

Abstract
Following acute injury, the concerted action of resident and non-resident cell populations evokes wound healing responses that entail a temporary increase in inflammation, extracellular matrix production, and proliferation to ultimately restore normal organ architecture. However, chronic injury evokes a perpetuating wound healing response promoting the development of fibrosis, organ failure, and cancer. Recent evidence points towards toll-like receptors (TLRs) as important regulators of inflammatory signals in wound healing. Here we will review the activation of TLRs by different endogenous and bacterial TLR ligands during wound healing, and the contribution of TLR-induced signals to injury, fibrogenesis, regeneration and carcinogenesis. We will discuss the hypothesis that TLRs act as sensors of danger signals in injured tissue to switch the wound healing response towards fibrogenesis and regeneration as a protective response to imminent danger at the cost of an increased long-term risk of developing scars and cancer.
Background
The ability to respond to injury is a fundamental property of all multicellular organisms. Injury threatens organ function and integrity, and induces a rapid but highly complex wound healing response that serves to protect against imminent danger on the one hand and to restore tissue architecture on the other hand. In some species such as amphibians wound healing responses can achieve both goals as demonstrated by almost complete regeneration after the loss of complete limbs. However, this regenerative capacity has largely disappeared in mammals, with large and chronic wounds often healing by fibrotic scarring rather than regeneration. Development of fibrosis in organs such as the liver, lung, heart and kidney is accompanied by a progressive loss of function and represents a common and clinically highly relevant problem [1-4]. Moreover, chronic and uncontrolled wound healing provides a microenvironment that gives rise to cancer supporting the hypothesis that certain types of cancers represent “wounds that do not heal” and a final stage of abnormal and uncontrolled tissue repair processes [5]. Understanding the signaling mechanisms that promote fibrogenesis and carcinogenesis in chronically injured tissues is therefore of considerable clinical interest. In addition to the size and chronicity of a wound, scarring is also determined by the presence of inflammation. Infection is an obvious cause for inflammation and may promote scarring. However, in many injured organs inflammatory signals are activated even in the absence of infection. Signals induced by “sterile inflammation” are believed to play an important role in scar formation as evidenced by reduced scarring in mice that lack inflammatory cell types such as macrophages and neutrophils [6].
Wound healing responses can be divided into three distinct yet sometimes overlapping phases [7]: A first inflammatory and fibrogenic phase, a second regenerative phase and a third remodeling phase. In most organs, injury affects primarily the epithelial and endothelial compartments and triggers the release of anti-fibrinolytic mediators to induce blood clot formation, and of proinflammatory mediators to initiate the infiltration of leukocytes. Infiltration of leukocytes is the dominant event in the first phase of wound healing responses and serves at least two distinct functions: (i) Removal of dead cells by phagocytosis and (ii) promotion of ECM deposition by releasing profibrogenic mediators that activate fibroblasts. In several organs, a subset of white blood cells can also differentiate into fibrocytes to promote ECM deposition [8]. Newly synthesized extracellular matrix provides mechanic stability and serves as a scaffold for organ regeneration. In the second phase, macrophages, fibroblasts and pericytes secrete growth factors that promote epithelial proliferation and angiogenesis to restore organ mass and vessel formation to provide for nutritional requirements of newly formed tissue, respectively. In the final third phase, ECM is degraded and ideally normal organ architecture is restored. In chronic injury, ECM production outweighs its degradation thus favoring progressive ECM accumulation. Moreover, accumulating ECM is prone to undergo secondary changes such as cross-linking thus making it more resistant to degradation and favoring scar formation.
On a molecular level, a plethora of mediators are involved in the regulation of fibrogenic and regenerative signals following injury. The probably best-characterized promoters of fibrogenic signals are transforming growth factor beta (TGFβ) and platelet-derived growth factor (PDGF). TGFβ is mainly produced by macrophages, and signals through the Smad pathway to promote fibroblast activation and collagen production [9]. PDGF isoforms primarily promote the proliferation myofibroblasts and thus expand the pool of ECM-producing cells [10]. The crucial role of PDGF and TGFβ in fibrogenesis has been demonstrated in various organs by modulating the levels of bioactive PDGF and TGFβ in vivo. Overexpression of PDGF and TGFβ promotes spontaneous fibrosis of the lung, liver, kidney and pancreas [11-14] whereas inhibition of PDGF or TGFβ prevents hepatic and lung fibrosis [15-18]. In addition to PDGF and TFGβ, a number of other mediators such as chemokines, angiotensin, leptin, IL-4, IL-6 and IL-13 play important roles in fibrogenesis [19]. A number of growth factors such as HGF, TGFα, EGF, epiregulin, amphiregulin are elevated following tissue injury and promote epithelial regeneration.
Despite their prominent role in fibrogenesis, the effects of TGFβ and PDGF cannot account for the inflammatory component of wound healing. Recent evidence points towards Toll-like receptors (TLRs), a family of receptors that regulate innate and adaptive immune responses, as important modulators of inflammation during wound healing responses. Toll-like receptors recognize pathogen associated molecular patterns (PAMPs) and activate innate immune responses to eliminate pathogens. In addition to recognizing PAMPs, TLRs also recognize a class of endogenous molecules that are released from necrotic tissue termed damage associated molecular patterns (DAMPs). Thus, TLRs appear to regulate inflammatory responses during wound healing under both sterile and non-sterile conditions. Here we will review the role of TLRs in tissue injury, fibrogenesis and regeneration. We will discuss the hypothesis that TLRs serve as danger sensors in injured tissue, and that activation of TLRs protects against imminent danger such as infection or organ failure, but carries an increased risk of scar formation and cancer development.
I. TLRs and TLR signaling
Toll-like receptors are a group of highly conserved molecules that allow the immune system to sense molecules that are present in most classes of pathogens such as bacteria and viruses, but not the host, and to coordinate defense mechanisms against these pathogens (see Figure 1). The recognition of pathogen-associated molecular patterns (PAMPs) by TLRs is a cornerstone of innate immunity and provides a quick and highly efficient response to pathogens in both vertebrate and invertebrate species [20]. The human TLR family consists of currently 10 members, which are structurally characterized by the presence of a leucine-rich repeat (LRR) domain in their extracellular domain and a Toll/interleukin (IL)-1 receptor (TIR) domain in their intracellular domain [21]. The existence of a large number of TLRs enables the innate immune system to discriminate between PAMPs that are characteristic of different microbial classes and launch specific defense mechanisms. TLR4 senses gram-negative bacteria by binding lipopolysaccharide (LPS), a membrane component of gram-negative bacteria. TLR2 heterodimers recognize cell membrane components of gram-positive bacteria. TLR3 and TLR7 sense viral infections by recognizing double-stranded and single-stranded RNA, respectively. TLR9 recognizes non-methylated CpG-containing DNA from bacteria and viruses. TLRs that mainly serve to detect bacterial LPS and lipoproteins are located on the cell surface. TLRs such as TLR3, TLR7, TLR8, and TLR9 that mainly recognize viral RNA and bacterial DNA are located in late endosome-lysosomes in which these materials are processed and host DNA is not present, thus avoiding aberrant self-recognition. Some TLRs such as TLR2 and TLR4 have been suggested to also detect endogenous ligands termed DAMPs that are released from injured and inflamed tissue. A wide range of DAMPs including HMGB1, hyaluronan, S100 proteins, heat shock protein 60 and the alternatively spliced extra domain A of fibronectin have been suggested to activate TLRs [22]. However, there is still ongoing controversy in regards to several endogenous ligands as many of these ligands have either been purified in bacterial systems or have a high affinity to bacterial products such as LPS suggesting that bacterial products rather than the suggested ligands themselves mediate their TLR activating effect [23]. Although each TLR detects specific ligands, many of the signaling molecules that mediate intracellular response are shared by the TLRs (see Figure 1). All TLRs signal through one or two adapter molecules termed MyD88 and TRIF [21]. Whereas MyD88 is part of the signaling cascade of all TLRs except for TLR3, Trif only interacts with TLR3 and TLR4. MyD88- and TRIF-dependent pathways initiate the transcription of a specific set of genes involved in proinflammatory, antiviral, and antibacterial responses. MyD88 activates NF-κB, AP-1, p38, and IRF-7 signaling cascades while TRIF activates NF-κB and IRF-3. MyD88 and TRIF mediate NF-κB activation through different mechanisms and kinetics thus leading to the upregulation of distinct target genes.
