

Abstract
This study describes the photochemical deposition of Co-based oxygen evolution catalysts on a semiconductor photoanode for use in solar oxygen evolution. In the photodeposition process, electron-hole pairs are generated in a semiconductor upon illumination and the photogenerated holes are used to oxidize Co2+ ions to Co3+ ions, resulting in the precipitation of Co3+-based catalysts on the semiconductor surface. Both photodeposition of the catalyst and solar O2 evolution are photo-oxidation reactions using the photogenerated holes. Therefore, photodeposition provides an efficient way to couple oxygen evolution catalysts with photoanodes by naturally placing catalysts at the locations where the holes are most readily available for solar O2 evolution. In this study Co-based catalysts were photochemically deposited as 10–30 nm nanoparticles on the ZnO surface. The comparison of the photocurrent-voltage characteristics of the ZnO electrodes with and without the presence of the Co-based catalyst demonstrated that the catalyst generally enhanced the anodic photocurrent of the ZnO electrode with its effect more pronounced when the band bending is less significant. The presence of Co-based catalyst on the ZnO photoanode also shifted the onset potential of the photocurrent by 0.23 V to the negative direction, closer to the flat band potential. These results demonstrated that the cobalt-based catalyst can efficiently use the photogenerated holes in ZnO to enhance solar O2 evolution. The photodeposition method described in this study can be used as a general route to deposit the Co-based catalysts on any semiconductor electrode with a valence band edge located at a more positive potential than the oxidation potential of Co2+ ions.
Keywords: photocurrent, photoelectrochemical cell, solar energy conversion, zinc oxide, photodeposition
Recently, Nocera and coworkers reported a simple anodic electrodeposition route to produce an oxygen-evolving catalyst (OEC) by using an aqueous neutral phosphate medium containing Co2+ ions (1). The deposition mechanism involves oxidation of Co2+ ions to Co3+ ions that have a limited solubility in a pH 7 aqueous solution, resulting in precipitation of Co3+ ions on the working electrode as oxide or hydroxide containing a significant amount of phosphates (1–4). These deposits are amorphous and their structure and the exact catalytic mechanism are not yet fully understood. However, this OEC provides many attractive features (e.g., simple synthesis procedure, low cost, mild operation condition, and self-repair mechanism) and, therefore, further studies to better understand and exploit this system will be highly beneficial (1–4).
Nocera and coworkers deposited the Co-based catalyst on a conducting substrate [e.g., indium doped tin oxide (ITO)], which serves as a dark anode (anode that is not coupled with light) to evolve O2 under anodic bias (1–4). When this Co-based catalyst is deposited on an n-type semiconductor electrode (photoanode), it can directly use photogenerated holes to evolve O2 without applying an external bias if the valence band edge of the photoanode is located at a more positive potential than the O2 evolution potential (5). The photogenerated electrons can be used for photoreduction reactions (e.g., H2 evolution) at the dark cathode or photocathode coupled to the photoanode (6–8). This photoelectrochemical cell can combine a solar cell and an electrolyzer into one device, and has the potential of significantly reducing the production cost for the photoelectolysis of water (6–8).
In this study we demonstrate a photochemical method to place Co-based catalysts on a photoanode to enhance solar O2 evolution. In photochemical deposition, no external bias is used to produce the catalyst. Instead photogenerated holes in a semiconductor are used to oxidize Co2+ ions to Co3+ ions to precipitate the Co-based catalyst onto the semiconductor surface. Photochemical deposition offers a distinctive advantage over electrodeposition for combining OECs with a photoanode. Because both photodeposition of the Co-based catalyst and solar O2 evolution are photo-oxidation reactions using the photogenerated holes, photodeposition inherently places Co-based OECs at the locations where the holes are most readily available for solar O2 evolution. This ensures the most efficient use of the catalyst resulting in enhanced O2 evolution with a minimum amount of catalysts. This is highly beneficial because reducing the interference of the catalyst with photon absorption by the photoanode is an important issue for producing efficient semiconductor/catalyst composite photoanodes. Here, we report photochemical deposition of the Co-based catalyst on a ZnO photoanode and discuss the resulting effect on the photoelectrochemical properties. The method described here can be used as a general route to deposit the Co-based catalyst on any semiconductor electrode that has a valence band edge located at a more positive potential than the oxidation potential of Co2+ ions (Fig. 1).
Fig. 1.

