

Abstract
Identifying points of control in inflammation is essential to discovering safe and effective antiinflammatory medicines. Palmitoylethanolamide (PEA) is a naturally occurring lipid amide that, when administered as a drug, inhibits inflammatory responses by engaging peroxisome proliferator-activated receptor-α (PPAR-α). PEA is preferentially hydrolyzed by the cysteine amidase N-acylethanolamine-hydrolyzing acid amidase (NAAA), which is highly expressed in macrophages. Here we report the discovery of a potent and selective NAAA inhibitor, N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide [(S)-OOPP], and show that this inhibitor increases PEA levels in activated leukocytes and blunts responses induced by inflammatory stimuli both in vitro and in vivo. These effects are stereoselective, mimicked by exogenous PEA, and abolished by PPAR-α deletion. (S)-OOPP also attenuates inflammation and tissue damage and improves recovery of motor function in mice subjected to spinal cord trauma. The results suggest that PEA activation of PPAR-α in leukocytes serves as an early stop signal that contrasts the progress of inflammation. The PEA-hydrolyzing amidase NAAA may provide a previously undescribed target for antiinflammatory medicines.
Keywords: NAAA, oleoylethanolamide, PPAR-α
Soon after its isolation from plant tissues (1), palmitoylethanolamide (PEA) was shown to reduce allergic reactions and inflammation in animals (2) and was briefly used to treat influenza symptoms in humans. Interest in the properties of this lipid amide lessened, however, until the characterization of its antiinflammatory (3), analgesic (4), and neuroprotective (5) effects and the identification of peroxisome proliferator-activated receptor-α (PPAR-α) as its primary molecular target (6, 7). Like other members of the nuclear receptor superfamily, PPAR-α is activated through ligand binding, which promotes heterodimer formation with the 9-cis-retinoic acid receptor, recruitment of coactivators into a multiprotein complex, and regulated expression of responsive genes. PPAR-α controls transcriptional programs involved in the development of inflammation through mechanisms that include direct interactions with the proinflammatory transcription factors NF-κB and AP1, and modulation of IκB function (8). Pharmacologic studies have demonstrated that PPAR-α agonists are therapeutically effective in rodent models of inflammatory and autoimmune diseases (9). Furthermore, mutant PPAR-α-deficient mice seem to be vulnerable to various inflammatory stimuli (9), suggesting that endogenous PPAR-α activity negatively regulates the initiation of acute inflammatory responses.
The multifunctional ligand-binding pocket of PPAR-α allows this protein to recognize 3 distinct classes of natural agonists: nonesterified fatty acids, which activate PPAR-α with potencies in the midmicromolar range (median effective concentration, EC50, 2–20 μM) (10); oxygenated fatty acids, such as (8S)-hydroxy-eicosatetraenoic acid (11) and (8R)-hydroxy-11(R),12(R)-epoxyeicosa-5Z,9E,14Z-trienoic acid (12) (EC50 0.3–1 μM); and lipid amides such as PEA and its analog oleoylethanolamide (OEA) (EC50 0.1–3 μM) (6, 13). Innate immune cells produce significant amounts of PEA and OEA (14) and express a selective phospholipase D (PLD) that releases these endogenous PPAR-α agonists from their membrane phospholipid precursor, N-acylphosphatidylethanolamine (NAPE) (15). Inflammatory cells also express 2 intracellular amidases that have been implicated in lipid amide degradation: fatty-acid amide hydrolase (FAAH) (16) and N-acylethanolamine-hydrolyzing acid amidase (NAAA) (17, 18). Potent FAAH inhibitors, such as the compound URB597 (19), have been used to unmask the functions of the substrate preferred by FAAH, the endogenous cannabinoid agonist anandamide (20, 21). By contrast, effective inhibitors of intracellular NAAA activity, which preferentially recognizes PEA (17), have not been reported yet, and the roles played by endogenous PEA in the control of inflammatory responses remain unknown.
Results
Discovery of a Potent and Selective NAAA Inhibitor.