Figure 1. TLR signaling.
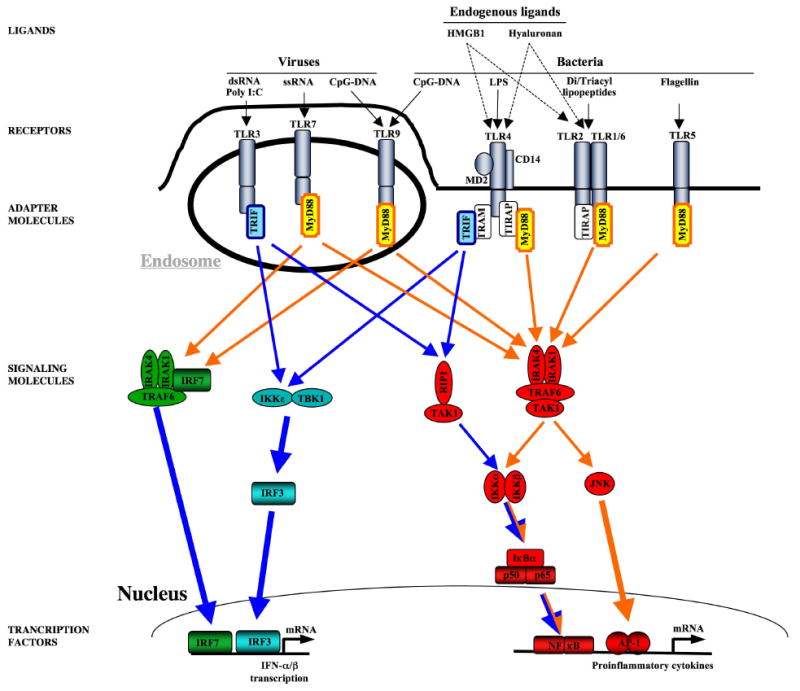
Viral PAMPS activate TLR3, TLR7, and TLR9, whereas bacterial PAMPs activate TLR2/1, TLR2/6, TLR4, and TLR9. Several endogenous mediators including hyaluronan and HMGB1 have been suggested to activate TLR2 and TLR4. TLRs mediate their signaling through to adapater molecules, MyD88 and TRIF to induce up-regulation proinflammatory and antiviral genes. MyD88-induced signals (marked in orange) predominantly activate NF-κB, IRF-7 and JNK, Trif-dependent signals (marked in blue) predominantly activate NF-κB and IRF-3.
II. TLRs and TLR ligands in wound healing response
TLR activation in wound healing appears to be mediated by two entirely different classes of ligands: (i) In organs such as the intestine, the skin and the liver that are in direct contact with microbiota and microbial products, tissue injury leads to a breakdown of protective barriers and subsequent TLR activation by bacterial PAMPs. (ii) In many organs such as the liver, heart and kidney, tissue injury leads to the release of DAMPs from dead and dying cells resulting in the activation of TLRs and “sterile inflammation”. The release of endogenous TLR ligands predominantly occurs after massive tissue injury, especially under circumstances where a significant percentage of cells undergo necrosis such as in ischemia-reperfusion injury [24]. It is likely that the perturbation of the cellular environment either induces a conformational change in some molecules that results in TLR agonistic activity, or that TLR agonistic molecules that are usually confined to a certain intracellular compartment are released into other compartments or the extracellular space and come into proximity of TLRs [24, 25]. According to their wide range of biological actions, TLRs have been implied in different phases of wound healing: (i) Activation of TLRs modifies tissue injury in positive or negative fashion either by recruiting inflammatory cells that release cytotoxic mediators or by activating cytoprotective signals. (ii) Activation of TLRs enhances fibrogenic responses in fibroblasts through direct and indirect mechanisms. (iii) Activation of TLRs promotes regenerative responses.
Prevention of epithelial injury by TLRs
The lung and the intestine are the prototypical organs in which TLRs exert a cytoprotective role and prevent tissue injury under conditions of stress. In bleomycin-induced lung injury, TLR- and MyD88-deficient mice displaye an increased degree of lung injury despite reduced recruitment of inflammatory cells [26]. This was attributed to a lack of cytoprotective signals in the absence of TLR2/4 or MyD88. Blocking the endogenous TLR ligand hyaluronan by a peptide-based approach resulted in a pattern of lung injury that was highly similar to that of MyD88 and TLR2-TLR4-deficient mice. Lung-specific overexpression of hyaluronan synthase decrease lung injury [26]. In the intestine, the TLR4-MyD88 pathway plays a similar role and prevents intestinal epithelial injury following application of dextran sulfate sodium (DSS), as seen by the increased injury in TLR4- and MyD88-deficient mice [27, 28]. Interestingly, gut-sterilized mice display a similar increase in intestinal injury and DSS-induced death as MyD88-deficient mice suggesting that PAMPs from the commensal intestinal microbiota stimulate cytoprotective pathways in the intestine to prevent epithelial injury [28].
Promotion of epithelial injury by TLRs
Ischemia-reperfusion injury represents the scenario in which a profound injury-promoting role of TLR2 and TLR4 has been most thoroughly established. The injury promoting effects of TLR4 manifest in almost all organs as seen by the protection of TLR4-mutant or deficient mice after hepatic, renal, cardiac and cerebral ischemia-reperfusion [29-32] suggesting a common mechanism by which TLR4 promotes injury. However, studies in chimeric mice have shown that the contribution of bone marrow to TLR4-dependent ischemia-reperfusion injury differs between organs. In hepatic ischemia reperfusion injury, the injury-promoting effects of TLR4 are mediated by a bone marrow-derived cell population [33]. In contrast, TLR4-mediated injury in renal ischemia-reperfusion injury depends on resident renal cell populations [30]. The chemotherapeutic agent cisplatin also induces renal injury in a TLR4-dependent manner [34]. Similar to renal-ischemia reperfusion injury, the TLR4-mediated cytotoxic effect in cisplatin-treated mice depends on resident cells of the kidney [34]. Thus, TLR4 appears to promote tissue injury through multiple bone marrow and non-bone marrow cell populations, possibly in an organ-specific manner. In the kidney, TLR2-deficiency protects from ischemia-reperfusion injury more efficiently than deficiency of MyD88 [35]. Since ischemia-reperfusion injury and cisplatin-induced cytotoxicity are primarily sterile, non-bacterial TLR ligands have been suggested to mediate TLR4 activation in this setting. The putative TLR4 ligand HMGB1 has been postulated to be an important mediator of ischemia-reperfusion injury. HMGB1 is rapidly upregulated in hepatic and renal ischemia-reperfusion injury [29, 30]. Antibody-mediated blockade of HMGB1 potently reduces hepatic injury after ischemia-reperfusion. Notably, HMGB1 blockade could not further reduce injury in TLR2-TLR4 double knockout mice suggesting that HMGB1 mediates its effects in ischemia-reperfusion injury through TLR2 and TLR4 [29]. Several other putative TLR4 ligands including hyaluronan and biglycan are upregulated in ischemia reperfusion injury of the kidney but their functional contribution to injury has not yet been characterized [30]. Alcoholic liver injury also depends on TLR4, but is mediated by LPS. The elevated levels of LPS in the portal circulation are due to an ethanol-induced disruption of the intestinal epithelial barrier and a subsequently enhanced intestinal permeability [36]. In animals continuously exposed to ethanol, liver injury is strongly reduced when the gram-negative gut microflora is eliminated by antibiotics [37] or when TLR4 signaling is blocked genetically [38]. It is unclear whether the effects of chronic alcohol on other organs such as the heart are also mediated by LPS-TLR4-dependent signals or whether the unique anatomical location of the liver predisposes to LPS-mediated damage in this scenario.
