

Abstract
The structure of the proline amino acid allows folded polyproline peptides to exist as both left- (PPII) and right-handed (PPI) helices. We have characterized the free energy landscapes of hexamer, nanomer, and tridecamer polyproline peptides in gas phase and implicit water as well as explicit hexane and 1-propanol for the nanomer. To enhance the sampling provided by regular molecular dynamics, we used the recently developed adaptively biased molecular dynamics method, which describes Landau free energy maps in terms of relevant collective variables. These maps, as a function of the collective variables of handedness, radius of gyration, and three others based on the peptide torsion angle ω, were used to determine the relative stability of the different structures, along with an estimate of the transition pathways connecting the different minima. Results show the existence of several metastable isomers and therefore provide a complementary view to experimental conclusions based on photo-induced electron transfer experiments with regard to the existence of stable heterogeneous subpopulations in PPII polyproline.
Keywords: cis-trans isomerization, left-handed helix, molecular dynamics, PPI, PPII
The concept of molecular chirality is used to describe molecular structures that are not superposable on their mirror images. Chiral molecules are quite prevalent in biological systems, which are primarily homochiral systems. For example, most proteins contain only L-amino acids, while DNA is made up primarily of D-deoxyribose. The relationship between chirality and helical polypeptide structure was first mentioned by Pauling (1) in reference to the α-helix. The naturally occurring L-amino acids predominantly form right-handed helices, whereas their stereoisomers D-amino acids favor left-handed helices. Indeed, right-handed helices are prevalent in biology, not only in peptides (with structural motifs such as α-helix, 310 helix, and π helix) but also in the double-helical structure of B- and A-DNA. Although much less common, left-handed helices with the same chiral units as right-handed helices also exist, such as those found in PPII and in Z-DNA.
In this paper, we investigate the free energy landscape of several short polyproline peptides. Proline is unique among the natural amino acids in that its side chain is cyclized to the backbone, restricting its backbone dihedral angle to ϕ = −75°, giving proline an exceptional rigidity and a considerably restricted conformational space. Polyproline is known to form helical structures with two well-characterized conformations: a left-handed polyproline helix (PPII) is formed when the sequential residues all adopt backbone dihedral angles (ϕ,ψ) of (−75°,146°), with all prolyl bonds in the trans-isomer conformation (i.e. backbone dihedral angle ω = 180°) with 3 residues per turn; and a more compact right-handed polyproline helix (PPI) is formed with all sequential residues adopting dihedral angles of roughly (−75°, 160°) and all prolyl bonds assume a cis-isomer conformation (i.e. backbone dihedral angle ω = 0°) with 3.3 residues per turn. Of the 20 natural amino acids, only proline is “comfortable” in the cis-isomer conformation, and proline seems to be quite effective in stabilizing left-handed helices. The probability distribution for cis-trans prolyl bonds is affected by neighboring amino acids (2, 3), pH and ionic strength (4), solvent (5 –11) and chain length (12). It has been noted experimentally that the PPII structure is favored in water, benzyl alcohol, and most of the other solvents, while the PPI structure is favored in the presence of aliphatic alcohols like propanol (12 –15). The two forms can reversibly interconvert by means of changes in solvent composition (5, 7, 10, 16, 17), as analyzed via circular dichronism (CD) spectroscopy experiments (12, 18 –20). It is also known that the five-membered pyrrolidine ring may adopt distinct up- and down-puckered conformations (21), which were deemed to be almost equally probable based on X-ray analyses of peptides (22) and proteins (23). Although these studies also suggest a correlation between puckering and the distribution of cis-trans bonds, it is now known that the prolyl ring can flip between its two states irrespective of the cis-trans nature of the peptide bond (24). The puckering preference of both the PPII and PPI structures has also been investigated in refs. (9) and (25 –27).
The structural role of proline depends on the position it occupies in a protein. When in the middle of α helices and β sheets, proline acts as a structural disruptor, but it is generally found at the beginning of α helices and edge of β sheets, as well as in turns. It has been noted that cis-trans isomerization of X-Pro peptide groups is one of the rate-determining steps for folding and unfolding of various proteins (28 –32) and several studies have investigated the cis-trans isomerization of proline-containing peptides (33 –37). The role that PPII conformations play in the nonstructured states of polypeptides has also been much discussed (38 –41), and experimental and theoretical evidence shows that the presence of nonproline residues decreases the PPII helix content in a pro-rich environment (39, 40). Proline oligomers also have considerable interest in their own right. Traditionally, the relatively rigid structure of PPII has been used as a “molecular ruler” in structural molecular biology, especially for the validation of spectroscopic rulers in F_ster resonance energy transfer (FRET) experiments (42). However, both FRET and photoinduced-electron transfer (PET) studies show deviations of experimentally observed end-to-end distances of polyproline from theoretical predictions. Recently, Doose et al. used PET techniques to probe the structure and dynamics of PPII with 1–10 residues in aqueous solution (43). The authors showed that “polyproline samples exhibit static structural heterogeneity with subpopulations of distinct end-to-end distances that do not interconvert on the scales from nano- to milliseconds”. This heterogeneity was attributed to interspersed cis isomers that disrupt the otherwise ideal all trans PPII structure, in agreement with theoretical studies based on conformational energy calculations (44, 45) and previous simulation results (40). The authors concluded that the stability of these heterogeneous subpopulations, caused by prolyl cis-trans isomerization, requires characterization of the cis isomers in order for polyproline to be used as a molecular ruler, for instance to calibrate FRET efficiency measurements. From a computational point of view, such a characterization is rather difficult because cis-trans isomerization in polyproline is much slower than the formation and rupture of hydrogen bonds in an α -helix-coil transition. Indeed, characteristic time scales for prolyl isomerization range from tens to hundreds of seconds at room temperature (2, 46), implying solvent-dependent barriers of the order of 10 to 20 kcal/mol (24, 27, 47 –49).