NAAA is a cysteine hydrolase that belongs to the N-terminal nucleophile (Ntn) family of enzymes (17, 22). To discover new NAAA inhibitors, we generated a 3-dimensional model of the catalytic site of NAAA (Fig. 1A) using the crystallographic coordinates of conjugated bile acid hydrolase (CBAH), another Ntn cysteine hydrolase that shares with NAAA a highly conserved sequence in the catalytic N-terminal region (Fig. S1) (23–26). According to our model, the tetrahedral intermediate formed through attack of catalytic cysteine 131 (17) on PEA is stabilized by electrostatic interactions between the carbonyl oxygen of PEA and the enzyme oxyanion hole, which includes the side-chain amide of the conserved asparagine 292 and the backbone amide of asparagine 209. Aspartate 150 is involved in the stabilization of C131, whereas a hydrophobic pocket lined by tyrosine 151, among other residues, accommodates the flexible acyl chain of PEA. Site-directed mutagenesis experiments confirmed that C131, N292, and D150 are essential for NAAA function because the mutants C131A, C131S, N292A, and D150E were devoid of enzyme activity (Fig. 1B). Moreover, the negative impact of mutations targeting Y151 (Y151A and Y151F) supports a contribution of the presumptive acyl chain-binding pocket to enzyme function (Fig. 1B). Control mutations distal to the active site, such as alanine 176 (A176G), had no effect on NAAA activity (Fig. 1B). We used the active-site properties predicted by our model to select a set of commercial molecules, which were then tested for their ability to inhibit NAAA. Although the enzyme was insensitive to most agents—including the FAAH inhibitor URB597 (Fig. 2A) and the compound N-(cyclohexylcarbonyl)pentadecylamine (17, 27) (Table S1)—the screening identified the serine β-lactone 1 as an NAAA inhibitor of low micromolar potency (Fig. 2A and Table 1).
Fig. 1.

Properties of the active site of NAAA. (A) Computational model illustrating the tetrahedral intermediate of PEA (green) within the active site of rat NAAA (gray). Red, oxygen; fuchsia, nitrogen; blue, hydrogen. Hydrogen bonds are symbolized by orange lines. (B) Site-directed mutagenesis of rat NAAA; NAAA activity is expressed as percentage of wild-type control (w.t.). ***, P < 0.001 vs. wild-type.
Fig. 2.

Characterization of the NAAA inhibitor (S)-OOPP. (A) Concentration-dependent inhibition of NAAA activity by (S)-OOPP (filled triangles), compound 1 (filled squares), (R)-OOPP (open triangles), and URB597 (filled circles). (B) Kinetic analysis of NAAA inhibition by (S)-OOPP (1 μM). Maximal reaction velocity (Vmax) and Michaelis constant are expressed in pmol·min−l·mg protein−1 and μmol/L, respectively. Vehicle, open squares; (S)-OOPP, filled squares. (C and D) Effects of (S)-OOPP (S) and its enantiomer (R)-OOPP (R) on (C) carrageenan (Carr)-induced (0.1 mL, 1%) reduction in PEA levels in infiltrating leukocytes in vivo; and (D) LPS-induced reduction in PEA levels in RAW264.7 macrophages in vitro. **, P < 0.01 vs. vehicle/vehicle; ##, P < 0.01 vs. carrageenan/vehicle; ###, P < 0.001 vs. LPS/vehicle (n = 3–8).
Table 1.
Structures of selected NAAA inhibitors
| IC50 (nM) | |||
|---|---|---|---|
| R | R1 | ||
| 1 | 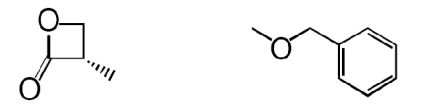 |
2,960 ± 300 | |
| 2 | 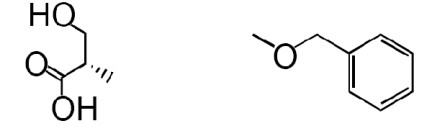 |
>100,000 | |
| 3 (S)-OOPP | 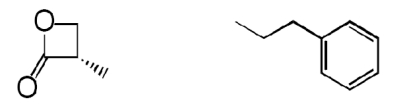 |
420 ± 20 | |
| 4 | 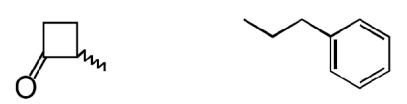 |
>100,000 | |
| 5 | 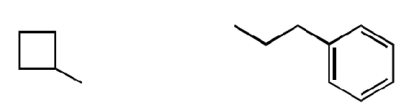 |
>100,000 | |
| 6 (R)-OOPP | 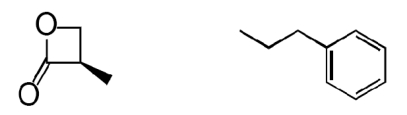 |
6000 ± 600 | |
Values reported are the concentrations required to inhibit NAAA activity by 50% (IC50) in nM and are expressed as mean ± SEM of at least three independent experiments. They were calculated from concentration response curves using nonlinear regression analysis in the Prism 4.0 software package.