Schematic representation. (A) photochemical deposition of the Co-based catalyst on ZnO and (B) relevant energy levels.
Results and Discussion
ZnO electrodes used in this study were electrochemically deposited on FTO (fluorine doped tin oxide) in an aqueous solution containing 0.005 M Zn(NO3)2 and 0.05 M NaNO3 at 70 °C. Reduction of NO3− ions to NO2− ions elevated the local pH on the working electrode (NO3− + H2O + 2e− → NO2− + 2OH−), which resulted in a decrease in solubility of Zn2+ ions and therefore precipitation of ZnO on the working electrode (9–10). The resulting electrodes contained ZnO rods with an average diameter and length of ≈100 nm and 600 nm, respectively (Fig. 2A).
Fig. 2.

SEM images. (A) ZnO electrode used for this study, (B) Co-based catalyst nanoparticles photochemically deposited on ZnO for 30 min, and (C) Co-based catalyst layer electrochemically deposited on ZnO for 10 min.
The Co-based catalyst was photochemically deposited on the ZnO surface by placing a ZnO electrode in a 0.1 M potassium phosphate solution (pH 7) containing 0.5 mM CoCl2 and illuminating with a UV light (λ = 302 nm, 1.5 μW/cm2). Because the valence band edge of ZnO is located at ≈2.6 V vs. NHE, the photogenerated holes have enough overpotential to oxidize Co2+ ions to Co3+ ions to produce the Co-based catalyst on the ZnO surface (Fig. 1). For example, the oxidation potential of Co2+ to form Co2O3 is 0.70 V vs. NHE when [Co2+] = 0.5 mM at pH 7 (Eqs. 1 and 2) (11). Formation of the Co-based catalyst, which is also based on Co2+/Co3+ oxidation, is expected to require a similar potential (1–2). During this deposition, the photogenerated electrons are consumed by the reduction of water or dissolved O2 in the solution.
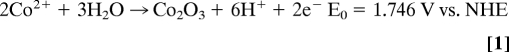 |
Fig. 2B shows the SEM image of the photochemically deposited Co-based catalyst on ZnO rods after 30 min of UV irradiation. The catalysts were deposited as 10–30 nm nanoparticles evenly distributed on the ZnO surface. This nanoparticulate morphology increases the surface area of catalysts per unit mass while minimizing the blockage of the ZnO surface. The electrochemically deposited Co-based catalyst on ZnO rods is also shown in Fig. 2C for comparison, which formed a featureless dense layer. The dissolution of ZnO noticeable in Fig. 2C is due to the instability of ZnO under the anodic deposition condition of the catalyst. This suggests that for a semiconductor electrode that dissolves or decomposes with application of anodic bias, photodeposition provides an alternative route to produce Co-based catalysts. It may also be worthwhile to point out that when the Co-based catalyst was electrochemically deposited on the ZnO electrode, significant deposition of the catalyst occurred on the more conductive FTO surface (Fig. 2C). However, photochemical deposition resulted in the deposition of the catalyst selectively on the ZnO surface because only ZnO can photogenerate holes (Fig. 2B). Considering that the catalyst on the FTO substrate cannot participate in solar O2 evolution, photochemical deposition prevents unnecessary catalyst deposition.
The photochemically deposited Co-based catalyst is X-ray amorphous as in the case of electrochemically deposited catalyst. The X-ray photoelectron spectroscopy (XPS) study showed that the cobalt 2p XPS of the photochemically and electrochemically deposited catalysts were identical, suggesting that they are composed of the same Co catalytic centers (Fig. 3A). The phosphorous 2p XPS of both the photochemically and electrochemically deposited Co-based catalysts show a peak at 133 eV due to the phosphate contained in the catalysis but the photochemically deposited catalysis generated an additional peak located at 140 eV (Fig. 3B). This peak is due to the phosphate directly adsorbed on the ZnO surface that is not a part of the OEC (12).
Fig. 3.

XPS data of the Co-based catalyst electrodeposited on FTO (dotted line) and photochemically deposited on ZnO electrode (solid line). (A) shows the Co 2p and (B) P 2p peaks.