Given these high barriers, traditional molecular dynamics (MD) simulations cannot explore the relevant conformational space for the wide range of cis-trans transitions. Rather, based on the recently developed adaptively biased molecular dynamics (ABMD) method (50), accurate free energy landscapes of short polyproline peptides in a few selected environments, as a function of several relevant-order parameters or collective variables, have been calculated. The ABMD method belongs to the general category of umbrella sampling methods with a time-dependent potential and has recently been implemented in the AMBER-10 modeling package (51). Different sets of collective variables have been used to study not only the two helical conformations of the polyproline peptide but also the transition pathways and the many conformations that are neither PPI nor PPII. The results provide a complementary view to the conclusions of Doose et al. (43) about the existence of stable heterogeneous subpopulations. Finally, all relevant simulation details are provided in the SI Text with clarifying Figs. S1–S3.
Free Energy Methods and Collective Variables
To calculate accurate free energy maps, we used the ABMD method (50), with supplementary equilibrium umbrella sampling runs (52). In addition, steered molecular dynamics (SMD) runs were used to investigate specific pathways on these free energy maps (53). The ABMD method belongs to the general category of umbrella sampling methods with a time-dependent potential and provides for an elegant way of computing the potential of mean force or free energy as a function of a collective variable σ(r 1,…,r N):
where p(ξ) =<δ[ξ−σ(r 1,…,r N)] > is the probability density of the collective variable (the angular brackets denote an ensemble average; k B, the Boltzmann constant; T, temperature). The method estimates the Landau free energy of a reaction coordinate from an evolving ensemble of realizations and uses that estimate to bias the system dynamics to flatten an effective free energy surface (which may not be exactly the same as the total conformational energy). Finally, all relevant simulation details are provided in the SI Text.
The ABMD method was used to calculate the free energy landscapes of short polyproline peptides [Ace-(Pro)n-Nme, n = 6,9,13], as a function of several sets of collective variables. These collective variables are carefully chosen as to reflect the “slow modes” of the system, which are ultimately responsible for the large-scale molecular structure—i.e., the changes associated with the cis-trans isomerization. The “fast modes” of the system, which take place on a relatively short time scale, are properly sampled and de facto integrated out when calculating the Landau free energy. An important example of a fast mode is provided by the puckering of the pyrrolidine ring. The solvated transition barrier between the puckered up and down states of the system is of the order of a few k B T (54), so that at T = 300 K, the ring oscillates rapidly between the two puckering conformations (see SI Text and Fig. S2), which are then integrated in the ensemble average of the slow modes. As a first set, we considered the collective variables of handedness (H) and the radius of gyration (R g), which are natural because polyproline peptides are characterized by not only PPII and PPI but also by more compact “globular” conformations. The definition of H is illustrated in Fig. 1. Moving through the helical turns, a sequence of four points A,…,D is defined. These points determine the vectors and . The midpoints of these vectors (E and F) form the vector . The handedness of these points is then defined as
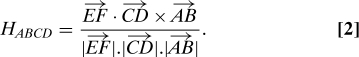 |
Fig. 1.

Geometrical construction involved in the definition of handedness: H < 0 for left-handed and H > 0 for right-handed structures.
Such a definition discriminates between PPI (H ABCD > 0) helices and PPII (H ABCD < 0) helices. To complete the definition, the four-point contributions are then summed along the backbone, thereby defining the collective variable H for the entire peptide: H = ∑i = 1 n−3 H i,i+1,i+2,i+3. Roughly speaking, the magnitude of H is a measure of the number of turns associated with the helices. For polyproline, the position of the nitrogen atoms was found to be a good choice for the location of the A,…,D points.
Even though the handedness is enough to differentiate between PPI and PPII, many compact structures are characterized by the same value of H. To remove this degeneracy, we considered the free energies associated with the radius of gyration R g of the heavy atoms: Here, R Σ = Σa(m a/m Σ)r a is the center of mass, m Σ = ∑a m a, and the sums run over all atoms except hydrogen. Note that R g is not the only quantity that can be used to characterize the spatial extent of a peptide's conformation. Experimentally, results are often discussed in terms of the peptide's “end-to-end” (d end) distance. Free energy maps based on either of these two variables capture essentially the same physical content, as demonstrated in the SI Text where several maps based on d end are shown in Figs. S4–S6.
We have also defined other collective variables specifically associated with the cis-trans isomerization. The torsion angle that describes this transition has value ω = 0° for the cis (C) isomer, and ω = 180° for the trans (T) isomer. Thus, we define Ω as the sum of the cosines of the ω angles, which for an n-mer peptide is
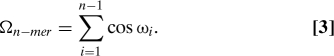 |
For a cis-trans prolyl bond, cosωi is + 1 (−1) and, therefore, Ωn−mer can take any of the following values: −n + 1, −n + 3,…, n − 3, or n − 1. The collective variable Ω, like H, is degenerate. For a sequence with n b = n − 1 bonds, the number of conformations with n C cis bonds and n T trans bonds (with n b = n C + n T) is n b!/(n C!n T!). Naturally, perfect PPI is characterized by Ωn−mer = n − 1 and perfect PPII by Ωn-mer = −(n − 1).
