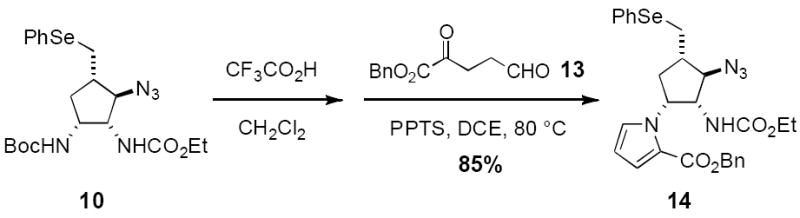
Paal-Knorr condensation installs pyrrole unit.
To complete the agelastatin synthesis, azide 14 is reduced chemoselectively under Staudinger conditions (Me3P, THF/H2O, Scheme 1). Once reduction is complete, MeNCO is added to the reaction flask to produce urea 15. This compound is easily purified by normal-phase silica gel chromatography in spite of the presence of the polar N-methyl urea moiety. Exposure of 15 to m-CPBA induces selenide to selenoxide conversion and subsequent elimination to afford alkene 16. While attempts to cleave the C5 exo-methylene unit under ozonolytic conditions gave only intractable mixtures of products, successful installation of the C5 carbonyl was realized using a combination of 2.5 mol% OsO4 and NaIO4. Once the C5-ketone is exposed, addition of the urea is highly favored and the product is isolated exclusively as hemi-aminal 17.
Scheme 1.
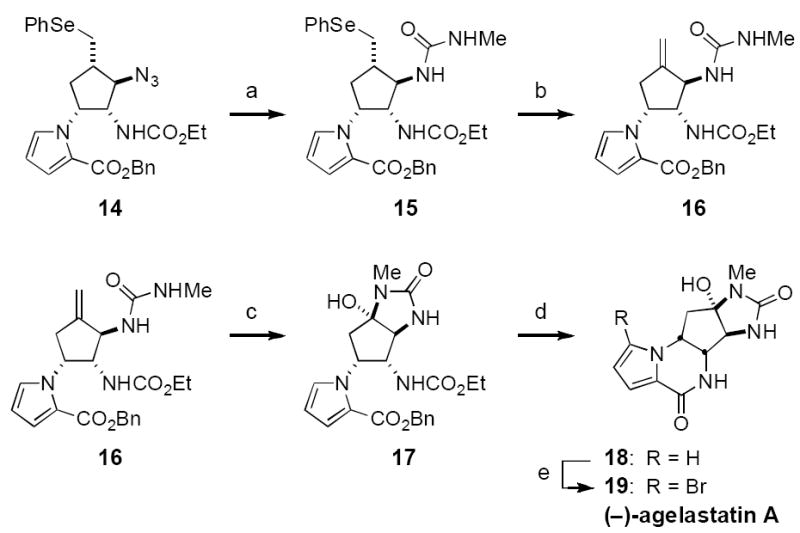
a) Me3P, THF/H2O; then MeNCO, 81%; b) m-CPBA, DCE, 0 °C; then Et3N, 80 °C, 89%; c) 2.5 mol % OsO4, NaIO4, THF/H2O, 45 °C, 81%; d) KOtBu, tAmOH, 45 °C, 77%; e) NBS, THF/MeOH, 0→25 °C, 75%. m-CPBA = meta-chloroperbenzoic acid, NBS = N-bromosuccinimide.
The stability of the hemi-aminal in 17 obviates protection as the N,O-acetal. As such, assembly of the final target can be accomplished by first exposing 17 to KOtBu in t-amyl alcohol (Scheme 1).[22] This protocol generates the desired six-membered lactam with concomitant cleavage of the ethyl carbamate. Having intercepted the penultimate intermediate formed in prior syntheses of agelastatin A, a literature procedure using N-bromosuccinimde smoothly and selectively brominates the pyrrole unit and gives the natural product as a white, crystalline solid.[6e] This material matches reported spectral and optical rotation data in all respects.[2,6e] Starting from commercial 5, the 11-step sequence has been executed in a single pass to prepare >200 mg of the natural product (15% overall yield).