Energy dispersive spectroscopy (EDS) shows that the ratio of P/Co in photochemically deposited Co-based catalyst is ≈1.8/1 after correcting for the amount of phosphate directly adsorbed on the ZnO surface. This P/Co ratio is significantly higher than that of the electrochemically deposited catalyst (P/Co = 1/2) (1). This difference is most likely because of the difference in the morphology of photochemically and electrochemically deposited catalysts [10–30 nm nanoparticles vs. ≈3 μ thick layers (1, 3)], which in turn affects the fraction of surface Co atoms in the catalyst. Although the structure of amorphous Co-based catalyst is not fully understood yet, the surface of the catalyst particles or layers is most likely terminated with phosphates (13). Therefore, the nanoparticle morphology that allows for a much higher fraction of surface Co atoms than the layer morphology is expected to contain a greater amount of phosphates per cobalt atom. The presence of phosphates at the catalyst interface is currently considered important to assist the proton coupled electron transfer for O2 evolution, but the exact P/Co ratio does not appear to be critical for demonstrating catalytic activity (2–4). Further structural analysis of the Co-base catalysts will be necessary to better understand the structural arrangements and the role of the phosphates in these catalysts.
The effect of the photodeposited Co-based catalyst on photocurrent generation of ZnO was investigated by comparing photocurrent-potential characteristics of ZnO electrodes with and without Co-based catalyst (Fig. 4A). Photocurrent was measured in a three-electrode setup in a 0.1 M potassium phosphate solution (pH 11.5) bubbled with Ar. A pH 11.5 medium was chosen because the ZnO electrode is chemically and photochemically the most stable in this pH (11, 14). Fig. 4A shows that both electrodes generated anodic photocurrents upon illumination due to the n-type nature of ZnO. However, the photocurrent onset potential of the ZnO/Co-based catalyst electrode is shifted to the negative direction by 0.23 V compared to that of the ZnO electrode. The Mott-Schottky plots show that these two electrodes have comparable flat band potentials (−0.90 V vs. Ag/AgCl) (Fig. S1) (15). Therefore, the observed shift of the photocurrent onset potential can be attributed to the Co-based catalyst decreasing the electron-hole recombination near the flat band potential.
Fig. 4.

Photocurrent measurement. (A) Photocurrent-potential characteristics of ZnO (black) and ZnO/Co-based catalyst electrodes (gray) measured (scan rate, 10 mV/s) with chopped light (100 mW/cm2, 1Hz). Arrows indicate the onset potential of photocurrent. Photocurrent of ZnO (black) and ZnO/Co-based catalyst electrodes (gray) measured at (B) 0.0 V and (C) 0.2 V against the Ag/AgCl reference electrode.
The effect of Co-based catalyst is also evident in the general enhancement of anodic photocurrent observed in the entire potential range shown in Fig. 4A. However, the photocurrent enhancement by Co-based catalyst becomes less significant as the potential is scanned to the positive direction because the more pronounced band bending helps the electron-hole pair separation and photo-oxidation reactions even without the assistance of the catalyst. The photocurrents measured at a constant bias of 0.0 V and + 0.2 V against the Ag/AgCl reference electrode show that the presence of Co-based catalyst enhanced the steady-state photocurrent by 2.6 and 1.5 times, respectively (Fig. 4 B and C). The photocurrent of the ZnO/catalyst composite electrode after 7 h of illumination remained the same without any sign of catalyst dissolution or degradation.
In summary, we successfully demonstrated the photochemical deposition of Co-based catalysts as an efficient route to couple an oxygen evolution catalyst and a photoanode. The photodeposition method ensures placing the catalysts where they can best use photogenerated holes for solar O2 production. The Co-based catalysts deposited on the ZnO surface clearly exhibited their ability to use photogenerated holes in ZnO for enhancing solar O2 evolution. This result encourages further studies on coupling the Co-based catalyst with more efficient photoanode systems via photochemical deposition.
Methods
Synthesis.
A standard three-electrode setup in an undivided cell was used. The working electrode was fluorine-doped tin oxide (FTO). The counter electrode consisted of 1,000 Å of platinum deposited on 300 Å of titanium on a glass slide by sputter coating. The reference electrode was an Ag/AgCl electrode in 4 M KCl solution. Rod shaped ZnO crystals were obtained when a potentiostatic deposition was carried out at 70 °C in an aqueous solution containing 0.005 M Zn(NO3)2·6H2O and 0.05 M NaNO3. A nucleation pulse (E = −0.9 V against Ag/AgCl in 4 M KCl) was applied for 10 s, followed by a growth potential (E = −0.5 V against Ag/AgCl) until 0.18 C passed.