Another possibility is to consider the “interface” between bonds and define the collective variable Λ as
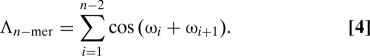 |
For an n-mer, there are n − 1 prolyl bonds and n − 2 interfaces. If two neighboring bonds have the same dihedral angle ω, then their interface contributes cos(ωi + ω i+1) = +1; otherwise it gives −1. Correspondingly, Λn−mer can take on any of the values −n + 2, −n + 4,…,n − 4,n − 2. Λn−mer removes some of the degeneracy associated with Ω. For |Ω| = n − 1, there is only one value of Λ(Λ = n − 2); for Ω = 0 (which exists only for odd values of n), there are (n − 2) values of Λ; for |Ω| = m (with m ≠ n − 1 and m ≠ 0), there are n − 1 − m values of Ω.
Finally, given the binary nature of this torsion angle and considering that for an n-mer there are 2n−1 possible configurations of the bonds, it is possible to define a collective variable that avoids the degeneracy problem entirely. First, one maps the cis-trans bond conformations into binary numbers by using b i = (cosωi + 1)/2 and then converts the sequences {b i} into a decimal number:
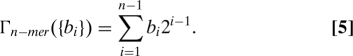 |
Therefore Γ is a number ranging from 0 (PPII) to 2n−1 − 1 (PPI). This new collective variable acts like a label for each possible conformation. It can be related to Ω via Ωn−mer = ∑i=1 n−1 (2b i − 1). Notice that the decimal number thus assigned does not have any physical meaning: It is just a label for the different isomers. The collective variable Γ is only convenient for the investigation of relatively short polypeptides because the number of minima associated with Γ increases exponentially with peptide length.
Results and Discussion
We now discuss the free energy landscapes of Ace-(Pro)n-Nme peptides (n = 6,9,13) both in vacuo and for selected solvated environments (implicit water, hexane, and 1-propanol).
Fig. 2 A shows the (H,R g) free energy map for the peptides in vacuo. For discussion purposes, we focus primarily on the nanomer free energy map. It displays three sets of minima: Three minima (f, g, h) with H > 2 correspond to PPI; a broad basin with several minima (d, e) between −1.5 < H < 2 with R g < 7 correspond to twisted and globular structures; and three minima (a, b, c) with H < −0.95 and R g > 7, correspond to PPII. A ribbon representation of the typical structures in these minima is also shown in Fig. 2 A. A quantitative description of the minima (location, depth) and characteristic isomer is given in Table S1. The isomer associated with minimum (a) corresponds most closely to the ideal PPII structure (RMSD = 0.4 Å), whereas that associated with (h) is closest to PPI (RMSD = 1.0 Å). There are qualitative changes in the free energy maps as the length of polyproline is increased. Although the same three classes of minima are found on all three maps, their relative importance changes. For short 6-mers, the free energy favors the formation of PPII structures, as opposed to PPI. For the longer 13-mer, the trend is reversed, and PPI is favored. The 9-mer appears to correspond to an intermediate case, such that PPI and PPII structures are approximately balanced. Such a change with peptide length has also been noted experimentally in alcohol environments. Free energy maps for polyproline in implicit water (Fig. 2 B) are qualitatively similar, except that PPII is favored over PPI for all chain lengths, in agreement with experimental results (12). In implicit water, the free energy minima associated with PPII are of the same magnitude as those associated with the compact structures, and they form a “broad valley” with relatively low barriers.
Fig. 2.

Free energy landscapes (kcal/mol) as a function of collective variables (H,R g). Landscapes are shown for a 6-mer (Top), 9-mer (Middle), and 13-mer (Bottom) polyproline peptide: in vacuo (A) and in implicit water (B). Examples of transition paths between PPI and PPII are shown: The solid line passes through the global minimum whereas the dashed line avoids it. A ribbon representation is used for some of the structures associated with some of the major minima, with cis-trans prolyl bonds highlighted in red (blue), respectively. In the 13-mer case, all the prolyl bonds of both E and F structures are cis, but the prolyl amide bond in the amidated terminal of the peptide (not shown) is trans in E and cis in F.
The (Ω,R g) and (Ω,Λ) free energy maps for n -mers in implicit water are shown in Fig. 3 (gas phase results are qualitatively similar and shown in the SI Text and Fig. S7). Because for an n-mer polypeptide there are n − 1 peptide bonds, Ω runs between ±5, ±8 and ±12 for the different length peptides. Allowed values for Ω are centered on even values for the 9,13-mers, and about odd values for the 6-mer. The free energy minima associated with the Ω values are also elongated in the R g direction, forming narrow valleys, which is a reflection of the underlying structures seen in the ribbon representations of Fig. 3 A. For example, the flipping of a single bond within an all-trans helix does not greatly modify the helical structure if the bond is near the end, but it produces a sharp turn when it is in the center. For the nanomer, the deepest minima correspond to globular and PPII structures. Because the PPII-like helices are more stretched, they have more flexibility in terms of R g, unlike PPI-like structures which are more rigid. This flexibility translates into a net decrease of the elongation of the free energy contours as one moves from PPII to PPI. Finally, because for n − 1 bonds there are n − 2 possible “interfaces” between them, the possible values of Λ vary between ±4, ±7, and ±11 for the 6−, 9−, and 13-mer, respectively. For the nanomer, Λ = 7 corresponds to either a complete PPI or PPII helix because both of these structures have no cis-trans interfaces. In contrast, Ω = 0 and Λ = −7 correspond to a situation of all alternating cis-trans bonds.