An efficient, easily scaled, and flexible route to (–)-agelstatin A has been made possible following the development and application of a selective Rh-catalyzed aziridination method. With the aid of Rh2(esp)2, this reaction is made to proceed in high yield at negligible catalyst loadings. The resulting tricyclic product 7 represents a unique heterocyclic structure that is efficiently transformed into a differentially protected cyclopentyltriamine. New protocols for manipulating the intermediate oxathiazepane and for crafting the pyrrole lactam also distill from this work. Overall, the preparation of agelastatin A is illustrative of the manner in which modern oxidative methods for C–N bond formation can alter the retrosynthetic logic of complex chemical synthesis.[23]
Supplementary Material
Footnotes
This work was funded by a grant from the National Institutes of Health. We are grateful to Pfizer for additional support of our program.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Paul M. Wehn, Amgen, Inc., 1120 Veterans Blvd., South San Francisco, California 94080
J. Du Bois, Department of Chemistry, Stanford University, Stanford, California 94305-5080, Fax: (+1) 650-725-0259, jdubois@stanford.edu
References
- 1.For reviews on bromopyrrole natural products, see: Dembitsky VM. Russ Bioorg Chem. 2002;19:170–182.Jacquot DEN, Lindel T. Curr Org Chem. 2005;9:1551–1565.Weinreb SM. Nat Prod Rpts. 2007;24:931–948. doi: 10.1039/b700206h.Köck M, Grube A, Seiple AB, Baran PS. Angew Chem Int Ed. 2007;46:6586–6594. doi: 10.1002/anie.200701798.
- 2.a) D’Ambrosio M, Guerriero A, Debitus C, Ribes O, Pusset J, Leroy S, Pietra F. J Chem Soc Chem Comm. 1993:1305–1306. [Google Scholar]; D’Ambrosio M, Guerriero A, Chiasera G, Pietra F. Helv Chim Acta. 1994;77:1895–1902. [Google Scholar]
- 3.Hong TW, Jimenez DR, Molinski TF. J Nat Prod. 1998;61:158–161. doi: 10.1021/np9703813. [DOI] [PubMed] [Google Scholar]
- 4.a) D’Ambrosio M, Guerriero A, Ripamonti M, Debitus C, Waikedre J, Pietra F. Helv Chim Acta. 1996;79:727–735. [Google Scholar]; b) Pettit GR, Ducki S, Herald DL, Doubek DL, Schmidt JM, Chapuis JC. Oncology Res. 2005;15:11–20. doi: 10.3727/096504005775082075. [DOI] [PubMed] [Google Scholar]; c) Mason CK, McFarlane S, Johnston PG, Crowe P, Erwin PJ, Domostoj MM, Campbell FC, Manaviazar S, Hale KJ, El-Tanani M. Mol Cancer Ther. 2008;7:548–558. doi: 10.1158/1535-7163.MCT-07-2251. [DOI] [PubMed] [Google Scholar]
- 5.It has also been suggested that agelastatin A inhibits GSK-3β, an enzyme implicated in the development of the neurofibrillary tangles associated with Alzheimer’s disease, see: Meijer L, Thunnissen AMWH, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkow EM, Kim SH, Pettit GR. Chem Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6.
