

Abstract
The endocannabinoid system comprises specific cannabinoid receptors such as Cb1 and Cb2, the endogenous ligands (anandamide and 2-arachidonyl glycerol among others) and the proteins responsible for their synthesis and degradation. This system has become the focus of research in recent years because of its potential therapeutic value several disease states. The following review describes our current knowledge of the changes that occur in the endocannabinoid system during carcinogenesis and then focuses on the effects of anandamide on various aspects of the carcinogenic process such as growth, migration, and angiogenesis in tumors from various origins.
I. Introduction
Marijuana and its derivatives have been used in medicine for centuries, however, it was not until the isolation of the psychoactive component of Cannabis sativa (Δ9-tetrahydrocannabinol; Δ9-THC) and the subsequent discovery of the endogenous cannabinoid signaling system that research into the therapeutic value of this system reemerged. Ongoing research is determining that regulation of the endocannabinoid system may be effective in the treatment of pain (Calignano et al., 1998; Manzanares et al., 1999), glaucoma (Voth and Schwartz, 1997), and neurodegenerative disorders such as Parkinson's disease (Piomelli et al., 2000) and multiple sclerosis (Baker et al., 2000). In addition, cannabinoids might be effective anti-tumoral agents because of their ability to inhibit the growth of various types of cancer cell lines in culture (De Petrocellis et al., 1998; Ruiz et al., 1999; Sanchez et al., 1998, 2001) and in laboratory animals (Galve-Roperh et al., 2000).
The endogenous cannabinoid system (endocannabinoid) consists of the cannabinoid receptors, their endogenous ligands (endocannabinoids) and the proteins for their synthesis and inactivation (Bisogno et al., 2005). The cannabinoid receptors are seven-transmembrane-domain proteins coupled to Gi/o type of G-proteins (Bisogno et al., 2005). To date, there are two definitive cannabinoid receptors, Cb1 and Cb2, as well as a putative involvement of the vanilloid receptor VR1. More recently, the orphan receptor GPR55 was shown to function as a novel cannabinoid receptor (Ryberg et al., 2007). Cb1 receptors are found predominantly in the central nervous system, but also in most peripheral tissues including immune cells, the reproductive system, the gastrointestinal tract and the lungs (Devane et al., 1988; Matsuda et al., 1990; Munro et al., 1993). Cb2 receptors are found predominantly in the immune system, that is, in tonsils, spleen, macrophages, and lymphocytes (Devane et al., 1988; Matsuda et al., 1990; Munro et al., 1993).
To date, many endocannabinoids have been identified with varying affinities for the receptors and all of which are lipid molecules. Anandamide (AEA) was the first endogenous ligand to be identified (Devane et al., 1988), which acts as a partial Cb1 agonist and weak Cb2 agonist. It has also been shown to activate the GPR55 (Ryberg et al., 2007). While the physiological roles of many of the other ligands have not yet been fully clarified, AEA has been implicated in a wide variety of physiological and pathological processes.
Currently, there are two biosynthesis pathways for AEA. The first is via the remodeling of an existing membrane phosphoglyceride, that is, the calcium-dependent N-transacylation of phosphotidylethanolamine with arachidonic acid to form N-arachidonyl-phosphatidyl-ethanolamine, which is then hydrolyzed to AEA (Bisogno et al., 2005; Di Marzo and Deutsch, 1998). The enzyme responsible for the catalysis of this pathway is phospholipase D (Bisogno et al., 2005). The second pathway is via the de novo synthesis of AEA from arachidonic acid and ethanolamine by the enzyme anandamide amidohydrolase catalyzing the reverse reaction from high levels of ethanolamine (Di Marzo and Deutsch, 1998). After synthesis, AEA is rapidly inactivated via a tightly controlled series of events involving sequestration by cells and enzymatic hydrolysis. The mechanism of AEA uptake is largely unknown with data suggesting that it is via passive diffusion and others suggesting that it is through the presence of an active transporter (Glaser et al., 2005). Regardless of the mechanism, this uptake is a rapid event with a half-life of approximately 2.5 min (Di Marzo and Deutsch, 1998). After uptake, AEA is hydrolyzed and degraded by the enzyme anandamide amidohydrolase (also called fatty acid amide hydrolase or FAAH) (Di Marzo and Deutsch, 1998).