Fig. 3.

Shown are (R g,Ω) and (Λ,Ω) free energy landscapes (kcal/mol) for a 6-mer (Top), 9-mer (Middle), and 13-mer (Bottom) polyproline in implicit water.
The 1D free energy profile as a function of Γ for a hexamer in vacuo is shown as an inset in Fig. 4. The label Γ runs between 0 (PPII) and 31 (PPI) because there are 32 possible isomers involved. To make the connection with this collective variable and other ABMD variables, we have calculated the corresponding values of Ω and Λ for each of the possible combination of b i's. Because of the inherent degeneracy, multiple Γ 's may have the same value for Ω and Λ. Suppose that Γ1, Γ2,…,Γm all have the same values of Ω and Λ. In such a case, the probability of finding a structure with a given value of Ω and Λ is the sum of the probabilities related to Γ1,Γ2,…,Γm. We can therefore define a new phase space—labeled with N Ω,Λ—and relate the free energies in this space to those in Γ space via
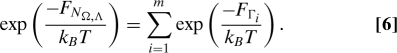 |
To illustrate this explicitly, consider the (Ω,Λ) free energy plot for a 6-mer shown in Fig. 3. This map is characterized by 14 distinct minima, which we map onto the single variable N Ω,Λ, which now runs from 1 to 14. Moving from left to right, and then bottom to top, we label each minima in (Ω,Λ) space by a number N Ω,Λ, i.e., N −5,4 = 1 (PPII), N −3,0 = 2, N −3,2 = 3,…,N 5,4 = 14 (PPI). Table 1 gives the Γ, Ω, Λ, and N Ω,Λ values for all 32 possible configurations of a 6-mer polyproline, and Fig. 4 plots the free energy as computed from the variable Γ for the 14 different states N Ω,Λ and compares it with the ABMD runs with (Ω,Λ). The correspondence between them is quite good, with only small, expected free energy differences (see also Fig. S8).
Fig. 4.
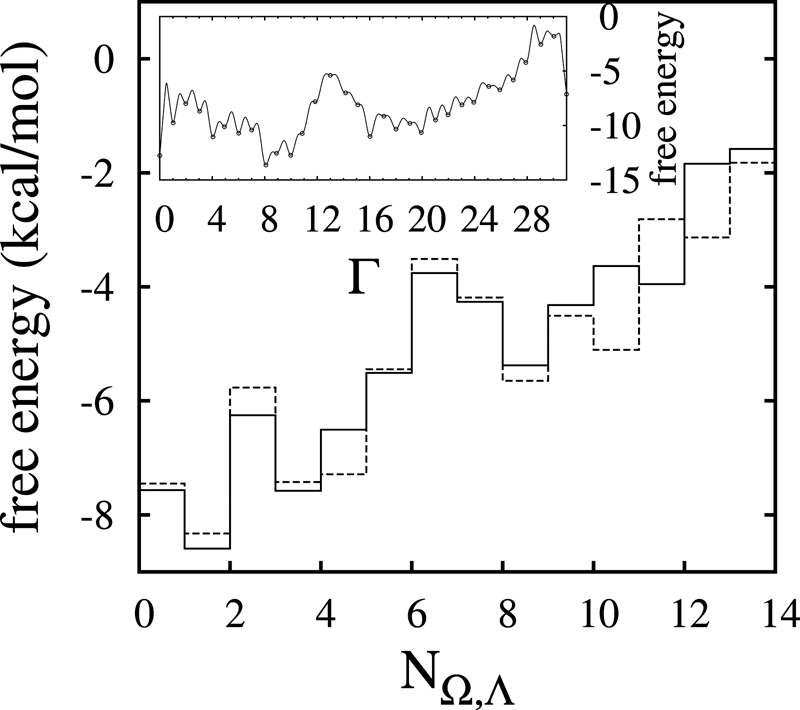
Free energy in the N Ω,Λ representation. The solid line is obtained from the (Ω,Λ) map by enumerating the minima and plotting the corresponding free energy values; the dotted line is obtained by using the Γ results combined with Eq. 6. (Inset). Free energy profile (kcal/mol) of a hexamer polyproline in vacuo, as a function of the single collective variable Γ.
Table 1.
The values of the order parameters Γ, Ω, Λ, and N Ω Λ for different configurations of a 6-mer polyproline peptide
| NΩ Λ | Ω | Λ | Γ | Sequence |
|---|---|---|---|---|
| 1 | −5 | 4 | 0 | TTTTT |
| 2 | −3 | 0 | 2,4,8 | TCTTT, TTCTT, TTTCT |
| 3 | −3 | 2 | 1, 16 | CTTTT, TTTTC |
| 4 | −1 | −4 | 10 | TCTCT |
| 5 | −1 | −2 | 5, 9, 18, 20 | CTCTT, CTTCT, TCTTC, TTCTC |
| 6 | −1 | 0 | 6, 12, 17 | TCCTT, TTCCT, CTTTC |
| 7 | −1 | 2 | 3,24 | CCTTT, TTTCC |
| 8 | 1 | −4 | 21 | CTCTC |
| 9 | 1 | −2 | 11, 13, 22, 26 | CCTCT, CTCCT, TCCTC, TCTCC |
| 10 | 1 | 0 | 14, 19, 25 | TCCCT, CCTTC, CTTCC |
| 11 | 1 | 2 | 7, 28 | CCCTT, TTCCC |
| 12 | 3 | 0 | 23, 27, 29 | CCCTC, CCTCC, CTCCC |
| 13 | 3 | 2 | 15, 30 | CCCCT, TCCCC |
| 14 | 5 | 4 | 31 | CCCCC |
All 32 = 25 possible conformations of the prolyl bonds are presented. The acetylated end is at the left and the amidated end at the right.