Promotion of epithelial regeneration by TLRs
There is evidence from different organs that TLR ligands promote epithelial regeneration under many circumstances, but that this effect may be dose-dependent with higher concentrations mediating growth-suppressive responses and lower concentrations mediating regeneration. The liver has an astounding ability to regenerate, and after 70% partial hepatectomy can restore mass within 7 to 10 days. Following partial hepatectomy, MyD88 is required for restoration of liver mass at early time points in one study [39]. However, the contribution of MyD88-dependent signals to regeneration appears to be moderate as MyD88-deficient mice displayed the same liver/body weight ratio as wild-type mice 96 hours after post partial-hepatectomy in one study [39] and did not display a decreased hepatocyte proliferation at any time point when compared to heterozygous or wild-type mice in another study [40]. Interestingly, the most likely candidates to mediate MyD88-dependent regenerative signals such as TLR2, TLR4, TLR9 as well as IL-1R and IL-18R are not involved in liver regeneration [39, 40]. It is likely that the growth-promoting TLR ligand is derived from the intestinal microbiota as germ-free mice also display suppressed liver regeneration [41]. On the other hand, TLR4 activation by higher LPS doses can suppress liver regeneration suggesting that that TLR signaling can modulate liver regeneration in both directions depending on the degree of TLR activation [41-43]. In the intestine, a TLR2/TLR4/MyD88 signaling cascade is required for the regeneration of epithelia following DSS-induced injury [27, 44] (see Figure 2). This MyD88-dependent growth-promoting signal is transmitted through macrophages which following injury migrate towards intestinal crypts to stimulate the proliferation of epithelial progenitor cells [44, 45]. The key role of MyD88 in wound healing is further demonstrated by a study that found a markedly slower healing of excisional skin wounds in MyD88-deficient than in wild-type mice [46]. This delayed healing is characterized by slower contraction, a decreased rate of formation of granulation tissue, and a decreased density of blood vessels in the newly forming granulation tissue. However, the authors did not investigate whether there was simply less granulation tissue or whether re-epithelization also decreased in MyD88-deficient mice.
Figure 2. TLR-mediated epithelial regeneration after DSS-mediated colonic injury.
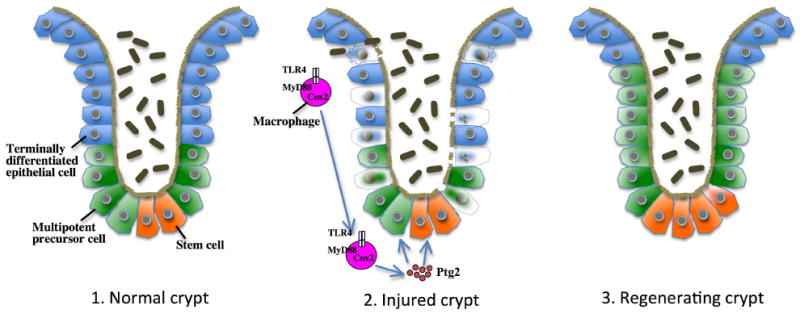
In the colonic crypt, stem cells generate multipotent precursor cells which then differentiate into different types of terminally differentiated epithelial cell including enteroabsorptive, paneth and goblet cells to chronically replace the epithelial layer of crypts. Following DSS-induced injury, terminally differentiated epithelial cells as well as multipotent precursor cells undergo cell death. TLR4- and MyD88-dependent signals are required to induce Cox2-mediated generation of prostaglandin2 (ptg2) and to stimulate epithelial cell proliferation. The MyD88-Cox2 signal that promotes regeneration is largely provided by macrophages which migrate towards the site of injury to stimulate proliferation in Cox2-dependent manner. (Figure adapted from Rakoff-Nahoum et al. [82].
Promotion of fibrogenesis by TLRs
A number of in vivo and in vitro observations support a role for TLRs in promoting fibrogenic and wound healing responses. The fibrogenic response is primarily mediated by fibroblasts, but involves the cross-talk with many other cell populations such as macrophages and epithelial cells. Thus, effects of TLR signaling on fibrogenesis may be mediated by several target cell populations. Mouse embryonic fibroblasts as well as fibroblasts and fibroblast precursors such as hepatic stellate cells and mesangial cells express TLR4 [47-50]. Hepatic stellate cells and synovial fibroblasts also express TLR2 [51,100]. However, the role of TLRs in fibroblast activation and wound healing in vivo remains unknown for the majority of organs. The liver represents the only organ in which the role of TLRs in fibroblast activation and subsequent wound healing responses has been investigated thoroughly. Due to its direct link through the portal vein, the liver is a main target of bacterial products from the intestine. In the course of chronic liver injury, portal and systemic LPS levels increase, most likely due to changes in the intestinal microbiota and changes in the intestinal barrier that promote increase bacterial translocation [52, 53]. TLR4-deficient mice display significantly decreased hepatic fibrogenesis in three different models of experimental fibrogenesis induced by bile duct ligation, CCl4 injection or thioacetamide treatment [53, 54]. The importance of bacterial TLR ligands in promoting liver fibrogenesis was demonstrated by a strong reduction of fibrogenesis in gut-sterilized mice [54]. Hepatic stellate cells are a direct target of LPS and are responsible for TLR4-dependent fibrogenesis as shown by in vivo LPS challenge and in vivo fibrogenesis studies in TLR-chimeric mice [54]. Notably, a recent study has identified a single nucleotide polymorphism that results in a T399I substitution and confers a significantly reduced risk for fibrosis progression in patients with chronic hepatitis C virus infection [55]. This polymorphism is associated with a reduced TLR4 responsiveness thus confirming the profibrogenic role of TLR4 in a clinically relevant setting. In contrast to TLR4, TLR2 has not major role in the promotion of hepatic fibrogenesis [54]. Mice lacking TLR9 also display a reduction of CCl4-induced liver fibrosis [56]. TLR9 is expressed in hepatic stellate cells and upregulates TGFβ1 and collagen expression in response to stimulation by CpG-DNA or apoptotic hepatocytes. Although it was concluded that TLR9 is activated by apopotic hepatocytes to promote stellate cell activation and liver fibrosis, it cannot be excluded that TLR9 is also the target of profibrogenic bacterial products released from the intestinal microbiota. In the heart, TLR4 deficiency results in a reduction of left ventricular hypertrophy, cardiac fibrosis, and TGF-β expression after myocardial infarction [57]. In the vasculature, atherosclerosis entails remodeling processes that display similarities with wound healing and fibrogenesis in other organs such as infiltration of inflammatory cells, activation of myofibroblast-like cells and accumulation as well as changes in the composition of ECM. Recent genetic studies in atherosclerosis-prone apoE- or low-density lipoprotein-receptor–deficient mice have revealed a central role for TLR-signaling in the development of atherosclerosis. Deletion of MyD88 results in a reduction in plaque burden of 60% [58] while deletion of TLR4 or TLR2 alone also leads to reductions in aortic lesion area of around 30% to 60%, respectively [58, 59]. Conversely, administrations of the TLR4 agonist LPS [60] or TLR2 agonist Pam3CSK4, accelerate atherogenesis significantly [59, 61]. A number of groups have described increased TLR4 expression in intimal lesions, predominantly on macrophages and endothelial cells. One group reported that human adventitial fibroblasts express TLR4, and that TLR4 activation induces the production of proinflammatory cytokines suggesting that activation of TLR4 in the adventitia may augment neointima formation [62].
Macrophages not only express high levels of TLRs but also promote fibrogenesis in the liver [53, 63, 64] and the kidney [65], and play a key role during early and late stage of atherosclerosis [66]. Thus, macrophages are prime candidates to promote fibrogenesis in response to TLR ligands. Overloading with free cholesterol causes cell death of macrophages within atheromas and has been suggested to be a critical event during later stages of atherogenesis [66]. Deficiency of TLR4 or MyD88 reduces free cholesterol-induced macrophage apoptosis, supporting a proatherogenic role of the TLR4-MyD88 signaling axis in atherosclerosis [67]. TLR2 expression on macrophages was required to mediate the atherogenic effects of the TLR2 ligand Pam3CSK4. However, in response to high fat diet, non-bone marrow-derived cells such as endothelial cells contribute to the proatherogenic effects of TLR2 whereas BM-derived cells do not promote TLR2-dependent atherosclerosis [59]. It was suggested that endothelial expression of TLR2 contributes to endothelial cell activation at lesion-prone sites in the vasculature in early phases to promote macrophage infiltration, whereas TLR2 activation in macrophages exacerbates the proinflammatory milieu within the lesion [59]. A number of studies have also reported single nucleotide polymorphisms that are associated with a lower-responsiveness of TLR4 to confer protection to atherosclerosis, but results between different studies have been contradictory and the degree to which these polymorphisms protect from atherosclerosis still needs to be established [68]. In other organs such as the kidney and the lung, the role of TLRs has been investigated in tissue injury but its potential role in fibrogenesis has not yet been addressed.