The β-lactone group of compound 1 binds covalently the catalytic cysteine of hepatitis A virus proteinase 3C, blocking its activity (28). Focused structure–activity relationship studies revealed that the ability of 1 to inhibit NAAA also depended on the β-lactone ring, rather than the carbamate fragment, because opening the β-lactone abrogated inhibitory activity (Table 1, compound 2), whereas removing the carbamate group yielded a compound [N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide; (S)-OOPP] that was significantly more potent than 1 at inhibiting NAAA (Fig. 2A and Table 1, compound 3). The role of the β-lactone was confirmed by the inactivity of the cyclobutanone and cyclobutane analogs of (S)-OOPP, compounds 4 and 5 (Table 1). Moreover, the markedly diminished potency displayed by the enantiomer, (R)-OOPP (Fig. 2A and Table 1, compound 6), highlighted the selective recognition of (S)-OOPP by NAAA. Such selectivity might account for the weak interaction of the inhibitor with acid ceramidase (IC50 = 10.9 ± 3.1 μM; n = 4), a cysteine amidase that is both structurally and functionally related to NAAA (17). In addition, (S)-OOPP did not inhibit FAAH (IC50 >100 μM) or several other serine hydrolases that use lipids as substrates (Table S2). Kinetic analyses revealed that (S)-OOPP blocks NAAA through a noncompetitive mechanism (Fig. 2B). The results indicate that (S)-OOPP is a potent, selective, and noncompetitive NAAA inhibitor.
NAAA Inhibition Normalizes PEA Levels in Activated Inflammatory Cells.
To test whether (S)-OOPP inhibits NAAA activity in intact cells we first induced a localized inflammatory response in mice by implanting s.c. polyethylene sponges instilled with the proinflammatory polysaccharide carrageenan. Three days after surgery, we collected infiltrating cells—mostly neutrophils (90.3% ± 2.0%) and macrophages (6.1% ± 0.9%, mean ± SEM; n = 4)—and analyzed their lipid content by liquid chromatography/mass spectrometry (LC/MS). Consistent with previous results (29, 30), the chemoattractant caused a marked decrease in cellular PEA (Fig. 2C). The limited plasma stability of (S)-OOPP (half-life <1 min) precluded its systemic administration in vivo, but when the compound was instilled into the sponges (25 μg per sponge) it prevented in a stereoselective manner the carrageenan-induced reduction in leukocyte PEA levels (Fig. 2C), suggesting that it effectively inhibited NAAA activity in these cells. In additional experiments, we stimulated RAW264.7 macrophages in cultures with LPS, which also caused a reduction in cellular PEA content (Fig. 2D). Incubation with (S)-OOPP, but not (R)-OOPP (each at 10 μM), inhibited NAAA activity in these cells, as assessed ex vivo using a standard enzyme assay (Fig. S2A), and blocked the LPS-induced reduction of PEA levels (Fig. 2D). Furthermore, (S)-OOPP (3–30 μM) increased PEA levels in NAAA-overexpressing HEK293 cells exposed to the calcium ionophore ionomycin (Fig. S2B). That the effects of (S)-OOPP on PEA levels were restricted to cells stimulated with LPS or ionomycin suggests that inflammatory triggers might activate NAAA (through autoproteolysis or other posttranslation modifications) or facilitate its access to cellular pools of PEA. Finally, treatment with (S)-OOPP did not affect the levels of the endocannabinoid anandamide, which were increased by LPS (Fig. S2C) (15), or those of ceramide (Fig. S3), confirming that (S)-OOPP preferentially inhibits NAAA over other lipid amidases, such as FAAH and acid ceramidase.
NAAA Inhibition Reduces Neutrophil Migration.
The discovery of (S)-OOPP allowed us to ask whether preventing PEA hydrolysis by NAAA might modulate inflammatory responses. Addition of (S)-OOPP to carrageenan-containing sponges increased PEA levels and inhibited leukocyte migration in a dose-dependent and stereoselective manner (Fig. 3 A and B). Histologic analyses showed that (S)-OOPP selectively reduced the infiltration of neutrophils into the sponges (Fig. S4). (S)-OOPP also inhibited plasma extravasation induced by carrageenan (Fig. S5A). The ability of (S)-OOPP to decrease inflammatory responses was likely mediated by endogenous PEA acting as an agonist at PPAR-α. Consistent with this idea, we found that the antiinflammatory effects of (S)-OOPP were abrogated by targeted PPAR-α deletion (Fig. 3C), which did not influence PEA production (Fig. 3D), and were mimicked either by exogenous PEA or the synthetic PPAR-α agonist GW7647 in a PPAR-α-dependent manner (Fig. 4). Notably, (S)-OOPP did not directly stimulate PPAR-α in a cellular transactivation assay (EC50 >100 μM) and did not inhibit the eicosanoid-synthesizing enzymes cyclooxygenase-2 and 5-lipoxygenase (IC50 >100 μM). We have not yet identified the precise cellular site of action of (S)-OOPP. Experiments in RAW264.7 cells show, however, that the compound reduces LPS-induced expression of iNOS, but not TNF-α (Fig. S5 B and C), which suggests that NAAA inhibition in activated macrophages may normalize PEA levels and blunt signaling downstream of TNF-α, as previously suggested for direct-acting PPAR-α agonists (31).