Now we turn to explicit solvent results. We investigated a nanomer in explicit nonpolar hexane and in 1-propanol, with free energy maps shown in Fig. 5. In hexane, the phase space associated with the PPI helices is reduced, as compared with the gas-phase and implicit water results: The only minima identified correspond to PPII and globular structures. By contrast, 9-mer polyproline in 1-propanol shows minima for the compact and PPI structures. Again, these results, agree with trends observed in experiments. What is the origin of this differing behavior of polyproline in the two different solvents? First, the characteristic ring of polyproline precludes the nitrogen atom of the prolyl bond from engaging in hydrogen bonding. This feature not only affects the structure of the helices but also the interaction with solvents. The cis-trans conformations are linked to the orientation of the carboxyl groups C = O: In PPI, these groups are almost parallel to the axis of the compact helix and are shielded from the solvent by proline rings; in PPII, the C = O groups are mainly perpendicular to the axis of the helix, and because the helix is more elongated, the carboxyl oxygen is more exposed to solvents. Indeed, results for different radial distributions linking the carboxyl oxygen and different atoms in the solvent showed the following: (i) the distances between oxygen in C = O and hexane atoms are shorter in PPII than in PPI (therefore hexane favors PPII); (ii) the distances between O and propanol atoms are shorter in PPI than PPII (therefore propanol favors PPI); and (iii) the distances related to the hexane atoms in both cases are shorter than the distances related to propanol (therefore the effects of hexane are stronger than the effects of propanol).
Fig. 5.

Free energy landscapes (kcal/mol) of a 9-mer polyproline in hexane (A) and 1-propanol (B), as obtained from ABMD runs followed up with umbrella corrections.
As a short summary, Table 2 gives the calculated free energy differences between the PPI and PPII structures, as obtained from the different runs with different collective variables. There is good agreement between all the different numerical values.
Table 2.
Free energy difference (in kcal = mol) of all cis PPI and all trans PPII [f(PPI)−f(PPII)], for 6−, 9−, and 13−mer Ace-(Pro)n-Nme, in vacuo, in implicit water, and in explicit solvents hexane and propanol (for 9-mer), obtained from ABMD simulations with different sets of collective variables
| Environment | (H,Rg) | (Ω,Rg) | (Ω,Λ) | Γ |
|---|---|---|---|---|
| 6-mer | ||||
| In vacuo | 5.12 | 5.22 | 6.02 | 5.63 |
| Water | 4.88 | 5.20 | 5.33 | 5.15 |
| 9-mer | ||||
| In vacuo | 1.05 | 1.42 | 1.24 | 1.52 |
| Water | 4.53 | 5.32 | 6.08 | 6.43 |
| Hexane | 18.13 | 19.68 | 20.12 | – |
| Propanol | −6.98 | −6.13 | −8.19 | – |
| 13-mer | ||||
| In vacuo | −7.21 | −6.22 | −6.65 | – |
| Water | 6.47 | 9.60 | 8.05 | – |
The fields marked by blank lines were not computed.
To further characterize the ABMD-based free energy maps, we performed SMD runs that use the topology of these landscapes to steer the polyproline peptide between PPI and PPII. The aim here is not to compute accurate free energy differences but rather to compare pathways associated with different mechanisms in a qualitative way. Here, we choose selected trajectories, such as those given by the lowest free energy path (LFEP) method (55) in the (H,R g) plane, paths on the (Ω, Λ) plane, paths where Ω is the only variable, etc.
Two LFEP paths identified between PPI and PPII are shown in Fig. 2. One such path passes through the global minimum (solid line), but a second path avoids it (dotted line). This second path is characterized by considerably lower free energy barriers, as shown in Fig. 6 A. It is plausible that the two different paths are associated with different transition mechanisms. Specifically, for the path avoiding the global minimum, sampling of intermediate conformations along the path is mostly consistent with a zipper-like mechanism, in which changes take place via the successive switching of neighboring prolyl bonds (i.e., for 6-mer undergoing a PPII → PPI transition, this would take the form TTTTT → CTTTT → CCTTT… → CCCCC). Nucleation begins primarily at the ends or in their near vicinity, with a rate that appears to be length dependent. In contrast, for the path through the global minima, bond flipping takes place in a less-ordered fashion. The helix seems to undergo melting before reassembling again. This view is supported by examining the structures along the path. The corresponding work done by carrying out SMD along the two paths from PPI to PPII is shown in Fig. 6 B. There is less work done along the path that avoids the global minimum. There are many degeneracies associated with the H and R g collective variables, and as a result the work function is just monotonically increasing. It is possible to overcome some of the ambiguities related to the degeneracy in the minima by steering the system from a pure PPI structure to a pure PPII structure by using different collective variables. We have tried this both by using the 1D variable Ω and also by choosing a path in the (Ω,Λ) phase space by keeping Λ at its highest possible value (Fig. 6 C). In both cases, the total work is less than in the other two trajectories, indicating that it is much more natural for the system to move along such a path. The work as a function of time (Fig. 6 D) shows eight periodic looking barriers that correspond to the flipping of each of the bonds and that can be interpreted as a more clear signature of a zipper-like mechanism. We have studied these pathways and others not shown here. The SMD runs can give a qualitative description of different pathways as well as possible accompanying mechanisms, but unless all trajectories are examined, it is difficult to make quantitative assertions. The zipper-like mechanism is a highly probable mechanism for helix conversion but not necessarily the most probable mechanism. As another example, the work for the zipper-like mechanism is compared in Fig. 6 D with that corresponding to an extreme case where all the bonds (except one) flip simultaneously. The cosines of each of the prolyl bonds for both mechanisms are shown in Fig. 6 E and 6F. The huge barrier linked to the “en masse” mechanism would make this transition rather improbable.