The available data suggest that the presence of TLR ligands during wound healing responses promotes healing by scarring rather than healing by regeneration. It is likely that the fibrogenic response serves as defense mechanism against imminent danger, and that TLRs in fibroblasts function to sense this danger. Increases in fibrogenesis allow a faster wound closure and/or preservation of organ integrity, and thus protect from infection and organ failure. However, these beneficial short-term responses come at the cost of increased long-term problems, especially when injury is chronic.
III. TLRs and TLR ligands in cancer
Already in the 19th century Rudolf Virchow, one of the founding fathers of modern pathology, postulated a link between chronic inflammation and cancer [69]. This link is especially apparent in organs such as the liver and the stomach in which chronic inflammation induced by HBC and HCV, or Helicobacter pylori, respectively, leads to chronic injury, wound healing and cancer. Accordingly, cancer has also been suggested to be “a wound that does not heal” [5]. This concept is further supported by the finding that a wound-like gene expression signature was present in many common human cancers [70]. Due to the important role in inflammation, tissue regeneration and fibrogenesis, TLRs are likely candidates to mediate effects of the innate immune system on carcinogenesis. Although the role of TLRs in cancer is far from being completely understood, current results suggest a dual role of TLRs in cancer: Exceptionally high doses of TLR agonists appear to have an anti-cancer effect whereas low doses of TLR agonists promote cancer growth.
Anti-tumor effects of TLRs
Anti-tumor effects of bacterial infections were first recognized by Busch and Bruns in the 18th and 19th century in patients with advanced cancers [71, 72]. Coley observed a considerable rate of tumor regression and even complete cure in larger sets of patients with sarcomas of the bone using “Coley's toxin”, a mixture of heat-killed Streptococci and heat-killed Serratia marracens [73-75]. However, the mechanisms through which Coley's toxin induces tumor regression are still not understood, and it remains to be established whether TLRs are indeed required for the anti-tumor effect of this toxin, and whether it acts on TLRs in the immune system, the tumor or the tumor vasculature. A recent study suggests that TLR4 and MyD88 play an important role in anti-tumor responses following chemotherapy and irradiation [76]. TLR4-deficient mice have significantly larger tumors after doxorubicin and oxaliplatin treatment or irradiation than wild-type mice, yet displayed similar growth rates in the absence of treatment [76]. Data from this study suggests that cell death induction by chemotherapy or irradiation induces the release of HMGB1 to subsequently trigger TLR4 activation in dendritic cells, enhance antigen presentation and promote cytotoxic T cell responses. This hypothesis is further confirmed by the finding that a mutation in the human TLR4 gene is associated with an increased frequency of metastasis in breast cancer patients after conventional chemotherapy [76].
Tumor-promoting effects of TLRs
In contrast to the anti-tumor effects of bacterial infection or high dose TLR agonists, low doses of LPS have been shown to enhance tumor growth and to decrease tumor apoptosis in adoptive transfer tumor models. Balb/c mice receiving a tail vein injection of 4T1 mouse mammary carcinoma cells show an increase in lung metastases when simultaneous injected with LPS [77, 78]. Similarly, tumor nodule numbers and lung weights are increased in response to LPS in mice that had been injected with colon adenocarcinoma cell line CT26 [79]. In this study, the tumor-promoting effects of LPS were not a result of direct actions of LPS in tumor cells, but depended on the presence of the LPS receptor TLR4 in the host as there was no LPS-induced NF-κB activation in tumors when CT26 cells were injected into mice expressing a non-functional TLR4. Using TNFα-chimeric mice, the authors demonstrated that LPS induces the release of TNFα in bone marrow-derived cells to induce NF-κB and tumor growth [79]. In another study, injection of L. monocytogenes promoted tumor growth in mice that were subcutaneously injected with the hepatocarcinoma cell line H22 [80]. Knockdown of TLR2 but not TLR4 in H22 tumor cells abrogated the growth promoting effects of L. monocytogenes. These studies strongly suggest a role for TLR ligands in promoting carcinogenesis, but have a number of limitations such as the unphysiologic tumor and tumor microenvironment of adoptive transfer models as well as the focus on only one or two TLR ligands.
A number of recent studies have investigated carcinogenesis in mice deficient in TLRs or TLR adapter molecules using models of inflammation-associated cancer. These models cover all stages from tumor initiation to tumor promotion, and provide a physiological tumor environment thus accounting for a possible role of TLRs in stroma-tumor interactions, possibly mediated by endogenous TLR ligands released from necrotic tumor cells. In patients with ulcerative colitis and colon cancer as well as in a murine model of inflammation-associated colon cancer, azoxymethane plus DSS, TLR4 expression is strongly upregulated in the epithelial compartment of tumors [81]. Moreover, TLR4-deficient mice display a profoundly reduced dysplasia, number and size of tumors [81]. Similar results were reported in a study that investigated the role of MyD88 in colon cancers. MyD88-deficiency leads to decreased carcinogenesis in ApcMin/+ mice, a non-inflammatory model of colon cancer, as well as in the azoxymethane model [82]. Absence of MyD88 reduces the size and number of polyps. Although polyps showed the same degree of proliferation in Myd88-deficient and wt mice, there was more apoptosis in polyps from the MyD88-deficient mice. In the liver MyD88- and IL-6-deficiency significantly reduce tumor load in male but not female mice in the model of diethylnitrosamine (DEN)-induced hepatocellular carcinomas [83]. The authors of this study demonstrated that necrotic hepatocytes stimulate Kupffer cells through a MyD88-dependent pathway to induce IL-6 production which promotes liver injury and compensatory proliferation. However, a recent follow-up study identified the IL-1R as upstream mediator of MyD88-dependent tumor-promoting signals as demonstrated by the decreased IL-6 levels and tumor load in IL-1R-deficient mice [84]. In the liver, TLR4 and MyD88 are required for the development of fibrosis [53], a first stage towards the development of hepatocellular cancer in patients with chronic hepatitis. Thus, it is likely that fibrosis-associated HCC is promoted by the TLR4-MyD88 pathway. As the DEN infant mouse model of hepatocellular cancer only induces a small degree of inflammation and no overt fibrosis, it is possible that non-fibrotic HCC and fibrosis-associated HCCs may both be MyD88-dependent but mediated by distinct yet not mutually exclusive molecular mechanisms, i.e. an IL-1R-dependent pathway in non-fibrotic HCC and a TLR4-dependent pathway in fibrosis-associated HCC.
IV. Signaling mechanisms by which TLRs modulate injury and wound healing
Anti-apoptotic signals
Toll-like receptors are potent activators of the NF-κB pathway [21]. NF-κB regulates the transcription of several hundred genes with κB sites among them a number of anti-apoptotic genes such as Bcl-2, iNOS, c-Flip, IAPs and Traf molecules [85]. There is increasing evidence that TLRs provide signals to promote the survival of epithelial cells under stress conditions. However, it is not entirely clear whether the anti-apoptotic effects of TLRs are directly activated in epithelial cells, or whether TLRs stimulate other cell populations which then secrete factors to activate NF-κB and prevent apoptosis in epithelial cells. The lung and the intestine are the prime examples in which a cytoprotective effect of TLRs in epithelial cells has been demonstrated. The activation of NF-κB by a TLR2/TLR4/MyD88-dependent pathway is required to protect from bleomycin-induced apoptosis as demonstrated by an increase in epithelial cell apoptosis in the lung of TLR2/TLR4 double-deficient and MyD88-deficient mice [26]. In the lung, the endogenous TLR2/TLR4 ligand hyaluronan provides an important signal to activate this TLR-dependent NF-κB response [26]. In the intestine, TLR signalling is required for the maintenance of intestinal epithelial homeostasis and protection from injury. TLR2-, TLR4- and MyD88-deficient mice show an increased mortality and increased epithelial injury in response to DSS [27] [28]. In comparison to wild-type mice, MyD88-deficient mice display a strong decrease of cytoprotective factors such as IL-6, KC-1, hsp25 and hsp72 [28]. Moreover, Cox2 levels are decreased in MyD88- and TLR4-deficient mice. The decreased level of Cox2 is correlated with an increased level of apoptosis after DSS. As Cox2 is know to be an NF-κB target and a mediator of anti-apoptotic, proliferative and tumor-promoting effects in the colon, it appears that the a TLR4-MyD88-NF-κB-Cox2 axis is involved in protection from apoptosis in normal as well as premalignant cells of the colon [27].