Fig. 3.

Effects of (S)-OOPP on carrageenan (Carr)-induced neutrophil migration in mice. (A and B), Dose-dependent effects of (S)-OOPP (filled squares) and its enantiomer (R)-OOPP (open circles) on (A) cellular PEA levels and (B) leukocyte infiltration. Vehicle, open triangle. (C and D) Effects of (S)-OOPP (25 μg per sponge) on (C) leukocyte infiltration and (D) cellular PEA levels in wild-type (+/+) and PPAR-α−/− (−/−) mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. vehicle; #, P < 0.01 vs. carrageenan/vehicle; ##, P < 0.001 vs. carrageenan/vehicle (n = 5–10).
Fig. 4.

Antiinflammatory effects of PEA and synthetic PPAR-α agonist GW7647. Effects of (A) PEA or (B) GW7647 (each at 50 μg per sponge) on leukocyte infiltration in s.c. sponges instilled with vehicle (sterile saline, 0.1 mL) or carrageenan (Carr, 1%) in wild-type C57BL/6J (+/+) and PPAR-α−/− (−/−) mice. **, P < 0.01 vs. vehicle; ***, P < 0.001 vs. vehicle; ###, P < 0.001 vs. carrageenan/vehicle (n = 5–6).
Effects of FAAH Inhibition.
PEA is a preferred substrate for NAAA but can also be hydrolyzed by FAAH (16). To assess the contribution of FAAH to PEA hydrolysis in inflammatory cells, we tested the effects of the FAAH inhibitor URB597 (20). Even though leukocytes express FAAH (32), administration of URB597 neither affected PEA levels nor counteracted the proinflammatory effects of carrageenan in vivo (Fig. S6 A–C) or those of LPS in vitro (Fig. S6 D and E). Previous studies have shown that mutant mice lacking peripheral FAAH have a reduced sensitivity to various inflammatory challenges (33). We found, however, that FAAH−/− mice display normal suppression of PEA levels (Fig. S6F) and normal neutrophil infiltration and plasma extravasation in response to carrageenan (Fig. S6 G and H). We interpret these results to indicate that, under our experimental conditions, FAAH does not play an obligatory role in the degradation of PEA by activated leukocytes.
NAAA Inhibition Reduces Spinal Cord Injury.
To further characterize the antiinflammatory properties of (S)-OOPP, we determined the effects of this compound on the tissue response produced by traumatic spinal cord injury (SCI) in mice, which is known to be accompanied by profound inflammation (34) and changes in PEA levels (35). We induced SCI by applying vascular clips to the dura mater via a 4-level T5–T8 laminectomy (36). This resulted in marked neutrophil infiltration, production of inflammatory mediators, apoptosis, and edema (Fig. 5 A–G and Fig. S7). Previous studies have shown that these effects are reduced by administration of exogenous PEA and are magnified in PPAR-α−/− mice (36). Two consecutive intrathecal injections of (S)-OOPP (30 μg per mouse, 1 and 6 h after SCI induction) strongly reduced tissue injury (Fig. 5 A–D) and inhibited expression of inflammation and apoptosis markers, such as iNOS, tissue nitrotyrosine, and TUNEL staining (Fig. 5 E–G). Furthermore, administration of (S)-OOPP produced a marked improvement in the recovery of motor limb function (Fig. 5H), suggesting that the treatment attenuates the neural inflammation and motor deficits produced by SCI in mice.
Fig. 5.

Antiinflammatory effects of (S)-OOPP in SCI mice. (A–C) Immunohistochemical staining of sections of spinal cord tissue for iNOS in (A) sham-operated mice treated with vehicle (saline, intrathecal); (B) SCI mice treated with vehicle; and (C) SCI mice treated with (S)-OOPP (30 μg per mouse, 1 and 6 h after SCI, intrathecal). (D–G) Effects of (S)-OOPP on (D) histologic score; (E) iNOS immunoreactivity; (F) nitrotyrosine immunoreactivity; and (G) TUNEL staining. (H) Effect of (S)-OOPP (filled squares; open squares, vehicle) on recovery from motor impairment, as assessed daily using the modified murine Basso, Beatty, and Bresnahan hind limb locomotor rating scale. *, P < 0.05 vs. vehicle; #, P < 0.05 vs. SCI/vehicle; (n = 6–10); gm, gray matter; wm, white matter; nd, not detectable.