Photochemical deposition of Co-based catalyst on ZnO was carried out by immersing the ZnO electrode in a Petri dish in 0.5 mM CoCl2 and 0.1 M potassium phosphate electrolyte (pH 7), and illuminating UV light from the top for 30 min by using a handheld UV light (UVM-57, λ = 302 nm) with an output voltage of 1.5 μW/cm2. The catalyst loading achieved by 30 min deposition (Fig. 2B) was optimum for enhancing the photocurrent of ZnO. Full coverage of the ZnO surface with Co-based catalyst can be achieved when the deposition time is elongated to 3 h.
Electrodeposition of Co-based catalyst on ZnO was carried out by using the ZnO electrode as the working electrode and applying + 1.2 V against Ag/AgCl in 4 M KCl in an electrolyte containing 0.5 mM CoCl2 and 0.1 M potassium phosphate buffer (pH 7) (1). The counter electrode consisted of 1,000 Å of platinum deposited on 300 Å of titanium on a glass slide by sputter coating. An SEM image of the ZnO electrode after 10 min of depositing the OEC shows that severe dissolution of ZnO occurred during deposition (Fig. 2C).
Characterization.
Scanning electron microscopy (SEM) images were obtained by using a FEI-NOVA NanoSEM scanning electron microscope operated at 5 kV. The ZnO films were coated with Pt by thermal evaporator before imaging to minimize charging problems. Quantitative microprobe analysis was performed with a FEI-NOVA NanoSEM scanning electron microscope equipped with an Oxford Inca 250 EDS energy-dispersive spectroscopy detector to access the amount of cobalt, potassium, and phosphate present in the final samples. The samples for EDS were prepared by scraping the films and piling them onto carbon tape on a SEM stub. The average EDS results of the Co-based catalyst photodeposited on the ZnO surface show that the Co:P ratio is 1:3.3. However, the amount of phosphorous in this result contains the contribution of phosphate ions that are not a part of the Co-based catalyst but directly adsorbed on the ZnO surface. To measure the amount of phosphate directly adsorbed on the ZnO surface, a ZnO electrode was immersed in the 0.1 M phosphate solution with no Co2+ ions for 30 min of UV illumination (identical condition used for Co-based catalyst deposition). The average EDS results show that the Zn to P ratio is 40:2.2. This indicates the amount of phosphate adsorbed on the ZnO surface is equivalent to 5.5% of the total zinc content. Subtracting this amount from the original Co:P ratio, the Co:P ratio considering only phosphorus present in the Co-based catalyst becomes 1:1.8. XPS measurements were made directly from the prepared electrodes by using a Kratos Axis ULTRA spectrometer. The XPS spectra was processed by using the CasaXPS software and the binding energies were calibrated with respect to the residual C (1s) peak at 284.6 eV.
Photocurrent Measurements.
Photoelectrochemical performance of ZnO electrodes was tested by using a 300 W Xe arc lamp and light was illuminated through fiber optics with the output power of 100 mW/cm2 and spot size of 0.196 cm2 at the surface of ZnO. The electrolyte was 0.1 M potassium phosphate, with pH adjusted to 11.5 with KOH. All solutions were bubbled with Ar gas before photocurrent measurements to purge the dissolved oxygen. The counter electrode was oversized platinum on glass and the reference electrode was Ag/AgCl in 4 M KCl. For linear sweep voltammetry, potential was swept to the negative direction with a scan rate of 10 mV/s and was reported against Ag/AgCl in 4 M KCl. A chopped light (chopping frequency = 1Hz) was used to record both the dark and photocurrent during a single scan. For an n-type photoelectrode, sweeping to the negative direction reduces band bending of ZnO at the interface. Therefore, a negative scan allows for finding the photocurrent onset potential with applying a minimally necessary negative bias. When a positive scan is used, a more negative potential than the onset potential should be used as the starting potential, which may cause a significant reduction of ZnO to Zn and alter the photoelectrochemical property of ZnO for the entire scan range. The photocurrent onset potential shown in Fig. 4A was determined as the potential where the photocurrent and dark current merged when (photocurrent density)2 was plotted against the potential.
Capacitance-Potential Characteristics (Mott-Schottky Plots).