Cell death promoting signals
While TLRs promote protection from injury under some circumstances, they can also promote tissue injury under different circumstances, mostly under conditions of extreme stress. Very high doses of LPS alone, e.g. in patients with septic shock, or high doses of LPS in combination with priming events, e.g. infection with certain bacterial strains such as Propionibacterium acnes, result in significant cell death [86]. Necrotic tissue injury, especially after ischemia-reperfusion injury, is another setting in which TLRs have been implicated in the promotion of cell death. In this setting, cytotoxic effects of TLRs appear to be the result of an increased recruitment of bone marrow-derived inflammatory cells which further promote injury. In contrast to apoptosis, necrosis triggers a strong inflammatory response which may, at least in part, be mediated by TLRs and the putative TLR ligand HMGB1. Scaffidi and colleagues reported that challenge of monocytes with necrotic HMGB1 -/- cells reduces TNFα production by 75% in comparison to challenge with necrotic HMGB1 +/+ cells [87]. HMGB1 also has a moderate effect on neutrophil recruitment as seen by a 37% reduction of neutrophil recruitment in mice treated with a neutralizing HMGB1 antibody after acetaminophen-induced liver injury [87], and an approximately 30% reduction of neutrophil recruitment after intraperitoneal injection with HMGB1-/- cells in comparison to mice injected with HMGB1+/+ cells. [88]. Ischemia-reperfusion injury is the best characterized setting in which TLR4 acts as a central mediator of injury as demonstrated by the protection of TLR4-mutant or TLR4-deficient mice after hepatic, renal, cardiac and cerebral ischemia-reperfusion [29-32]. In ischemia-reperfusion injury, the endogenous TLR4 agonist HMGB1 seems to be the predominant activator of TLR4 [29]. HMGB1 levels are increased shortly after ischemia-reperfusion and blockade of HMGB1 by a neutralizing antibody reduces injury in wild-type but not TLR4-deficient mice. In hepatic ischemia-reperfusion injury, TLR ligands such as HMGB1 do not directly activate pro-apoptotic signaling pathways in target cells but through bone marrow-derived inflammatory cells as elegantly shown in chimeric TLR4 mice [29]. Dendritic cells are the most likely candidate to mediate injury following ischemia-reperfusion injury. After hepatic ischemia-reperfusion, wild-type but not TLR4-mutant dendritic cells displayed a strong increase of TNFα production [89], a well-known mediator of hepatic IR injury that is rapidly and potently released from monocytic cells such as macrophages and dendritic cells following TLR activation [90, 91]. Therefore, the most likely scenario is that HMGB1 is released from necrotic parenchymal cells to activate TLR4 on dendritic cells which in turn release TNFα to promote tissue injury. The role of the HMGB1 and dendritic cell-dependent pathway in ischemia reperfusion injury has not been established for other organs and it is possible that other TLR4 ligands and cellular targets contribute to injury in these organs. In alcoholic liver injury, increased exposure to ethanol induces hepatocellular necrosis which is largely dependent on Kupffer cells as demonstrated by reduced injury in Kupffer cell-depleted mice [92]. Hepatocellular injury by ethanol appears to depend on NF-κB activation and subsequent release of toxic mediators such as TNFα by TLR4-activated Kupffer cells [38, 93]. As mice deficient in the NADP(H) oxidase component p47 display substantially reduced NF-κB activation, TNFα induction and liver injury, NADP(H) seems to link the LPS-induced TLR4 activation and the release of cytotoxic mediators such as TNFα [94]. It is likely that the effects of LPS synergize with direct effects of ethanol on the liver to promote injury.
Growth-promoting and angiogenic signals
There is evidence that TLR receptors are involved in the regulation of epithelial proliferation and angiogenesis following injury. The best example for growth promoting effects of the TLR-MyD88 signaling cascade is the intestine, where a TLR2/TLR4/MyD88 signaling cascade is required for the regeneration of epithelia following DSS-induced injury [27, 44]. This TLR4-MyD88-dependent growth-promoting signal is transmitted through cyclooxgynease 2 (Cox2)-expressing macrophages migrate towards intestinal crypts to stimulate the proliferation of epithelial progenitors [45]. Prostaglandin E2 appears to be one crucial growth-promoting signal provided by macrophages stimulated through the TLR4-MyD88 pathway. After DSS injury, Cox-2 expression was increased in wild-type, but not TLR4-deficient mice [27]. Moreover, exogenous prostaglandin 2 supplementation of TLR4-deficient mice completely restored the defective proliferation. The mechanism for improved epithelial repair may be through prostaglandin E2-dependent activation of the epidermal growth factor receptor (EGFR). It is known that prostaglandin E2 transactivates the EGF receptor [95]. Accordingly, inhibitors of Cox-2 and EGFR tyrosine activity decreased LPS-induced proliferation in a human intestinal cell line [27]. In the liver, deficiency of MyD88 delays the expression of growth promoting genes such as c-fos, c-jun, JunB and c-myc and suppresses the activation of NF-κB [39, 40]. It remains unclear which TLR mediates the above described MyD88-dependent regenerative responses as different TLR knockout mice as well as mice deficient in IL-1R and IL-18R receptor signaling did not display these defects [39].
The TLR4-MyD88 signaling axis is also involved in the regulation of angiogenesis to restore blood flow to the site of injured tissue, a prerequisite for mounting a successful repair response. In skin wounds, absence of MyD88 results in slower wound healing, decreased angiogenesis and decreased VEGF levels, albeit by a minor extent [46]. However, it seems unlikely that the rather small decrease in VEGF production is responsible for the striking reduction of skin wound healing in MyD88-deficient mice, and it is likely that other VEGF-independent mechanisms such decreased fibroblast activation or decreased re-epithelization are additionally involved. TLR4 activation alone is not sufficient for the induction of VEGF, but requires the presence of adenosine [96]. Promotion of angiogenesis by TLR4 therefore appears to be restricted to specific pathophysiological circumstances. TLR4-mediated increase of VEGF production is largely mediated by a transcriptional upregulation of HIF1α which binds to a known hypoxia response element in the VEGF promoter [96]. The potential regulation of angiogenesis by the TLR pathway is further supported by a study that has linked downstream mediators of TLRs such as TBK1, TRIF and IRF3, to hypoxia-induced VEGF, FGF1 and FGF2 expression and angiogenesis [97].
Profibrogenic signals
Fibrosis-promoting signals can either directly target fibroblasts or cells such as macrophages to indirectly promote fibroblast activation. Despite the high level of TLR expression in macrophages, there is accumulating evidence that TLRs directly target fibroblasts to induce activating signals. One main mechanism by which TLRs modulate fibrogenic responses is through the TGFβ signaling pathway. Hepatic stellate cells, the main precursor of fibroblasts in the liver, functionally express TLR4. Activation of TLR4 sensitizes hepatic stellate cells towards the effects of TGFβ, and thereby promotes TGFβ-dependent activation and collagen production [53]. This effect is mediated by the downregulation of an inhibitory TGFβ pseudoreceptor, Bambi. Bambi is a type I TGFβ receptor that lacks an intracellular kinase domain and thus acts an inhibitor of BMP, activin and TGFβ signaling [98]. A recent report has shown that Bambi is dispensable for normal development as demonstrated by the unaltered phenotype of Bambi-deleted mice [99] suggesting that Bambi's main function may not lie within the regulation of BMP family member signaling in development but in the regulation of BMP family member signaling in disease processes. In hepatic stellate cells of the normal liver, Bambi is highly expressed to presumably restrict TGFβ- and BMP-induced signals. TLR4-mediated downregulation of Bambi allows for unrestricted activation of the TGFβ pathway in hepatic stellate cells (see Figure 3). Bambi downregulation is mediated through a MyD88-dependent and Trif-independent pathway [53]. Accordingly, suppression of liver fibrosis is much stronger in MyD88- than in Trif-deficient mice. Interestingly, hepatic fibrogenesis strongly downregulates Bambi expression in hepatic stellate cells from wild-type mice but not TLR4-mutated mice [53]. These results are consistent with the hypothesis that increases in LPS during hepatic fibrogenesis induce a downregulation of Bambi. While hepatic stellate cells display a more than 50-fold higher expression of Bambi than whole liver (AM and RFS, unpublished data), the expression and role of Bambi in fibroblasts and fibroblast precursors is not known. It is conceivable that Bambi acts as a repressor of TGFβ signaling in all fibroblast populations, or that hepatic stellate cells have developed high Bambi expression as a result of their unique function and anatomical localisation.