Fig. 6.

Paths and work done via SMD for a nanomer in vacuo. (A) Free energy profiles for the LFEP paths shown in Fig. 2 A as function of normalized path length (s) in (H, R g) space, (solid line passes through the global minimum and dotted line avoids it). (B) Work done via SMD for the paths shown in A. (C) A trajectory associated with a zipper-like mechanism in the (Ω,Λ) space. (D) Work done via SMD for the path shown in C (dashed line) and for another trajectory associated with an “en masse” flipping of the cis-trans bonds (solid line). (E and F) Time evolution of the cosines for each individual prolyl bond. Here, (E) is associated with the zipper-like transition and (F) with the “en masse” flipping mechanism (except for one bond).
Summary
This work presents a comprehensive study of polyproline as a paradigm of molecules whose conformational phase space contains left-handed and/or right-handed helices. The free energies were calculated as a function of a variety of relevant collective variables: handedness H, radius of gyration R g, and the variables Ω, Λ, and Γ, associated specifically with the cis-trans isomerization of the peptide bond. The theoretical framework developed here may readily be used for a quantative description of other helical molecules involving bistable changes in the relevant molecular conformation (e.g., the anti\syn flipping of nucleotides in B- to Z-DNA transistions). Specifically, the free energy landscapes of polyproline n-mers (n = 6,9,13) in vacuo and in implicit water were investigated, along with the nanometer in explicit hexane and 1-propanol. We also considered minimum free energy pathways through these landscapes, which were further investigated via SMD runs. General trends are as follows: The stability of PPI is enhanced by increasing the length of the peptide chain, which means that cooperativity or interresidue interaction is more favorable in PPI than PPII. Pure PPI and PPII are just one of the minima (when present) in the phase space, with many other minima corresponding to other stable subpopulations whose representative structures, in terms of cis-trans bonds, are readily obtainable. Favored structures as a result of interactions with different solvents agree qualitatively with experiments. The position of the carboxyl oxygen (parallel to the helical axis and hidden by proline rings in compact PPI; perpendicular to the axis and more exposed to solvent in the more-open PPII) plays an important role in the interaction with solvents. Different mechanisms of transitions are associated with different pathways in the phase space, some extreme examples being the case where all bonds rotate together, a melting and reordering mechanism, and a more probable zipper-like mechanism. Our results provide a complementary view to recent PET experiments regarding the existence of stable heterogeneous subpopulations of polyproline peptide conformations.
Supplementary Material
Acknowledgments.
We thank Dr. Jung Goo Lee for many useful discussions. This research was supported by the National Science Foundation Career Grants DMR-0348039 and FRG-0804549. In addition, we thank the North Carolina State University High Performance Computing (HPC) center for extensive computational support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0807786105/DCSupplemental.
References
- 1.Pauling L, Corey RB, Branson HR. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA. 1951;37:205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reimer U, et al. Side-chain effects on peptidyl-prolyl cis/trans isomerisation. J Mol Biol. 1998;279:449–460. doi: 10.1006/jmbi.1998.1770. [DOI] [PubMed] [Google Scholar]
- 3.Grathwohl C, Wuthrich K. The x-pro peptide bond as an nmr probe for conformational studies of flexible linear peptides. Biopolymers. 1976;15:2025–2041. doi: 10.1002/bip.1976.360151012. [DOI] [PubMed] [Google Scholar]
- 4.Grathwohl C, Wuthrich K. Nmr studies of the molecular conformations in the linear oligopeptides h-(l-ala)n l-pro-oh. Biopolymers. 1976;15:2043–2057. doi: 10.1002/bip.1976.360151013. [DOI] [PubMed] [Google Scholar]
- 5.Steinberg IZ, Harrington WF, Berger A, Sela M, Katchalski E. The configurational changes of poly-l-proline in solution. J Am Chem Soc. 1960;82:5263–5279. [Google Scholar]
- 6.Gornick F, Mandelkern L, Diorio AF, Roberts DE. Evidence for a cooperative intramolecular transition in poly-l-proline. J Am Chem Soc. 1964;86:2549–2555. [Google Scholar]
- 7.Mandelkern L. Poly-L-proline. In: Fasman GD, editor. In Poly- -Amino Acids. New York: Marcel Dekker; 1967. pp. 675–724. [Google Scholar]
- 8.Strassmair H, Engel J, Zundel G. Binding of alcohols to the peptide co-group of poly-l-proline in the i and ii conformation. i. Demonstration of the binding by infrared spectroscopy and optical rotatory dispersion. Biopolymers. 1969;8:237–246. [Google Scholar]