- 6.For completed works, see: Stein D, Anderson GT, Chase CE, Koh Y, Weinreb SM. J Am Chem Soc. 1999;121:9574–9579.Feldman KS, Saunders JC. J Am Chem Soc. 2002;124:9060–9061. doi: 10.1021/ja027121e.Feldman KS, Saunders JC, Wrobleski ML. J Org Chem. 2002;67:7096–7109. doi: 10.1021/jo026287v.Hale KJ, Domostoj MM, Tocher DA, Irving E, Scheinmann F. Org Lett. 2003;5:2927–2930. doi: 10.1021/ol035036l.Domostoj MM, Irving E, Scheinmann F, Hale KJ. Org Lett. 2004;6:2615–2618. doi: 10.1021/ol0490476.Davis FA, Deng J. Org Lett. 2005;7:621–623. doi: 10.1021/ol047634l.Trost BM, Dong G. J Am Chem Soc. 2006;128:6054–6055. doi: 10.1021/ja061105q.Ichikawa Y, Yamaoka T, Nakano K, Kotsuki H. Org Lett. 2007;9:2989–2992. doi: 10.1021/ol0709735.Yoshimitsu T, Ino T, Tanaka T. Org Lett. 2008;10:5457–5460. doi: 10.1021/ol802225g.
- 7.For an approach to agelastatin A involving intermolecular aziridination, see: Baron E, O’Brien P, Towers TD. Tetrahedron Lett. 2002;43:723–726.. Trost and Dong have successfully applied an intermolecular aziridination reaction to complete a short, elegant synthesis of agelastatin, see reference 6g. Intramolecular alkene aziridination using an azidoformate has recently been employed, see reference 6i.
- 8.a) Wehn PM, Lee J, Du Bois J. Org Lett. 2003;5:4823–4826. doi: 10.1021/ol035776u. [DOI] [PubMed] [Google Scholar]; b) Wehn PM, Du Bois J. Org Lett. 2005;7:4685–4688. doi: 10.1021/ol051896l. [DOI] [PubMed] [Google Scholar]; c) Guthikonda K, Wehn PM, Caliando BJ, Du Bois J. Tetrahedron. 2006;62:11331–11342. [Google Scholar]
- 9.For additional examples of intramolecular aziridination in natural products synthesis, see: Fukuyama T, Yang L. J Am Chem Soc. 1989;111:8303–8304.Williams DR, Rojas CM, Bogen SL. J Org Chem. 1999;64:736–746. doi: 10.1021/jo981310r.Bergmeier SC, Stanchina DM. J Org Chem. 1999;64:2852–2859. doi: 10.1021/jo9823893.Bodner R, Marcellino BK, Severino A, Smenton AL, Rojas CM. J Org Chem. 2005;70:3988–3996. doi: 10.1021/jo0500129.Bräse S, Gil C, Knepper K, Zimmerman V. Angew Chem Int Ed Engl. 2005;44:5188–5240. doi: 10.1002/anie.200400657., and references therein.
- 10.This work is taken from Wehn PM. Ph.D. Thesis. Stanford University; 2006.
- 11.For examples of Rh-catalyzed intramolecular aziridination using sulfonamides and carbamates, see: Liang J-L, Yuan S-X, Chan PWH, Che C-M. Org Lett. 2002;4:4507–4510. doi: 10.1021/ol0270475.Liang J-L, Yuan S-X, Chan PWH, Che C-M. Tetrahedron Lett. 2003;44:5917–5920.Fruit C, Muller P. Tetrahedron: Asym. 2004;15:1019–1026.Padwa A, Flick AC, Leverett CA, Stengel T. J Org Chem. 2004;69:6377–6386. doi: 10.1021/jo048990k.Bodner R, Marcellino BK, Severino A, Smenton AL, Rojas CM. J Org Chem. 2005;70:3988–3996. doi: 10.1021/jo0500129.Catino AJ, Nichols JM, Forslund RE, Doyle MP. Org Lett. 2005;7:2787–2790. doi: 10.1021/ol0510973.Lorpitthaya R, Xie Z-Z, Kuo J-L, Liu X-W. Chem Eur J. 2008;14:1561–1570. doi: 10.1002/chem.200701288. Also, see: Liang J-L, Yuan S-X, Huang J-S, Che C-M. J Org Chem. 2004;69:3610–3619. doi: 10.1021/jo0358877.A related method uses N-tosyloxycarbamates for intramolecular aziridination, see: Lebel H, Huard K, Lectard S. J Am Chem Soc. 2005;127:14198–14199. doi: 10.1021/ja0552850.