Figure 3. Mechanisms by which TLR4 promotes hepatic stellate cell activation and liver fibrosis.
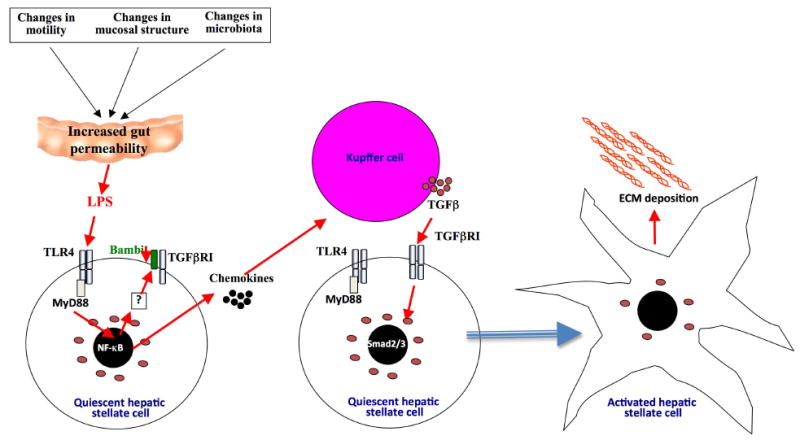
Changes in the intestinal motility, mucosa and microbiota promote release of LPS from the intestinal microbiota. These relatively low amounts of LPS do not induce any appreciable liver injury but target hepatic stellate cells to downregulate TGFβ pseudoreceptor Bambi and upregulate chemokines. These two signals work hand in hand to promote hepatic stellate cell activation: 1. Downregulation of Bambi through a TLR4-MyD88-NF-κB signaling cascade sensitizes hepatic stellate cells towards the effect of TGFβ. 2. Chemokines induce Kupffer cells, a main source of TGFβ in the injured liver, to migrate towards hepatic stellate cells. Thus, the LPS-TLR4-MyD88 signaling cascade mediates the cross-talk between HSCs and Kupffer cells resulting in unrestricted TGFβ-mediated activation of hepatic stellate cells, increased deposition of extracellular matrix and promotion of liver fibrosis.
In addition to sensitizing towards TGFβ-induced signals, TLRs promote proinflammatory and anti-apoptotic signals in fibroblast populations [50, 100]. NF-κB is a key proinflammatory and anti-apoptotic transcription factor and is activated by TLRs in fibroblast populations [50,100]. In the liver, NF-κB activation has been suggested to promote the survival of hepatic stellate cells and development of hepatic fibrosis [101, 102]. There is only little information on the role of TLRs in macrophages during fibrogenic responses. TLR4 on BM-derived cell populations does not promote hepatic fibrogenesis [39], but it is conceivable that TLRs on macrophages from other organs might promote fibrogenesis by either upregulating the secretion of TGFβ or other mediators.
V. Conclusions and outlook
Eleven years after the cloning of the first human TLR, TLRs have emerged as key regulators of inflammation with a broader role in acute and chronic inflammatory processes than initially perceived. In addition to their important role in innate immunity, TLRs are involved in the regulation of tissue injury and wound healing process even under sterile conditions. The ability of TLRs to recognize endogenous mediators appears to be key to their ability to regulate sterile inflammation after acute injury in many organs. Despite rapid progess in understanding the broad role of TLRs in the pathophysiology of disease, a number of questions remains to be answered: (i) There is no satisfying explanation why TLRs exert opposing effects on injury and wound healing in different organ systems. The most likely scenario is that there is already a sufficient activation of cytoprotective pathways in some organs, e.g. in the liver after ischemia-reperfusion, and that activation of cytoprotective pathways by TLRs does not provide additional benefit in these organs, whereas TLRs act as key mediators of cytoprotective signals in other organs and their absence thus leads to increased injury, even if the recruitment of inflammatory cells is reduced. (ii) The contribution of endogenous TLR ligands to sterile inflammation, injury and fibrogenesis needs to be further established. Current evidence for the role of these ligands is at best circumstantial. Genetic approaches such as conditional knock-out and knock-in are absolutely required to identify key actors and to define their cellular targets. It is likely that the effects of some endogenous TLR ligands have been overestimated, or that they are due to bacterial contaminants. (iii) The role of TLRs in carcinogenesis deserves further investigation and needs to be expanded to a larger number of organs. TLRs are likely to promote carcinogenesis in organs in which wound healing is mediated by TLRs, e.g. the liver. One could envision that targeting TLRs may help to redirect wound healing responses from healing by scarring towards healing by regeneration, and thus prevent the development of organ fibrosis and cancer. Better understanding of the TLR signaling pathways in wound healing responses will undoubtedly help to shape new concepts utilizing TLRs and their downstream signaling mediators as pharmacological targets for the treatment of chronic injury, fibrosis and cancer.
Acknowledgments
Johannes Kluwe was supported by a postdoctoral fellowship from the American Liver Foundation. Robert F. Schwabe was supported by grants R01DK076920-01A2 and U54CA126513 from the National Institutes of Health.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. DOI 5000054 [pii] [DOI] [PubMed] [Google Scholar]
- 3.Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE, Jr, Flaherty KR, Schwartz DA, Noble PW, Raghu G, Brown KK. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142:963–967. doi: 10.7326/0003-4819-142-12_part_1-200506210-00005. DOI 142/12_Part_1/963 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 5.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 6.Martin P, D'Souza D, Martin J, Grose R, Cooper L, Maki R, McKercher SR. Wound healing in the PU.1 null mouse--tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;13:1122–1128. doi: 10.1016/s0960-9822(03)00396-8. DOI S0960982203003968 [pii] [DOI] [PubMed] [Google Scholar]
- 7.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. DOI nature07039 [pii] [DOI] [PubMed] [Google Scholar]
- 8.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 9.Wells RG. Fibrogenesis. V. TGF-beta signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2000;279:G845–850. doi: 10.1152/ajpgi.2000.279.5.G845. [DOI] [PubMed] [Google Scholar]
- 10.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. S1359610104000164 [pii] [DOI] [PubMed] [Google Scholar]
- 11.Yoshida M, Sakuma J, Hayashi S, Abe K, Saito I, Harada S, Sakatani M, Yamamoto S, Matsumoto N, Kaneda Y, et al. A histologically distinctive interstitial pneumonia induced by overexpression of the interleukin 6, transforming growth factor beta 1, or platelet-derived growth factor B gene. Proc Natl Acad Sci U S A. 1995;92:9570–9574. doi: 10.1073/pnas.92.21.9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen HS, Yeh MM, Fausto N. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. DOI 0409722102 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, Schirmacher P, Biesterfeld S, Galle PR, Lohse AW, Kanzler S. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45:419–428. doi: 10.1016/j.jhep.2006.04.010. DOI S0168-8278(06)00245-5 [pii] [DOI] [PubMed] [Google Scholar]
- 14.Thieringer F, Maass T, Czochra P, Klopcic B, Conrad I, Friebe D, Schirmacher P, Lohse AW, Blessing M, Galle PR, Teufel A, Kanzler S. Spontaneous hepatic fibrosis in transgenic mice overexpressing PDGF-A. Gene. 2008 doi: 10.1016/j.gene.2008.05.022. DOI S0378-1119(08)00218-7 [pii] [DOI] [PubMed] [Google Scholar]
- 15.George J, Roulot D, Koteliansky VE, Bissell DM. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci U S A. 1999;96:12719–12724. doi: 10.1073/pnas.96.22.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi Z, Atsuchi N, Ooshima A, Takeshita A, Ueno H. Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci U S A. 1999;96:2345–2349. doi: 10.1073/pnas.96.5.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdollahi A, Li M, Ping G, Plathow C, Domhan S, Kiessling F, Lee LB, McMahon G, Grone HJ, Lipson KE, Huber PE. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med. 2005;201:925–935. doi: 10.1084/jem.20041393. DOI jem.20041393 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. 083001.084359 [pii] [DOI] [PubMed] [Google Scholar]