- 9.Tanaka S, Scheraga HA. Theory of the cooperative transition between two ordered conformations of poly(l-proline). ii. Molecular theory in the absence of solvent. Macromolecules. 1975;8:504–516. doi: 10.1021/ma60046a024. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka S, Scheraga HA. Theory of the cooperative transition between two ordered conformations of poly(l-proline). iii. Molecular theory in the presence of solvent. Macromolecules. 1975;8:516–521. doi: 10.1021/ma60046a025. [DOI] [PubMed] [Google Scholar]
- 11.Kofron JL, Kuzmic P, Kishore V, Colon-Bonilla E, Rich DH. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry. 1991;30:6127–6134. doi: 10.1021/bi00239a007. [DOI] [PubMed] [Google Scholar]
- 12.Kakinoki S, Hirano Y, Oka M. On the stability of polyproline-i and ii structures of proline oligopeptides. Poly Bul. 2005;53:109–115. [Google Scholar]
- 13.Traub W, Shmueli U. Structure of poly-l-proline i. Nature. 1963;198:1165–1166. [Google Scholar]
- 14.Cowan PM, McGavin S. Structure of poly-l-proline. Nature. 1955;176:501–503. [Google Scholar]
- 15.Mutter M, Wohr T, Gioria S, Keller M. Pseudo-prolines: Induction of cis/trans-conformational interconversion by decreased transition state barriers. Biopolymers. 1999;51:121–128. doi: 10.1002/(SICI)1097-0282(1999)51:2<121::AID-BIP2>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 16.Kurtz J, Berger A, Katchalski E. Mutarotation of poly-l-proline. Nature. 1956;178:1066–1067. [Google Scholar]
- 17.Steiberg IZ, Berger A, Katchalski E. Reverse mutarotation of poly-l-proline. Biochim Biophys Acta. 1958;28:647–648. doi: 10.1016/0006-3002(58)90537-7. [DOI] [PubMed] [Google Scholar]
- 18.Okabayashi H, Isemura T, Sakakibara S. Steric structure of l-proline oligopeptides. ii. Far-ultraviolet absorption spectra and optical rotations of l-proline oligopeptides. Biopolymers. 1968;6:323–330. doi: 10.1002/bip.1968.360060307. [DOI] [PubMed] [Google Scholar]
- 19.Helbecque N, Loucheux-Lefebvre MH. Critical chain length for polyproline-ii structure formation in h-gly-(pro)n-oh. Int J Pept Protein Res. 1982;19:94–101. doi: 10.1111/j.1399-3011.1982.tb03028.x. [DOI] [PubMed] [Google Scholar]
- 20.Crespo L, et al. Peptide dendrimers based on polyproline helices. J Am Chem Soc. 2002;124:8876–8883. doi: 10.1021/ja020364m. [DOI] [PubMed] [Google Scholar]
- 21.Momany FA, McGulre RF, Burgess AW, Scheraga HA. Energy parameters in polypeptides. vii. Geometric parameters, partial atomic charges, nonbonded interactions, hydrogen bond interactions, and intrinsic torsional potentials for the naturally occurring amino acids. J Phys Chem. 1975;79:2361–2381. [Google Scholar]
- 22.Madison V. Flexibility of the pyrrolidine ring in proline peptides. Biopolymers. 1977;16:2671–2692. [Google Scholar]
- 23.Vitagliano L, Berisio R, Mastrangelo A, Mazzarella L, Zagari A. Preferred proline puckerings in cis andtrans peptide groups: Implications for collagen stability. Protein Sci. 2001;10:2627–2632. doi: 10.1110/ps.ps.26601a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang YK, Choi HY. Cistrans isomerization and puckering of proline residue. Biophys Chem. 2004;111:135–142. doi: 10.1016/j.bpc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka S, Scheraga HA. Calculation of conformational properties of oligomers of l-proline. Macromolecules. 1974;7:698–705. doi: 10.1021/ma60041a029. [DOI] [PubMed] [Google Scholar]
- 26.Zhong H, Carlson HA. Conformational studies of polyprolines. J Chem Theory Comput. 2006;2:342–353. doi: 10.1021/ct050182t. [DOI] [PubMed] [Google Scholar]
- 27.Kang YK, Jhon JS, Park HS. Conformational preferences of proline oligopeptides. J Phys Chem B. 2006;110:17645–17655. doi: 10.1021/jp0629792. [DOI] [PubMed] [Google Scholar]
- 28.Brandts JF, Halvorson HR, Brennan M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry. 1975;14:4953–4963. doi: 10.1021/bi00693a026. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka S, Scheraga HA. Hypothesis about the mechanism of protein folding. Macromolecules. 1977;10:291–304. doi: 10.1021/ma60056a015. [DOI] [PubMed] [Google Scholar]
- 30.Schmid FX, Mayr LM, Mücke M, Schönbrunner ER. Prolyl isomerases: Role in protein folding. Adv Protein Chem. 1993;44:25–66. doi: 10.1016/s0065-3233(08)60563-x. [DOI] [PubMed] [Google Scholar]
- 31.Houry WA, Scheraga HA. Nature of the unfolded state of ribonuclease a: Effect of cis-trans x-pro peptide bond isomerization. Biochemistry. 1996;35:11719–11733. doi: 10.1021/bi960745a. [DOI] [PubMed] [Google Scholar]
- 32.Wedemeyer WJ, Welker E, Scheraga HA. Proline cis-trans isomerization and protein folding. Biochemistry. 2002;41:14637–14644. doi: 10.1021/bi020574b. [DOI] [PubMed] [Google Scholar]