- 12.For selected examples of Cu-catalyzed intramolecular azirdination with sulfonamide and sulfamate substrates, see: Dauban P, Dodd RH. Org Lett. 2000;2:2327–2329. doi: 10.1021/ol000130c.Dauban P, Sanière L, Tarrade A, Dodd RH. J Am Chem. 2001;123:7707–7708. doi: 10.1021/ja010968a.Duran F, Leman L, Ghini A, Burton G, Dauban P, Dodd RH. Org Lett. 2002;4:2481–2483. doi: 10.1021/ol0200899.Esteoule A, Duran F, Retailleau P, Dodd RH, Dauban P. Synthesis. 2007:1251–1260.
- 13.Taylor SJC, Brown RC, Keene PA, Taylor IN. Bioorg Med Chem. 1999;7:2163–2168. doi: 10.1016/s0968-0896(99)00146-7. and references therein.
- 14.a) Okada M, Iwashita S, Koizumi N. Tetrahedron Lett. 2000;41:7047–7051. [Google Scholar]; b) Espino CG, Wehn PM, Chow J, Du Bois J. J Am Chem Soc. 2001;123:6935–6936. [Google Scholar]
- 15.Espino CG, Fiori KW, Kim M, Du Bois J. J Am Chem Soc. 2004;126:15378–15379. doi: 10.1021/ja0446294.. This catalyst is commercially available from Aldrich Chemical Co.
- 16.We refer to this heterocycle as an oxathiazepane for convenience.
- 17.Opening of related bicyclic aziridines generally favors the oxathiazepane ring. For a complete discussion on this subject, see reference 8c.
- 18.Melèndez RE, Lubell WD. Tetrahedron. 2003;59:2581–2616.Bower JF, Szeto P, Gallagher T. Org Lett. 2007;9:4909–4912. doi: 10.1021/ol7022104., and references therein.
- 19.a) Gagneux A, Winstein S, Young WG. J Am Chem Soc. 1960;82:5956–5957. [Google Scholar]; b) Feldman AK, Colasson B, Sharpless KB, Fokin VV. J Am Chem Soc. 2005;127:13444–13445. doi: 10.1021/ja050622q. [DOI] [PubMed] [Google Scholar]
- 20.Tricarbonyl 13 is prepared in two steps [(1) mono-addition of H2C=CHCH2CH2MgBr to dibenzyl oxalate; (2) O3, CH2Cl2, –78 °C; then Ph3P] and used immediately following chromatographic purification.
- 21.For a general review on pyrrole synthesis, see: Ferreira VF, de Souza MCBV, Cunha AC, Pereira LOR, Ferreira MLG. Org Prep Proced Int. 2001;33:411–454.. Also, see: Tracey MR, Hsung RP, Lambeth RH. Synthesis. 2004:918–922.Iden HS, Lubell WD. Org Lett. 2006;8:3425–3428. doi: 10.1021/ol061036k.
- 22.To ensure the reaction was performed under anhydrous conditions, t-amyl alcohol was preferred over t-BuOH.
- 23.For additional examples, see: Wehn PM, Du Bois J. J Am Chem Soc. 2002;124:12950–12951. doi: 10.1021/ja028139s.Trost BM, Gunzner JL, Dirat O, Rhee YH. J Am Chem Soc. 2002;124:10396–10415. doi: 10.1021/ja0205232.Huang H, Panek JS. Org Lett. 2003;5:1991–1993. doi: 10.1021/ol034582b.Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510–11511. doi: 10.1021/ja0368305.Parker KA, Chang W. Org Lett. 2005;7:1785–1788. doi: 10.1021/ol050356l.Yakura T, Sato S, Yashimoto Y. Chem Pharm Bull. 2007;55:1284–1286. doi: 10.1248/cpb.55.1284.Mulcahy JV, Du Bois J. J Am Chem Soc. 2008;130:12630–12631. doi: 10.1021/ja805651g.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.