II. Changes in the Endocannabinoid System in Cancer
Evidence suggests that the endocannabinoid system may be dysregulated in a number of cancers (summarized in Table 18.1). Indeed, both AEA and 2-arachidonyl glycerol (2-AG) have been shown to be increased in human colorectal adenomatous polyps and carcinomas compared to normal colorectal mucosa (Ligresti et al., 2003), suggesting that these endocannabinoids increase when passing from normal to transformed mucosa. No consistent differences were observed in the expression levels of Cb1, Cb2, or FAAH as assessed by both RT-PCR and immunoblotting between normal and colorectal cancer tissue (Ligresti et al., 2003). Similarly, AEA levels were enhanced by 17-fold in glioblastomas whereas meningiomas were characterized by a massively enhanced level of 2-AG (Petersen et al., 2005). Coupled with these changes was a 60% reduction in the enzyme activities of the AEA degradation enzymes, N-acylphosphotidylethanolamine-hydrolyzing phospholipase D and fatty acid amide hydrolase in the glioblastoma tissue and an enhanced in vitro conversion of phosphotidyl choline to monoacyl glycerol in the meningioma tissue (Petersen et al., 2005). Similarly, the levels of AEA and 2-AG were found to be increased in human pituitary adenomas compared to normal pituitary gland (Pagotto et al., 2001). Moreover, endocannabinoid content in the different pituitary adenomas correlated with the presence of Cb1, being elevated in the tumoral samples positive for Cb1 and lower in the samples in which no or low levels of Cb1 were found (Pagotto et al., 2001). In another study, the levels of AEA were found to differ depending upon the source of the tumor (Schmid et al., 2002). For example, AEA was increased in prostate carcinoma, endometrial sarcoma, and thigh histiocytoma, with no change in ileum lymphoma, and bladder carcinoma and a significant decrease in stomach carcinoma (Schmid et al., 2002).
Table 18.1.
Changes in the endocannabinoid system in various types of human cancer
| Cancer type | In vivo/in vitro | Changes in the endocannabinoid system | References |
|---|---|---|---|
| Colorectal | In vivo | AEA ↑, 2-AG ↑ | Ligresti et al. (2003) |
| Glioblastomas | In vivo | AEA ↑, FAAH ↓, Phopholipase D ↓, VR1 ↑ | Contassot et al. (2004b) and Petersen et al. (2005) |
| Meningioma | In vivo | 2-AG ↑ | Petersen et al. (2005) |
| Pituitary adenomas | AEA ↑, 2-AG ↑, Cb1 ↑ | Pagotto et al. (2001) | |
| Prostate carcinoma | In vivo | AEA ↑, Cb1 ↑, Cb2 ↑ | Schmid et al. (2002) and Sarfaraz et al. (2005) |
| Endometrial sarcoma | In vivo | AEA ↑ | Schmid et al. (2002) |
| Thigh histiocytoma | In vivo | AEA ↑ | Schmid et al. (2002) |
| Stomach carcinoma | In vivo | AEA ↓ | Schmid et al. (2002) |
| Mantle cell lymphoma | In vivo | Cb1 ↑, Cb2 ↑ | Ek et al. (2002) and Islam et al. (2003) |
| Acute myeloid leukemia | In vitro | Cb2 ↑ | Alberich Jorda et al. (2004) |
| Breast cancer | In vivo | Cb1 ↓, Cb2 ↓ | Caffarel et al. (2006) |
Furthermore, the cannabinoid receptor system has also been shown to be altered during the carcinogenic process. Indeed, in Mantle cell lymphoma and prostate cancer cells, both Cb1 and Cb2 are upregulated compared to nonmalignant tissue (Ek et al., 2002; Islam et al., 2003; Sarfaraz et al., 2005), whereas in acute myeloid leukemia, only Cb2 is upregulated (Alberich Jorda et al., 2004). A differential effect on receptor expression is shown in breast cancer, with a marked suppression of Cb1 and an increased Cb2 expression (Caffarel et al., 2006). In another study using human brain tissue, the expression of VR1 was aberrantly expressed in glioma brain tumors compared to nonmalignant brain tissue sampled from epileptic patients undergoing surgery (Contassot et al., 2004b).
Dysregulation of the endocannabinoid system still needs to be examined in a number of other cancer types and the implications of a dysregulated endocannabinoid system on malignant transformation and tumor progression remains to be addressed.