Capacitance measurements were carried out to obtain Mott-Schottky plots (Fig. S1) by using an EG&G Princeton Applied Research Potentiostat/Galvanostat Model 263A interlinked with a Princeton Applied Research FRD100 frequency response detector using the PowerSine software program. A sinusoidal perturbation of 10 mV was applied at frequencies of 5 kHz and 500 Hz. The electrolyte was 0.1 potassium phosphate (pH 11.5, adjusted with KOH) that was purged with Ar gas before measurements. A standard three-electrode set-up was used with the sample of interest as the working electrode, while an oversized platinum on glass counter was used with Ag/AgCl in 4 M KCl as the reference electrode.
Supporting Information.
This article contains Mott-Schottky plots of ZnO and ZnO/Co-catalyst composite electrodes as Fig. S1.
Supplementary Material
Acknowledgments.
The XPS data were obtained by Dr. D. Zemlyanov of the Surface Analysis Laboratory, Birck Nanotechnology Center, Purdue University. This work was supported by a Center for Chemical Innovation of the National Science Foundation Grant CHE-0802907 (to K.-S.C.) and made use of the Life Science Microscopy Facility at Purdue University.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0910203106/DCSupplemental.
References
- 1.Kanan MW, Nocera DG. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+ Science. 2008;321:1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- 2.Surendranath Y, Dinca M, Nocera DG. Electrolyte-dependent electrosynthesis and activity of cobalt-based water oxidation catalysts. J Am Chem Soc. 2009;131:2615–2620. doi: 10.1021/ja807769r. [DOI] [PubMed] [Google Scholar]
- 3.Kanan MW, Surendranath Y, Nocera DG. Cobalt-phosphate oxygen-evolving compound. Chem Soc Rev. 2009;38:109–114. doi: 10.1039/b802885k. [DOI] [PubMed] [Google Scholar]
- 4.Lutterman DA, Surendranath Y, Nocera DG. A self-healing oxygen-evolving catalyst. J Am Chem Soc. 2009;131:3838–3839. doi: 10.1021/ja900023k. [DOI] [PubMed] [Google Scholar]
- 5.Zhong DK, Sun J, Inumaru H, Gamelin DR. Solar water oxidation by composite catalyst/α-Fe2O3 photoanodes. J Am Chem Soc. 2009;131:6086–6087. doi: 10.1021/ja9016478. [DOI] [PubMed] [Google Scholar]
- 6.Nozik AJ. Photoelectrochemistry: Applications to solar energy conversion. Ann Rev Phys Chem. 1978;29:189–222. [Google Scholar]
- 7.Bak T, Nowotny J, Rekas M, Sorrell CC. Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects. Int J Hydrogen Energ. 2002;27:991–1022. [Google Scholar]
- 8.Lewis NS, Nocera DG. Powering the planet: Chemical challenges in solar energy utilization. Proc Natl Acad Sci USA. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izaki M, Omi T. Transparent zinc oxide films prepared by electrochemical reaction. Appl Phys Lett. 1996;68:2439–2440. [Google Scholar]
- 10.Therese GHA, Kamath PV. Electrochemical synthesis of metal oxides and hydroxides. Chem Mater. 2000;12:1195–1204. [Google Scholar]
- 11.Pourbaix M. Atlas of Electrochemical Equilibria in Aqueous Solutions. 2nd English Ed. Houston: National Association of Corrosion Engineers; 1976. p. 324. [Google Scholar]
- 12.Heo YW, Ip K, Park SJ, Pearton SJ, Norton DP. Shallow donor formation in phosphorus-doped ZnO thin films. Appl Phys A: Mater Sci Process. 2004;78:53–57. [Google Scholar]
- 13.Risch M, Khare V, Zaharieva I, Gerencser L, Chernev P, Dau H. Cobalt-oxo core of a water-oxidizing catalyst film. J Am Chem Soc. 2009;131:6936–6937. doi: 10.1021/ja902121f. [DOI] [PubMed] [Google Scholar]
- 14.Limmer SJ, Kulp EA, Switzer JA. Epitaxial electrodeposition of ZnO on Au(111) from alkaline solution: Exploiting amphoterism in Zn(II) Langmuir. 2006;22:10535–10539. doi: 10.1021/la061185b. [DOI] [PubMed] [Google Scholar]
- 15.Cardon F, Gomes WP. On the determination of the flat-band potential of a semiconductor in contact with a metal or an electrolyte from the Mott-Shottky plot. J Phys D: Appl Phys. 1978;11:L63–L67. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.