Discussion
Previous studies have shown that PEA and other lipid amides activate the antiinflammatory nuclear receptor PPAR-α in a range of concentrations (0.1–3 μM) (6, 13) that occur normally in tissues (6, 14). Furthermore, exogenous PEA exerts profound antiinflammatory effects (3) that are mediated by PPAR-α (6), and mice lacking this receptor display a heightened sensitivity to inflammatory challenges and spinal cord trauma (37). The present report discloses a potent and selective inhibitor of the PEA-degrading enzyme, NAAA, and demonstrates that NAAA blockade normalizes PEA levels in activated inflammatory cells and dampens tissue reactions to various proinflammatory triggers. These results suggest that endogenous PEA acting at PPAR-α provides a no-go signal that hinders the development of acute inflammation. Such a role may be analogous to those documented for annexin-1 (38) and other antiinflammatory proteins (34, 39) but clearly differentiates PEA from known lipid mediators, which either incite the inflammatory process (e.g., prostaglandins) (31) or terminate it by promoting resolution and tissue healing (e.g., lipoxins and resolvins) (40, 41). Interestingly, a recent report suggests that circulating oleic acid may exert a tonic inhibitory tone on chemotaxis, although its mechanism of action has not been elucidated (42).
Is PEA signaling at PPAR-α relevant to human inflammatory states? An initial answer to this question, which will require further experimental confirmation, is provided by a recent report showing that the levels of PEA found in synovial fluid from rheumatoid arthritis and osteoarthritis patients are strikingly lower than those measured in healthy subjects (43). Because experimental inflammatory triggers also decrease PEA levels in animals, it is tempting to speculate that a deficit in PEA signaling may contribute to the pathogenesis of chronic inflammation. If this is the case, then pharmacologic strategies aimed at correcting a deficit in PEA/PPAR-α signaling might provide a new mechanism for the treatment of inflammatory disorders. One such strategy consists in activating PPAR-α by the use of potent and selective receptor agonists (9). However, the prolonged clinical use of these agents has been linked to a variety of untoward effects, which include oncogenesis, renal dysfunction, rhabdomyolysis, and cardiovascular toxicity (44). A possible alternative might be to magnify endogenous PEA activity at PPAR-α by protecting this lipid amide from NAAA-catalyzed degradation. The present findings, demonstrating that NAAA blockade reduces inflammatory responses in various experimental models, provisionally validate the latter approach. Notably, our results show that genetic or pharmacologic interruption of FAAH activity does not prevent the proinflammatory effects of LPS in vitro or those of carrageenan in vivo, implying that FAAH may not contribute to the termination of PEA signaling in those models. However, genetically modified mice that lack FAAH in peripheral tissues display a hypoinflammatory phenotype (33), and antiinflammatory effects of FAAH inhibitors have been reported in other models (45), which suggests that the roles played by FAAH and NAAA in regulating inflammation might be context dependent.
In conclusion, inhibition of NAAA activity by (S)-OOPP reveals a role for endogenous PEA as a negative regulator of tissue responses to proinflammatory stimuli. The decrease in synovial PEA levels observed in rheumatoid arthritis and osteoarthritis patients (43) suggests that this mechanism may be disrupted in human inflammatory pathologies. A second generation of systemically active NAAA inhibitors—possibly derived from the scaffold provided by (S)-OOPP—might help combat inflammation by correcting deficits in PEA signaling.
Materials and Methods
Compound Syntheses.
(S)-OOPP and (R)-OOPP were prepared in 3 steps from N-Boc-L-serine and N-Boc-D-serine, respectively. Mitsunobu (46) cyclization of these products gave lactone intermediates, which were deprotected and salified (47). Coupling of the resulting tosylates with 3-phenylpropionyl chloride according to standard procedures (48) afforded the desired compounds. Compound 5 was synthesized similarly from 3-phenylpropionyl chloride and cyclobutylamine. A published procedure (49) for the synthesis of benzyloxycarbonylaminocyclobutanone was applied to the synthesis of compound 4, which was obtained from the reaction of bis(trimethylsilyloxy)cyclobutene and 3-phenylpropionamide. Additional synthetic details will be provided elsewhere. A first batch of N-(cyclohexylcarbonyl)pentadecylamine was purchased from Cayman Chemicals, and a second one was prepared according to a published procedure (17) and purified by flash chromatography (silica gel; cyclohexane/ethyl acetate 8:2). The compound yielded white crystal needles, an Mp of 89 °C (in acetone) and a single peak (m/z 338) by LC/MS, positive electrospray ionization (M+H). The 1H NMR (CDCl3) spectrum revealed minor differences relative to the published data (17): δ = 0.88 (t, 3H, J = 6.5 Hz), 1.21–1.51 (m, 31H), 1.69–1.89 (m, 5H), 1.98–2.11 (m, 1H), 3.18–3.28 (m, 2H), 5.44 (br s, 1H) ppm. Neither batch significantly inhibited recombinant NAAA activity in our tests. Solvents were from Burdick and Jackson.