III. Antiproliferative Effects of Anandamide
There is a large volume of data indicating antiproliferative effects of AEA on various cancer types via a number or receptor-dependent and receptor-independent mechanisms. A schematic diagram of the proposed mechanisms is depicted in Fig. 18.1.
Figure 18.1.
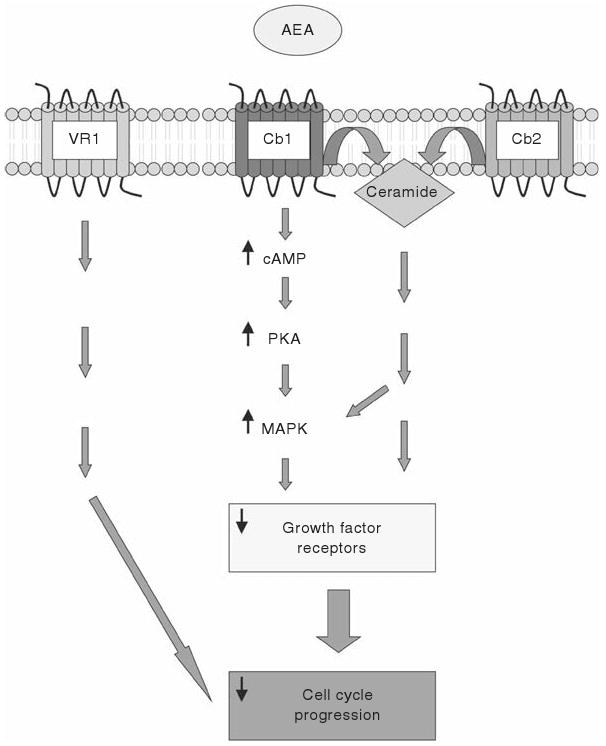
Schematic diagram of the cannabinoid receptor-dependent mechanisms whereby AEA leads to tumor growth suppression and/or cell death. AEA may act via the Cb1 receptor to activate the cAMP/PKA/MAPK pathway or to increase the production of ceramide. These effects ultimately result in a decrease in the expression of various growth factor receptors and decrease cell cycle progression. Alternatively, AEA may activate either Cb2 or VR1 to elicit a similar response, although the mechanism by which this occurs is not clear.
A. Receptor mediated effects
Cb1
AEA has been shown to decrease the proliferation of breast cancer cells in vitro by decreasing the levels of the long form of the prolactin receptor (De Petrocellis et al., 1998) and inhibiting the nerve growth factor (NGF)-mediated proliferation by decreasing the expression of the trk NGF receptor (Melck et al., 2000). This was shown to be via a Cb1-dependent mechanism and involves the activation of the cAMP/protein kinase A pathway and subsequent activation of the mitogen-activated protein kinase (MAPK) pathway (Melck et al., 1999). Furthermore, these growth suppressing effects of AEA are not due to toxicity or apoptosis, but by cell cycle arrest in the S phase which was correlated with an activation of Chk1 (an S phase checkpoint kinase), and a suppression of Cdk2 activity (Laezza et al., 2006).
Similar antiproliferative and apoptotic effects of AEA were found in human prostate cancer cell lines (Mimeault et al., 2003), which were also via the Cb1-dependent downregulation of growth factor signaling (Mimeault et al., 2003). Specifically, AEA decreased the expression of the epidermal growth factor receptor (EGFR) in several prostate cancer cell lines which was associated with a G1 arrest and cell death (Mimeault et al., 2003). Furthermore, this effect could be partially blocked by acidic ceramide inhibitors indicating that AEA might be induced via the cellular ceramide production (Mimeault et al., 2003).
Cb2
In contrast, the antiproliferative actions of AEA on glioma and asytocytoma cells are via the activation of the Cb2 receptor (Sanchez et al., 2001). Treatment of mice inoculated with human glioma tumor cells or human astrocytoma cells with the specific Cb2 agonist, JWH-133 completely blocked the tumor growth in vivo (Sanchez et al., 2001), and was, once again, associated with increased de novo ceramide synthesis in vitro (Sanchez et al., 2001). Furthermore, targeting the Cb2 receptor has been suggested as a therapy to treat malignant lymphoblastic diseases (McKallip et al., 2002). In these types of cancers, AEA induced apoptosis in vitro and in vivo and it is suggested that because the Cb2 agonists lack psychotropic effects, targeting of the Cb2 receptor would be preferential to targeting the Cb1 receptor in cancers of immune origin (McKallip et al., 2002). Another study demonstrated that interleukin-12 treatment and/or overexpression in thyroid carcinoma cells leads to an increase in Cb2 expression and that either overexpression of interleukin-12 or Cb2 resulted in tumor regression and increased sensitivity to chemotherapy (Shi et al., 2008). Taken together, these data indicate that targeting Cb2 may be of therapeutic value in certain tumor types and warrants further investigation.