- 21.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. nri1391 [pii] [DOI] [PubMed] [Google Scholar]
- 22.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. DOI S1044-5323(06)00122-9 [pii] [DOI] [PubMed] [Google Scholar]
- 23.Tsan MF, Baochong G. Pathogen-associated molecular pattern contamination as putative endogenous ligands of Toll-like receptors. J Endotoxin Res. 2007;13:6–14. doi: 10.1177/0968051907078604. DOI 13/1/6 [pii] [DOI] [PubMed] [Google Scholar]
- 24.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tor M, Billiar T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. DOI IMR579 [pii] [DOI] [PubMed] [Google Scholar]
- 25.Beutler B. Neo-ligands for innate immune receptors and the etiology of sterile inflammatory disease. Immunol Rev. 2007;220:113–128. doi: 10.1111/j.1600-065X.2007.00577.x. DOI IMR577 [pii] [DOI] [PubMed] [Google Scholar]
- 26.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. DOI nm1315 [pii] [DOI] [PubMed] [Google Scholar]
- 27.Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, Abreu MT. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862–877. doi: 10.1053/j.gastro.2006.06.017. DOI S0016-5085(06)01287-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. S0092867404006610 [pii] [DOI] [PubMed] [Google Scholar]
- 29.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. DOI jem.20042614 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. DOI 0702553104 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. 109/6/784 [pii] [DOI] [PubMed] [Google Scholar]
- 33.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, Geller DA, Billiar TR. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. DOI 175/11/7661 [pii] [DOI] [PubMed] [Google Scholar]
- 34.Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol. 2008;19:923–932. doi: 10.1681/ASN.2007090982. DOI ASN.2007090982 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007;178:6252–6258. doi: 10.4049/jimmunol.178.10.6252. DOI 178/10/6252 [pii] [DOI] [PubMed] [Google Scholar]
- 36.Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;1:179–182. doi: 10.1016/s0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- 37.Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108:218–224. doi: 10.1016/0016-5085(95)90027-6. DOI S0016508595000199 [pii] [DOI] [PubMed] [Google Scholar]
- 38.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. DOI S0270-9139(01)65119-X [pii] [DOI] [PubMed] [Google Scholar]
- 39.Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, Kishimoto T, Nakanishi K, Fujimoto J. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology. 2005;41:443–450. doi: 10.1002/hep.20603. [DOI] [PubMed] [Google Scholar]
- 40.Campbell JS, Riehle KJ, Brooling JT, Bauer RL, Mitchell C, Fausto N. Proinflammatory cytokine production in liver regeneration is Myd88-dependent, but independent of Cd14, Tlr2, and Tlr4. J Immunol. 2006;176:2522–2528. doi: 10.4049/jimmunol.176.4.2522. DOI 176/4/2522 [pii] [DOI] [PubMed] [Google Scholar]
- 41.Cornell RP, Liljequist BL, Bartizal KF. Depressed liver regeneration after partial hepatectomy of germ-free, athymic and lipopolysaccharide-resistant mice. Hepatology. 1990;11:916–922. doi: 10.1002/hep.1840110603. DOI S027091399000115X [pii] [DOI] [PubMed] [Google Scholar]
- 42.Akita K, Okuno M, Enya M, Imai S, Moriwaki H, Kawada N, Suzuki Y, Kojima S. Impaired liver regeneration in mice by lipopolysaccharide via TNF-alpha/kallikrein-mediated activation of latent TGF-beta. Gastroenterology. 2002;123:352–364. doi: 10.1053/gast.2002.34234. DOI S001650850200094X [pii] [DOI] [PubMed] [Google Scholar]
- 43.Sun R, Gao B. Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-gamma) Gastroenterology. 2004;127:1525–1539. doi: 10.1053/j.gastro.2004.08.055. DOI S0016508504015562 [pii] [DOI] [PubMed] [Google Scholar]
- 44.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A. 2005;102:99–104. doi: 10.1073/pnas.0405979102. DOI 0405979102 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF, Stappenbeck TS. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J Clin Invest. 2007;117:258–269. doi: 10.1172/JCI29159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macedo L, Pinhal-Enfield G, Alshits V, Elson G, Cronstein BN, Leibovich SJ. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am J Pathol. 2007;171:1774–1788. doi: 10.2353/ajpath.2007.061048. DOI ajpath.2007.061048 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurt-Jones EA, Sandor F, Ortiz Y, Bowen GN, Counter SL, Wang TC, Finberg RW. Use of murine embryonic fibroblasts to define Toll-like receptor activation and specificity. J Endotoxin Res. 2004;10:419–424. doi: 10.1179/096805104225006516. [DOI] [PubMed] [Google Scholar]
- 48.Wolf G, Bohlender J, Bondeva T, Roger T, Thaiss F, Wenzel UO. Angiotensin II upregulates toll-like receptor 4 on mesangial cells. J Am Soc Nephrol. 2006;17:1585–1593. doi: 10.1681/ASN.2005070699. DOI ASN.2005070699 [pii] [DOI] [PubMed] [Google Scholar]
- 49.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. DOI S0016508503004037 [pii] [DOI] [PubMed] [Google Scholar]
- 50.Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. S027091390300199X [pii] [DOI] [PubMed] [Google Scholar]
- 51.Pierer M, Rethage J, Seibl R, Lauener R, Brentano F, Wagner U, Hantzschel H, Michel BA, Gay RE, Gay S, Kyburz D. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. J Immunol. 2004;172:1256–1265. doi: 10.4049/jimmunol.172.2.1256. [DOI] [PubMed] [Google Scholar]
- 52.Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology. 2006;130:1886–1900. doi: 10.1053/j.gastro.2006.01.038. DOI S0016-5085(06)00065-5 [pii] [DOI] [PubMed] [Google Scholar]
- 53.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. DOI nm1663 [pii] [DOI] [PubMed] [Google Scholar]
- 54.Isayama F, Hines IN, Kremer M, Milton RJ, Byrd CL, Perry AW, McKim SE, Parsons C, Rippe RA, Wheeler MD. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1318–1328. doi: 10.1152/ajpgi.00405.2005. DOI 00405.2005 [pii] [DOI] [PubMed] [Google Scholar]
- 55.Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, Layden TJ, Wright TL, White T, Cheung RC. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007;46:297–306. doi: 10.1002/hep.21695. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, Mehal WZ. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 57.Riad A, Jager S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, Karatas A, Heimesaat MM, Bereswill S, Dragun D, Pauschinger M, Schultheiss HP, Tschope C. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol. 2008;180:6954–6961. doi: 10.4049/jimmunol.180.10.6954. DOI 180/10/6954 [pii] [DOI] [PubMed] [Google Scholar]
- 58.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. 0403249101 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lehr HA, Sagban TA, Ihling C, Zahringer U, Hungerer KD, Blumrich M, Reifenberg K, Bhakdi S. Immunopathogenesis of atherosclerosis: endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemic diet. Circulation. 2001;104:914–920. doi: 10.1161/hc3401.093153. [DOI] [PubMed] [Google Scholar]
- 61.Schoneveld AH, Oude Nijhuis MM, van Middelaar B, Laman JD, de Kleijn DP, Pasterkamp G. Toll-like receptor 2 stimulation induces intimal hyperplasia and atherosclerotic lesion development. Cardiovasc Res. 2005;66:162–169. doi: 10.1016/j.cardiores.2004.12.016. DOI S0008-6363(04)00588-7 [pii] [DOI] [PubMed] [Google Scholar]
- 62.Vink A, Schoneveld AH, van der Meer JJ, van Middelaar BJ, Sluijter JP, Smeets MB, Quax PH, Lim SK, Borst C, Pasterkamp G, de Kleijn DP. In vivo evidence for a role of toll-like receptor 4 in the development of intimal lesions. Circulation. 2002;106:1985–1990. doi: 10.1161/01.cir.0000032146.75113.ee. [DOI] [PubMed] [Google Scholar]