- 33.Zimmerman SS, Pottle MS, Némethy G, Scheraga HA. Conformational analysis of the 20 naturally occurring amino acid residues using ecepp. Macromolecules. 1977;10:1–9. doi: 10.1021/ma60055a001. [DOI] [PubMed] [Google Scholar]
- 34.Vásquez M, Némethy G, Scheraga HA. Computed conformational states of the 20 naturally occurring amino acid residues and of the prototype residue a-aminobutyric acid. Macromolecules. 1983;16:1043–1049. [Google Scholar]
- 35.Némethy G, et al. Energy parameters in polypeptides: 10. Improved geometrical parameters and nonbonded interactions for use in the ecepp/3 algorithm, with application to proline-containing peptides. J Phys Chem. 1992;96:6472–6484. [Google Scholar]
- 36.Stein RL. Mechanism of enzymatic and nonenzymatic prolyl cis-trans isomerization. Adv Protein Chem. 1993;44:1–24. doi: 10.1016/s0065-3233(08)60562-8. [DOI] [PubMed] [Google Scholar]
- 37.Kang YK, Jhon JS, Han SJ. Conformational study of ac-xaa-pro-nhme dipeptides: Proline puckering and trans/cis imide bond. J Pept Res. 1999;53:30–40. doi: 10.1111/j.1399-3011.1999.tb01614.x. [DOI] [PubMed] [Google Scholar]
- 38.Woody RW. Circular dichroism and conformation of unordered polypeptides. Adv Biophys Chem. 1992;2:37–39. [Google Scholar]
- 39.Kelly MA, et al. Host–guest study of left-handed polyproline ii helix formation. Biochemistry. 2001;40:14376–14383. doi: 10.1021/bi011043a. [DOI] [PubMed] [Google Scholar]
- 40.Vila JA, Baldoni HA, Ripoll DR, Ghosh A, Scheraga HA. Polyproline ii helix conformation in a proline-rich environment: A theoritical study. Biophys J. 2004;86:731–742. doi: 10.1016/S0006-3495(04)74151-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makowska J, et al. Polyproline ii conformation is one of many local conformational states and is not overall conformation of unfolded peptides and proteins. Proc Natl Acad Sci USA. 2006;103:1744–1749. doi: 10.1073/pnas.0510549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stryer L, Haugland RP. Probing polyproline structure and dynamics by photoinduced electron transfer provides evidence for deviations from a regular polyproline type ii helix. Proc Natl Acad Sci USA. 1967;58:719–726. doi: 10.1073/pnas.0705605104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doose S, Neuweiler H, Barsch H, Sauer M. Probing polyproline structure and dynamics by photoinduced electron transfer provides evidence for deviations from a regular polyproline type ii helix. Proc Natl Acad Sci USA. 2007;104:17400–17405. doi: 10.1073/pnas.0705605104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka S, Scheraga HA. Calculation of the characteristic ratio of randomly coiled poly(l-proline) Macromolecules. 1975;8:623–631. doi: 10.1021/ma60047a009. [DOI] [PubMed] [Google Scholar]
- 45.Zimmerman SS, Scheraga HA. Stability of cis, trans, and nonplanar peptide groups. Macromolecules. 1976;9:408–416. doi: 10.1021/ma60051a005. [DOI] [PubMed] [Google Scholar]
- 46.Grathwohl C, Wuthrich K. Nmr studies of the rates of proline cis-trans isomerization in oligopeptides. Biopolymers. 1981;20:2623–2633. [Google Scholar]
- 47.Venkatachalam CM, Price BJ, Krimm S. A theoretical estimate of the energy barriers between stable conformations of the proline dimer. Biopolymers. 2004;14:1121–1132. doi: 10.1002/bip.1975.360140602. [DOI] [PubMed] [Google Scholar]
- 48.Kang YK. Conformational preferences of non-prolyl and prolyl residues. J Phys Chem B. 2006;110:21338–21348. doi: 10.1021/jp0647481. [DOI] [PubMed] [Google Scholar]
- 49.Jhon JS, Kang YK. Imide cis-trans isomerization of n-acetyl-n′-methylprolineamide and solvent effects. J Phys Chem A. 1999;103:5436–5439. [Google Scholar]
- 50.Babin V, Roland C, Sagui C. Adaptively biased molecular dynamics for free energy calculations. J Chem Phys. 2008;128:134101. doi: 10.1063/1.2844595. [DOI] [PubMed] [Google Scholar]
- 51.Case DA, et al. AMBER 10. San Francisco: University of California; 2008. [Google Scholar]
- 52.Babin V, Roland C, Darden TA, Sagui C. The free energy landscape of small peptides as obtained from metadynamics with umbrella sampling corrections. J Chem Phys. 2006;125:2049096. doi: 10.1063/1.2393236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Izrailev S, et al. Steered Molecular Dynamics. Computational Molecular Dynamics: Challenges, Methods, Ideas. Berlin: Springer; pp. 39–65. [Google Scholar]
- 54.Kang YK. Puckering transition of proline residue in water. J Phys Chem B. 2007;111:10550–10556. doi: 10.1021/jp073411b. [DOI] [PubMed] [Google Scholar]
- 55.Ensing B, Laio A, Parrinello M, Klein M. A recipe in the computation of the free energy barrier and the lowest free energy path of concerted reactions. J Phys Chem B. 2005;109:6676–6687. doi: 10.1021/jp045571i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.