VR1
The actions of AEA on VR1 were shown to be antiproliferative in glioma cells (Contassot et al., 2004b) and in uterine cervix cancer cells (Contassot et al., 2004a). In both of these cell types, the VR1 was aberrantly expressed in the tumor cells and tissue compared to their nonmalignant counterparts (Contassot et al., 2004a,b). Furthermore, stimulation of Cb1 or Cb2 was protective against the VR1-mediated antiproliferative effects of AEA (Contassot et al., 2004a,b).
Combination of receptors
In addition to the above-mentioned findings, studies have shown an antiproliferative/growth suppressive effect of AEA that could not be attributable to just one receptor. Using rat C6 glioma cells, AEA was shown to have growth suppressing effects that were time and dose-dependent (Jacobsson et al., 2001). These effects could be partially blocked by the Cb1, Cb2, and VR1 antagonists alone, but was completely attenuated when the three receptor antagonists were added in combination (Jacobsson et al., 2001). In addition, in osteosarcoma cells, AEA induced apoptosis by increasing intracellular calcium levels, activation of p38 MAPK and subsequent activation of caspase 3 (Hsu et al., 2007). Unfortunately, the authors do not address the issue of receptor-dependence in this study, therefore for the purpose of this review we will assume potential involvement of all three receptors as none can be ruled out.
B. Receptor-independent effects
Cannabinoid receptor-independent actions of AEA have been described in several tumor cell types (DeMorrow et al., 2007; Hinz et al., 2004a,b; Patsos et al., 2005; Ramer et al., 2001). Furthermore, most of these effects are via the actions of AEA at the cell membrane (DeMorrow et al., 2007; Hinz et al., 2004a,b; Ramer et al., 2001). Treatment of human neuroglioma cells with the stable analogue of AEA (Met-AEA) resulted in the induction of apoptosis (Hinz et al., 2004a,b; Ramer et al., 2001). This effect could not be blocked by the coadministration of antagonists of the Cb1, Cb2, or VR1 receptors, nor the Gi/o protein inactivator pertussis toxin (Ramer et al., 2001). Coupled with the induction of apoptosis by Met-AEA, there was an increased synthesis of ceramide, expression of cyclooxygenase-2 (Cox-2) and subsequent prostaglandin E2 synthesis via a mechanism involving p38 and p42/44 MAPK activation (Ramer et al., 2001). Specific Cox-2 inhibitors, such as celecoxib and diclofenac, or the specific silencing of Cox-2 expression with small-interfering RNA blocked the Met-AEA-induced apoptosis (Hinz et al., 2004b). Furthermore, the lipid raft disruptor methyl-beta-cyclodextrin blocked the Met-AEA-induced effects of ceramide synthesis, phosphorylation of p38 and p42/44 MAPK, expression of Cox-2, and subsequent prostaglandin E2 synthesis (Hinz et al., 2004a). Together, these data suggest that Met-AEA, via lipid raft-mediated events, induces apoptosis of neuroglioma cells. A similar effect of AEA was observed in colorectal cancer cells (Patsos et al., 2005). Treatment of these cells with AEA increased the expression of Cox-2 and induced a subsequent cell death pathway (Patsos et al., 2005). Interestingly, inhibition of FAAH potentiated this cell death, suggesting that AEA-induced cell death was mediated via the metabolism of AEA by Cox-2 rather than through the classical AEA degradation pathway (Patsos et al., 2005).
More recently, we have shown that AEA can induce growth-suppressive/pro-apoptotic effects in cholangiocarcinoma cells which could not be blocked by any Cb1, Cb2, or VR1 antagonists nor the Gi/o protein inactivator pertussis toxin (DeMorrow et al., 2007). This is via the stabilization of the lipid raft structures within the plasma membrane, the increased production of ceramide, and the subsequent recruitment of the death receptor complex components into the lipid raft structures (DeMorrow et al., 2007). Interestingly, the other more prevalent endocannabinoid, 2-AG, had growth-promoting effects, which were shown to be via the complete disruption of the lipid raft structure (DeMorrow et al., 2007), an event which has previously been shown to result in growth-promoting effects in other cell types (Lambert et al., 2006; Mathay et al., 2008).