Molecular Modeling.
The amino acid sequence of the mature form of rat NAAA (rNAAA, amino acids 131–362 of Q5KTC7 in the SWISS-PROT/TrEMBL database) was used as a query for the automatic fold recognition server PHYRE (formerly known as 3D-PSSM). CBAH from Clostridium perfringens (2BJF in the Protein Data Bank) and resulted in the best reference template (identity score 11%, similarity score 23%). Limited modification of the 2D sequence alignment proposed by PHYRE allowed the superposition of 2 NAAA asparagines (N209 and N292 in the rat) to N82 and N175 of CBAH, which play a critical role in the catalytic activity of CBAH. The resulting alignment was used to build 3-dimensional models of NAAA using MODELLER 7.0 (50) and applying standard settings for loop modeling. The overall geometric quality of the structures was assessed by PROCHECK (51), and the NAAA model having the highest G-factor was selected and used for modeling purposes. Hydrogen atoms were added by the Biopolymer module of Sybyl 7.2 (Tripos), choosing the histidine tautomers that maximize the number of hydrogen bonds within the protein. An energy minimization was performed to optimize the geometry of the added hydrogen atoms using the force field MMFF94s (52) to an energy gradient of 0.05 kcal/(mol·Å). PEA was docked into the NAAA binding site, by choosing a pose consistent with bond formation between the carbonyl carbon of the substrate and the sulfur atom of C131, and accommodating the acyl chain within the lipophilic pocket corresponding to that occupied by the bile acid in the CBAH template. Position and conformation of PEA were then optimized by the Sybyl 7.2 Dock_minimize procedure and by energy minimization of the complex to an energy gradient of 0.2 kcal/(mol·Å). Starting from the Michaelis complex, the PEA-NAAA tetrahedral intermediate was built by imposing a covalent bond between the amide carbon atom and the sulfur atom of C131 and reassigning the atom types. The resulting structure was minimized to an energy gradient of 0.2 kcal/(mol·Å) and submitted to molecular dynamics simulation, using the force field MMFF94 implemented in Sybyl 7.2. A time step of 1 fs was applied with a nonbonded cutoff of 8 Å and dielectric constant set to 1. During the simulation, only the protein side chains and the ligand were allowed to move. A heating phase of 50 ps at 300 K was followed by 500 ps of simulation at the same temperature. The last snapshot structure was finally minimized using MMFF94s to an energy gradient of 0.2 kcal/(mol·Å) without restraints.
Animals and Cells.
Male Swiss mice (20–25 g) were from Charles River, C57BL/6J wild-type mice and C57BL/6J PPAR-α−/− mice (B6.129S2-Pparatm1GonzN12) (20–25 g) were from Jackson Laboratories. All procedures met the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the University of California-Irvine Institutional Animal Care and Use Committee. HEK293 and RAW264.7 cells were from American Type Culture Collection and were cultured in DMEM (Invitrogen) supplemented with FBS (10%; Invitrogen).
Animal Treatments.
Carrageenan-induced inflammation.
Sterile polyethylene sponges (1 cm3) were implanted under the dorsal skin of mice. Carrageenan (0.1–1.0% in 90 μL sterile water per sponge), drugs, or vehicles (DMSO, 10 μL per sponge) were instilled into the sponges, wounds were sutured, and mice were allowed to recover. After 6–72 h, mice were killed and sponges collected. Exudate volume was measured, and cells were counted using a hemocytometer.
Spinal cord injury.