- 63.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rivera CA, Bradford BU, Hunt KJ, Adachi Y, Schrum LW, Koop DR, Burchardt ER, Rippe RA, Thurman RG. Attenuation of CCl(4)-induced hepatic fibrosis by GdCl(3) treatment or dietary glycine. Am J Physiol Gastrointest Liver Physiol. 2001;281:G200–207. doi: 10.1152/ajpgi.2001.281.1.G200. [DOI] [PubMed] [Google Scholar]
- 65.Sung SA, Jo SK, Cho WY, Won NH, Kim HK. Reduction of renal fibrosis as a result of liposome encapsulated clodronate induced macrophage depletion after unilateral ureteral obstruction in rats. Nephron Exp Nephrol. 2007;105:e1–9. doi: 10.1159/000096859. DOI NEE2007105001001 [pii] [DOI] [PubMed] [Google Scholar]
- 66.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. DOI nature04399 [pii] [DOI] [PubMed] [Google Scholar]
- 67.Seimon TA, Obstfeld A, Moore KJ, Golenbock DT, Tabas I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc Natl Acad Sci U S A. 2006;103:19794–19799. doi: 10.1073/pnas.0609671104. DOI 0609671104 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–454. doi: 10.1038/ncpcardio0938. DOI ncpcardio0938 [pii] [DOI] [PubMed] [Google Scholar]
- 69.Virchow R. Aetiologie der neoplastischen Geschwülste/Pathogenie der neoplastischen Geschwülste. In: Virchow R, editor. Die krankhaften Geschwülste. Verlag von August von Hirschwald; Berlin: 1863. pp. 57–101. [Google Scholar]
- 70.Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Busch W. Aus der Stizung der medizinischen Section vom 13. November 1867. Berl Klin Wochenschr. 1868;5:137. [Google Scholar]
- 72.Bruns P. Die Heilwirkung des Erysipelas auf Geschwülste. Beitr Klin Chir. 1888;3:443. [Google Scholar]
- 73.Coley WB. Treatment of inoperable malignant tumors with the toxins of erysipelas and the Bacillus prodigiosus. Trans Amer Surg Assn. 1894;12:183–212. [Google Scholar]
- 74.Hobohm U. Fever and cancer in perspective. Cancer Immunol Immunother. 2001;50:391–396. doi: 10.1007/s002620100216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Starnes CO. Coley's toxins in perspective. Nature. 1992;357:11–12. doi: 10.1038/357011a0. [DOI] [PubMed] [Google Scholar]
- 76.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. DOI nm1622 [pii] [DOI] [PubMed] [Google Scholar]
- 77.Pidgeon GP, Harmey JH, Kay E, Da Costa M, Redmond HP, Bouchier-Hayes DJ. The role of endotoxin/lipopolysaccharide in surgically induced tumour growth in a murine model of metastatic disease. Br J Cancer. 1999;81:1311–1317. doi: 10.1038/sj.bjc.6694369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harmey JH, Bucana CD, Lu W, Byrne AM, McDonnell S, Lynch C, Bouchier-Hayes D, Dong Z. Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int J Cancer. 2002;101:415–422. doi: 10.1002/ijc.10632. [DOI] [PubMed] [Google Scholar]
- 79.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation-induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. S153561080400217X [pii] [DOI] [PubMed] [Google Scholar]
- 80.Huang B, Zhao J, Shen S, Li H, He KL, Shen GX, Mayer L, Unkeless J, Li D, Yuan Y, Zhang GM, Xiong H, Feng ZH. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007;67:4346–4352. doi: 10.1158/0008-5472.CAN-06-4067. DOI 67/9/4346 [pii] [DOI] [PubMed] [Google Scholar]
- 81.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–1881. doi: 10.1053/j.gastro.2007.09.008. DOI S0016-5085(07)01649-6 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. DOI 317/5834/124 [pii] [DOI] [PubMed] [Google Scholar]
- 83.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. DOI 317/5834/121 [pii] [DOI] [PubMed] [Google Scholar]
- 84.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte Necrosis Induced by Oxidative Stress and IL-1alpha Release Mediate Carcinogen-Induced Compensatory Proliferation and Liver Tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. DOI S1535-6108(08)00226-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. DOI S0092867402007031 [pii] [DOI] [PubMed] [Google Scholar]
- 86.Ferluga J, Allison AC. Role of mononuclear infiltrating cells in pathogenesis of hepatitis. Lancet. 1978;2:610–611. doi: 10.1016/s0140-6736(78)92828-3. [DOI] [PubMed] [Google Scholar]
- 87.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. nature00858 [pii] [DOI] [PubMed] [Google Scholar]
- 88.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. DOI nm1603 [pii] [DOI] [PubMed] [Google Scholar]
- 89.Tsung A, Zheng N, Jeyabalan G, Izuishi K, Klune JR, Geller DA, Lotze MT, Lu L, Billiar TR. Increasing numbers of hepatic dendritic cells promote HMGB1-mediated ischemia-reperfusion injury. J Leukoc Biol. 2007;81:119–128. doi: 10.1189/jlb.0706468. DOI jlb.0706468 [pii] [DOI] [PubMed] [Google Scholar]
- 90.Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA., Jr Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest. 1990;85:1936–1943. doi: 10.1172/JCI114656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rudiger HA, Clavien PA. Tumor necrosis factor alpha, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology. 2002;122:202–210. doi: 10.1053/gast.2002.30304. DOI S0016508502604792 [pii] [DOI] [PubMed] [Google Scholar]
- 92.Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. DOI 0270-9139(94)90199-6 [pii] [PubMed] [Google Scholar]
- 93.Uesugi T, Froh M, Arteel GE, Bradford BU, Gabele E, Wheeler MD, Thurman RG. Delivery of IkappaB superrepressor gene with adenovirus reduces early alcohol-induced liver injury in rats. Hepatology. 2001;34:1149–1157. doi: 10.1053/jhep.2001.29400. DOI S0270913901134634 [pii] ajhep0341149 [pii] [DOI] [PubMed] [Google Scholar]
- 94.Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8:289–293. doi: 10.1038/nm0302-289. nm0302-289 [pii] [DOI] [PubMed] [Google Scholar]
- 96.Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell. 2007;18:14–23. doi: 10.1091/mbc.E06-07-0596. DOI E06-07-0596 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Korherr C, Gille H, Schafer R, Koenig-Hoffmann K, Dixelius J, Egland KA, Pastan I, Brinkmann U. Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc Natl Acad Sci U S A. 2006;103:4240–4245. doi: 10.1073/pnas.0511319103. DOI 0511319103 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massague J, Niehrs C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature. 1999;401:480–485. doi: 10.1038/46794. [DOI] [PubMed] [Google Scholar]
- 99.Chen J, Bush JO, Ovitt CE, Lan Y, Jiang R. The TGF-beta pseudoreceptor gene Bambi is dispensable for mouse embryonic development and postnatal survival. Genesis. 2007;45:482–486. doi: 10.1002/dvg.20320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paik YH, Lee KS, Lee HJ, Yang KM, Lee SJ, Lee DK, Han KH, Chon CY, Lee SI, Moon YM, Brenner DA. Hepatic stellate cells primed with cytokines upregulate inflammation in response to peptidoglycan or lipoteichoic acid. Lab Invest. 2006;86:676–686. doi: 10.1038/labinvest.3700422. DOI 3700422 [pii] [DOI] [PubMed] [Google Scholar]
- 101.Oakley F, Meso M, Iredale JP, Green K, Marek CJ, Zhou X, May MJ, Millward-Sadler H, Wright MC, Mann DA. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128:108–120. doi: 10.1053/j.gastro.2004.10.003. DOI S0016508504018475 [pii] [DOI] [PubMed] [Google Scholar]
- 102.Watson MR, Wallace K, Gieling RG, Manas DM, Jaffray E, Hay RT, Mann DA, Oakley F. NF-kappaB is a critical regulator of the survival of rodent and human hepatic myofibroblasts. J Hepatol. 2008;48:589–597. doi: 10.1016/j.jhep.2007.12.019. DOI S0168-8278(08)00041-X [pii] [DOI] [PubMed] [Google Scholar]