These receptor-independent mechanisms are summarized in Fig. 18.2. These so-called “receptor-independent” effects of AEA must be taken with a note of caution. Most of these studies were performed prior to the identification of GPR55 as a putative cannabinoid receptor. Therefore, the possibility that AEA is exerting its effects through GPR55 or some other, as yet unidentified cannabinoid receptor cannot be ruled out.
Figure 18.2.
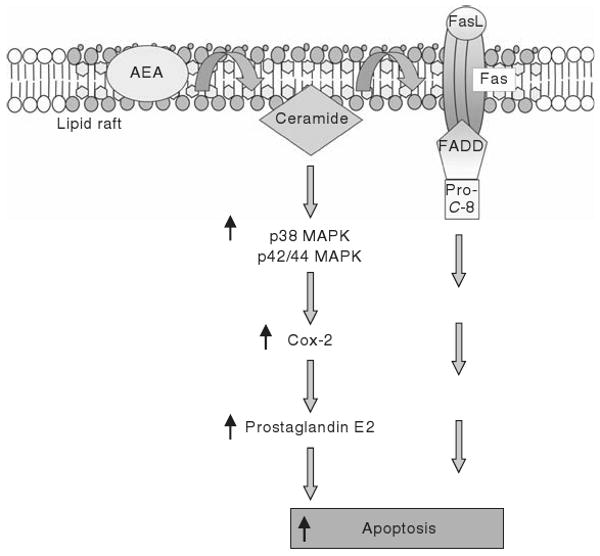
Schematic diagram of the cannabinoid receptor-independent mechanisms whereby AEA induces apoptosis. AEA stabilizes the lipid raft microdomains at the plasma membrane and increases ceramide production. This then has an effect on Cox-2 expression and prostaglandin E2 production with a subsequent increase in apoptosis. Alternatively, AEA-induced ceramide production facilitates the Fas/FasL death receptor complex within the lipid raft structure, which ultimately results in increased apoptosis.
IV. Effects of AEA on Migration, Invasion, and Angiogenesis
The acquisition of metastatic abilities by cancer cells often leads to clinically incurable disease. Metastasis consists of a series of sequential steps including detachment of cells from the primary tumor, survival of the cells in circulation, arrest in a new organ, initiation and maintenance of growth in the new tissue, and vascularization of the metastatic tumor (Fidler, 2002). AEA has been shown to have a regulatory role at each of these stages in the metastatic process (Fig. 18.3). Firstly, AEA was shown to be an endogenous inhibitor of migration of both colon carcinoma cells and nonmalignant T lymphocytes in vitro (Joseph et al., 2004). There was a differential mechanism involved in the regulation of the tumor versus immune cells, that is, tumor cell migration could be stimulated by specific agonists for Cb1 receptor, whereas the immune cell migration was inhibited by a Cb2-dependent mechanism (Joseph et al., 2004). Furthermore, in an in vivo model of metastatic spreading using breast cancer cell lines, the AEA analogue, met-AEA significantly reduced the number and dimension of metastatic nodes, an effect that was inhibited by specific Cb1 receptor antagonists (Grimaldi et al., 2006). Molecular changes in the organization and distribution of cytoskeleton proteins are necessary for focal adhesion; cell motility and cell invasion were then assessed. No changes in expression of any of the integrins were detected after Met-AEA treatment, however, there was a marked decrease in the phosphorylation of focal adhesion kinase and src, both of which are normally localized to the focal adhesions and are involved in the metastatic formation and development (Grimaldi et al., 2006).
Figure 18.3.