Mice were anesthetized using chloral hydrate (400 mg/kg). Using the clip compression model described by Rivlin and Tator, we produced SCI by extradural compression of a section of the spinal cord exposed via a 4-level T5–T8 laminectomy, in which the prominent spinous process of T5 was used as a surgical guide. A 6-level laminectomy was chosen to expedite timely harvest and to obtain enough tissue for biochemical examination. With the aneurysm clip applicator oriented in the bilateral direction, an aneurysm clip with a closing force of 24 g was applied extradurally at T5–T8 level. The clip was rapidly released with a clip applicator, which caused cord compression. In the injured groups, the cord was compressed for 1 min. After surgery, saline (1.0 mL) was administered s.c. After surgery, the mice were placed on a warm heating pad and were then singly housed in a temperature-controlled room at 27 °C for 10 days with food and water available ad libitum. During this time, the animals' bladders were manually voided twice per day until the mice were able to regain normal bladder function. Sham-injured animals were only subjected to laminectomy. Mice were randomized into groups of 10 each. Vehicle (saline) or (S)-OOPP (30 μg per mouse) was administered intrathecally 1 h and 6 h after surgery. Animals were killed 24 h after surgery.
Statistics.
Results are expressed as mean ± SEM of n observations. They were analyzed by one-way ANOVA followed by Bonferroni post hoc test for multiple comparisons. P values <0.05 were considered to be significant.
Other Methods.
See SI Materials and Methods for remaining methods, including plasmid preparation, RNA extraction and real-time PCR, expression of recombinant proteins, protein analyses, enzyme assays, PPAR-α transactivation assay, lipid analyses, immunohistochemistry, TUNEL assay, light microscopy, and grading of motor disturbances.
Supplementary Material
Acknowledgments.
We thank Drs. J. Fu and K. M. Jung for experimental advice; and N. Barsegyan, A. Der Mugrdechian, and C. Van for assistance. Financial support was provided by grants from the Sandler Program for Asthma Research, and the National Institute on Drug Abuse (to D.P.); and from the University of Urbino and Italian Ministry for Instruction, University and Research Grant 2005032713–002 (to G.T.).
Footnotes
Conflict of interest: D.P., G.T., A.D., A.T., and M. Mor are coinventors on a patent application filed by the Universities of California, Parma, and Urbino “Carlo Bo.”
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0907417106/DCSupplemental.
References
- 1.Long D, Martin A. Factors in arachis oil depressing sensitivity to tuberculin in B.C.G. -infected guinea pigs. Lancet. 1956;1:464–466. doi: 10.1016/s0140-6736(56)90529-3. [DOI] [PubMed] [Google Scholar]
- 2.Benvenuti F, Lattanzi F, De Gori A, Tarli P. Attivita' di alcuni derivati della palmitoiletanolamide sull'edema da carragenina nella zampa di ratto. Boll Soc It Biol Sper. 1968;44:809–813. [PubMed] [Google Scholar]
- 3.Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A. N-(2-hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur J Pharmacol. 1996;300:227–236. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- 4.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 5.Lambert DM, Vandevoorde S, Diependaele G, Govaerts SJ, Robert AR. Anticonvulsant activity of N-palmitoylethanolamide, a putative endocannabinoid, in mice. Epilepsia. 2001;42:321–327. doi: 10.1046/j.1528-1157.2001.41499.x. [DOI] [PubMed] [Google Scholar]
- 6.Lo Verme J, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 7.LoVerme J, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 8.Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- 9.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Kliewer SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci USA. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muga SJ, et al. 8S-lipoxygenase products activate peroxisome proliferator-activated receptor alpha and induce differentiation in murine keratinocytes. Cell Growth Differ. 2000;11:447–454. [PubMed] [Google Scholar]
- 12.Yu Z, Schneider C, Boeglin WE, Brash AR. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARalpha. Lipids. 2007;42:491–497. doi: 10.1007/s11745-007-3054-4. [DOI] [PubMed] [Google Scholar]
- 13.Fu J, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 14.Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, et al. A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 17.Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA) Chem Biodivers. 2007;4:1914–1925. doi: 10.1002/cbdv.200790159. [DOI] [PubMed] [Google Scholar]
- 18.Ueda N, Yamanaka K, Yamamoto S. Purification and characterization of an acid amidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J Biol Chem. 2001;276:35552–35557. doi: 10.1074/jbc.M106261200. [DOI] [PubMed] [Google Scholar]