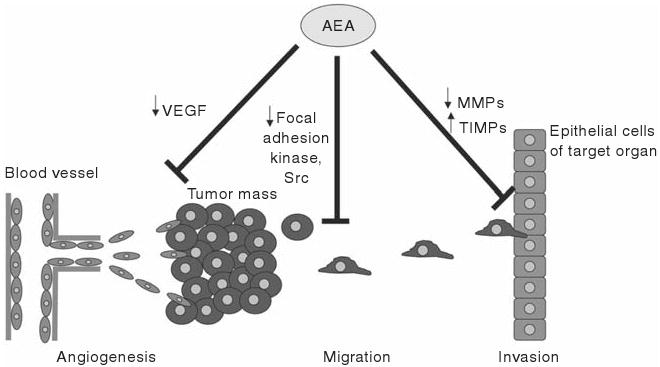
AEA inhibits other aspects of tumorigenesis such as angiogenesis, tumor cell migration, and tumor invasion. AEA inhibits angiogenesis via a decrease in VEGF expression, whereas the decrease in migration is thought to be via a decrease in the activation of focal adhesion kinases and src kinase, both of which are thought to be involved in cell migration and metastasis. Lastly, AEA inhibits tumor cell invasion by decreasing the expression of proteins responsible for breaking down the extracellular matrix of the target organ, such as MMPs and increasing the expression of the tissue inhibitors of MMPs.
In order for a migrating cancer cell to then invade another organ, the existing extracellular matrix components (e.g., collagens and proteoglycans) must be broken down and hence the rigid architecture of the target organ must be compromised. Matrix metalloproteinases (MMPs) are emerging as a family of enzymes that exerts important functions during tumor invasion (Curran and Murray, 2000; Stamenkovic, 2000). Tissue inhibitors of MMPs (TIMPs), and in particular TIMP-1, have also been shown to inhibit the proteolytic activity of MMPs and suppress vascular tumor growth and angiogenesis in xenograft animal models (Zacchigna et al., 2004). Furthermore, there appears to be a correlation between high cancer invasiveness and decreased TIMP-1 expression (Chan et al., 2005; Khokha et al., 1989). Recently, the effects of AEA on MMP and TIMP expression were evaluated in various cancer cell types. Using a cervical cancer cell line, Met-AEA as well as Δ9-THC, inhibited the invasive properties of these cells via the increased expression of TIMP-1 (Ramer and Hinz, 2008). The effects of the cannabinoids on invasion and TIMP-1 expression were inhibited by the pretreatment of the cells with Cb1 and Cb2 antagonists as well as specific inhibitors of the p38 and p42/44 MAPkinases (Ramer and Hinz, 2008). This effect of cannabinoids on TIMP-1 expression was mimicked by treatment of glioma cells with Δ9-THC (Blazquez et al., 2008a).
Conversely, in glioma cells, cannabinoid treatment selectively decreased the expression of MMP-2 via a Cb2-dependent mechanism and requiring the synthesis of ceramide (Blazquez et al., 2008b). By manipulating the levels of MMP-2 expression by siRNA and cDNA overexpression, the authors were able to show that the decrease in MMP-2 expression was critical for the cannabinoid-mediated inhibition of cell invasion (Blazquez et al., 2008b).
The last aspect of the metastatic process that is regulated by cannabinoids is angiogenesis. Met-AEA inhibited the basic fibroblast growth factor-stimulated endothelial cell proliferation and induced apoptosis in a Cb1-dependent manner (Pisanti et al., 2007). Furthermore, Met-AEA was able to inhibit bidimensional capillary-like tube formation and tumor-induced angiogenesis in a three-dimensional model of endothelial and thyroid tumor cell spheroid cocultures (Pisanti et al., 2007). In support of this, we have shown that AEA treatment of an in vivo xenograft model of cholangiocarcinoma also markedly inhibits the expression of members of the vascular endothelial growth factor family (DeMorrow et al., 2008), which are key regulators of both normal and abnormal angiogenesis (Ferrara, 2005; Ferrara and Kerbel, 2005).
Together, these data suggest that cannabinoids, and in particular anandamide, may be key endogenous inhibitors of various stages in metastatic processes, including migration, invasion, and angiogenesis. This further supports the notion that drugs directed at regulating the endocannabinoid system may prove to be valuable tools in the fight against various cancers.
V. Targeting Degradation Enzymes of Cannabinoids as an Anticancer Therapy
As mentioned previously, the degradation of endocannabinoids is an active and rapid process. Therefore, blocking the degradation pathway may enhance the antiproliferative effects of AEA and have beneficial effects in cancer treatment. Indeed, treatment of human breast cancer cells in vitro with palmitoylethanolamide enhances the antiproliferative effects of AEA (Di Marzo et al., 2001). This agent was shown to reduce the expression of FAAH up to 30–40% thereby allowing the accumulation of AEA and increasing its antiproliferative effects (Di Marzo et al., 2001). In addition, treatment of athymic mice with thyroid tumor xenografts with VDM-11 (a selective inhibitor of endocannabinoid cellular reuptake) or arachidonyl-serotonin (a selective blocker of endocannabinoid hydrolysis) increased the intratumoral levels of anandamide and significantly decreased tumor volume (Bifulco et al., 2004). The antiproliferative actions of these agents could be only partly inhibited by the pretreatment of the Cb1 receptor antagonist, suggesting that endocannabinoids tonically control tumor growth in vivo by both Cb1-mediated and non-Cb1-mediated mechanisms (Bifulco et al., 2004). Regardless of the molecular mechanism by which anandamide and other endocannbinoids regulate tumor growth, inhibitors of their inactivation might be useful for the development of novel anticancer drugs (Bifulco et al., 2004).
VI. Tumor Promoting Effects of Anandamide
The evidence supporting growth-promoting effect of AEA in tumors is pallid in comparison to the antitumoral effects described above. There is a greater volume of research indicating that the structurally similar, plant-derived cannabinoid, Δ9-THC stimulates growth in a number of cancer cell lines via Cb1 and Cb2 receptor-independent mechanisms (McKallip et al., 2005; Takeda et al., 2008). However, several cannabinoids, including AEA, have been shown to accelerate proliferation via the transactivation of the EGFR in a TACE/ADAM17 metalloprotease-dependent manner (Hart et al., 2004). This effect was observed in several cell lines from various origins including lung cancer, squamous cell carcinoma, bladder carcinoma, glioblastoma, astrocytoma, and kidney cancer (Hart et al., 2004). The cannabinoid-induced activation of the EGFR leads to the subsequent phosphorylation and activation of the adaptor protein Src homology 2 domain-containing (shc), and downstream activation of the ERK1/2 and Akt/PKB pathways (Hart et al., 2004). Thus, the cross-communication of cannabinoid receptors and EGFR may provide an explanation as to how cannabinoids may stimulate cancer cell proliferation (Hart et al., 2004).
In addition, the stable analogue of AEA, methanandamide, has a mitogenic effect on an androgen-dependent prostate cancer cell line that could be blocked by antagonists for either the Cb1 or Cb2 receptors as well as by the PI-3kinase inhibitor (Sanchez et al., 2003). The downstream consequence of activation of the endocannabinoid system was an increase in the expression of the androgen receptor, which is directly linked to the growth of these cells (Sanchez et al., 2003).
VII. Conclusions
In conclusion, the endocannabinoid system exerts a myriad of effects on tumor cell growth, progression, angiogenesis, and migration. With a notable few exceptions, targeting the endocannabinoid system with agents that activate cannabinoid receptors or increase the endogenous levels of AEA may prove to have therapeutic benefit in the treatment of various cancers. Further studies into the downstream consequences of AEA treatment are required and may illuminate other potential therapeutic targets.
Acknowledgments
This work was supported by an NIH K01 grant award (DK078532) to Dr DeMorrow and by the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White, the VA Research Scholar Award, a VA Merit Award and the NIH grant DK58411 and DK062975 to Dr. Alpini.
We acknowledge Glen Cryer of the Scott & White Hospital, Grants Administration Office for his assistance with proof reading and Bryan Moss of the Graphics Services, Biomedia Communications, Scott & White Hospital for his assistance with the figures.