- 19.Mor M, et al. Synthesis and quantitative structure-activity relationship of fatty acid amide hydrolase inhibitors: Modulation at the N-portion of biphenyl-3-yl alkylcarbamates. J Med Chem. 2008;51:3487–3498. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kathuria S, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 21.Di Marzo V. Targeting the endocannabinoid system: To enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 22.Tsuboi K, et al. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem. 2005;280:11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- 23.Brannigan JA, et al. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature. 1995;378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 24.Rossocha M, Schultz-Heienbrok R, von Moeller H, Coleman JP, Saenger W. Conjugated bile acid hydrolase is a tetrameric N-terminal thiol hydrolase with specific recognition of its cholyl but not of its tauryl product. Biochemistry. 2005;44:5739–5748. doi: 10.1021/bi0473206. [DOI] [PubMed] [Google Scholar]
- 25.Kumar RS, et al. Structural and functional analysis of a conjugated bile salt hydrolase from Bifidobacterium longum reveals an evolutionary relationship with penicillin V acylase. J Biol Chem. 2006;281:32516–32525. doi: 10.1074/jbc.M604172200. [DOI] [PubMed] [Google Scholar]
- 26.Shtraizent N, et al. Autoproteolytic cleavage and activation of human acid ceramidase. J Biol Chem. 2008;283:11253–11259. doi: 10.1074/jbc.M709166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vandevoorde S, et al. Esters, retroesters, and a retroamide of palmitic acid: Pool for the first selective inhibitors of N-palmitoylethanolamine-selective acid amidase. J Med Chem. 2003;46:4373–4376. doi: 10.1021/jm0340795. [DOI] [PubMed] [Google Scholar]
- 28.Lall MS, Karvellas C, Vederas JC. Beta-lactones as a new class of cysteine proteinase inhibitors: Inhibition of hepatitis A virus 3C proteinase by N-Cbz-serine beta-lactone. Org Lett. 1999;1:803–806. doi: 10.1021/ol990148r. [DOI] [PubMed] [Google Scholar]
- 29.Capasso R, et al. Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br J Pharmacol. 2001;134:945–950. doi: 10.1038/sj.bjp.0704339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D. The search for the palmitoylethanolamide receptor. Life Sci. 2005;77:1685–1698. doi: 10.1016/j.lfs.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Flower RJ. Prostaglandins, bioassay and inflammation. Br J Pharmacol. 2006;147(Suppl 1):S182–S192. doi: 10.1038/sj.bjp.0706506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun YX, et al. Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochim Biophys Acta. 2005;1736:211–220. doi: 10.1016/j.bbalip.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Cravatt BF, et al. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Natl Acad Sci USA. 2004;101:10821–10826. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Ovejero D, et al. The endocannabinoid system is modulated in response to spinal cord injury in rats. Neurobiol Dis. 2008;33:57–71. doi: 10.1016/j.nbd.2008.09.015. 2009; [DOI] [PubMed] [Google Scholar]
- 36.Genovese T, et al. Effects of palmitoylethanolamide on signaling pathways implicated in the development of spinal cord injury. J Pharmacol Exp Ther. 2008;326:12–23. doi: 10.1124/jpet.108.136903. 2008; [DOI] [PubMed] [Google Scholar]
- 37.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 38.D'Acquisto F, Perretti M, Flower RJ. Annexin-A1: A pivotal regulator of the innate and adaptive immune systems. Br J Pharmacol. 2008;155:152–169. doi: 10.1038/bjp.2008.252. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 40.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takano T, et al. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: Evidence for anti-inflammatory receptors. J Exp Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malawista SE, de Boisfleury Chevance A, van Damme J, Serhan CN. Tonic inhibition of chemotaxis in human plasma. Proc Natl Acad Sci USA. 2008;105:17949–17954. doi: 10.1073/pnas.0802572105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson D, et al. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res Ther. 2008;10:R43. doi: 10.1186/ar2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nissen SE, et al. Effects of a potent and selective PPAR-alpha agonist in patients with atherogenic dyslipidemia or hypercholesterolemia: Two randomized controlled trials. JAMA. 2007;297:1362–1373. doi: 10.1001/jama.297.12.1362. [DOI] [PubMed] [Google Scholar]
- 45.Sagar DR, Kendall DA, Chapman V. Inhibition of fatty acid amide hydrolase produces PPAR-alpha-mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol. 2008;155:1297–1306. doi: 10.1038/bjp.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnold LD, Kalantar TH, Vederas JC. Conversion of serine to stereochemically optically pure β-substituted α-amino acids via β-lactones. J Am Chem Soc. 1985;107:7105–7109. [Google Scholar]
- 47.Arnold LD, May RG, Vederas JC. Synthesis of optically pure α-amino-β-propiolactone. J Am Chem Soc. 1988;110:2237–2241. [Google Scholar]
- 48.Montalbetti CAGN, Falque V. Amide bond formation and peptide coupling. Tetrahedron. 2005;61:10827–10852. [Google Scholar]
- 49.Lall MS, Ramtohul YK, James MN, Vederas JC. Serine and threonine beta-lactones: a new class of hepatitis A virus 3C cysteine proteinase inhibitors. J Org Chem. 2002;67:1536–1547. doi: 10.1021/jo0109016. [DOI] [PubMed] [Google Scholar]
- 50.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 51.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 52.Halgren TA. Merck molecular force field. I. Basis, form, scope, parametrization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.