References
- Alberich Jorda M, Rayman N, Tas M, Verbakel SE, Battista N, van Lom K, Lowenberg B, Maccarrone M, Delwel R. The peripheral cannabinoid receptor Cb2, frequently expressed on AML blasts, either induces a neutrophilic differentiation block or confers abnormal migration properties in a ligand-dependent manner. Blood. 2004;104:526–534. doi: 10.1182/blood-2003-12-4357. [DOI] [PubMed] [Google Scholar]
- Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, Layward L. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature. 2000;404:84–87. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- Bifulco M, Laezza C, Valenti M, Ligresti A, Portella G, Di Marzo V. A new strategy to block tumor growth by inhibiting endocannabinoid inactivation. FASEB J. 2004;18:1606–1608. doi: 10.1096/fj.04-1754fje. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Ligresti A, Di Marzo V. The endocannabinoid signalling system: Biochemical aspects. Pharmacol Biochem Behav. 2005;81:224–238. doi: 10.1016/j.pbb.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Blazquez C, Carracedo A, Salazar M, Lorente M, Egia A, Gonzalez-Feria L, Haro A, Velasco G, Guzman M. Down-regulation of tissue inhibitor of metalloproteinases-1 in gliomas: A new marker of cannabinoid antitumoral activity? Neuropharmacology. 2008a;54:235–243. doi: 10.1016/j.neuropharm.2007.06.021. [DOI] [PubMed] [Google Scholar]
- Blazquez C, Salazar M, Carracedo A, Lorente M, Egia A, Gonzalez-Feria L, Haro A, Velasco G, Guzman M. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008b;68:1945–1952. doi: 10.1158/0008-5472.CAN-07-5176. [DOI] [PubMed] [Google Scholar]
- Caffarel MM, Sarrio D, Palacios J, Guzman M, Sanchez C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006;66:6615–6621. doi: 10.1158/0008-5472.CAN-05-4566. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Chan VY, Chan MW, Leung WK, Leung PS, Sung JJ, Chan FK. Intestinal trefoil factor promotes invasion in non-tumorigenic Rat-2 fibroblast cell. Regul Pept. 2005;127:87–94. doi: 10.1016/j.regpep.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Contassot E, Tenan M, Schnuriger V, Pelte MF, Dietrich PY. Arachidonyl ethanolamide induces apoptosis of uterine cervix cancer cells via aberrantly expressed vanilloid receptor-1. Gynecol Oncol. 2004a;93:182–188. doi: 10.1016/j.ygyno.2003.12.040. [DOI] [PubMed] [Google Scholar]
- Contassot E, Wilmotte R, Tenan M, Belkouch MC, Schnuriger V, de Tribolet N, Burkhardt K, Dietrich PY. Arachidonylethanolamide induces apoptosis of human glioma cells through vanilloid receptor-1. J Neuropathol Exp Neurol. 2004b;63:956–963. doi: 10.1093/jnen/63.9.956. [DOI] [PubMed] [Google Scholar]
- Curran S, Murray GI. Matrix metalloproteinases: Molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer. 2000;36:1621–1630. doi: 10.1016/s0959-8049(00)00156-8. [DOI] [PubMed] [Google Scholar]
- DeMorrow S, Glaser S, Francis H, Venter J, Vaculin B, Vaculin S, Alpini G. Opposing actions of endocannabinoids on cholangiocarcinoma growth: Recruitment of Fas and Fas ligand to lipid rafts. J Biol Chem. 2007;282:13098–13113. doi: 10.1074/jbc.M608238200. [DOI] [PubMed] [Google Scholar]
- DeMorrow S, Francis H, Gaudio E, Venter J, Franchitto A, Kopriva S, Onori P, Mancinelli R, Frampton G, Coufal M, Mitchell B, Vaculin B, Alpini G. The endocannabinoid anandamide inhibits cholangiocarcinoma growth via activation of the noncanonical Wnt signaling pathway. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1150–G1158. doi: 10.1152/ajpgi.90455.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Melck D, Palmisano A, Bisogno T, Laezza C, Bifulco M, Di Marzo V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci USA. 1998;95:8375–8380. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA, III, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- Di Marzo V, Deutsch DG. Biochemistry of the endogenous ligands of cannabinoid receptors. Neurobiol Dis. 1998;5:386–404. doi: 10.1006/nbdi.1998.0214. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Orlando P, Bisogno T, Zagoory O, Bifulco M, Vogel Z, De Petrocellis L. Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti-proliferative effect of anandamide in human breast cancer cells. Biochem J. 2001;358:249–255. doi: 10.1042/0264-6021:3580249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek S, Hogerkorp CM, Dictor M, Ehinger M, Borrebaeck CA. Mantle cell lymphomas express a distinct genetic signature affecting lymphocyte trafficking and growth regulation as compared with subpopulations of normal human B cells. Cancer Res. 2002;62:4398–4405. [PubMed] [Google Scholar]
- Ferrara N. VEGF as a therapeutic target in cancer. Oncology. 2005;69(Suppl 3):11–16. doi: 10.1159/000088479. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Critical determinants of metastasis. Semin Cancer Biol. 2002;12:89–96. doi: 10.1006/scbi.2001.0416. [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I, Sanchez C, Cortes ML, del Pulgar TG, Izquierdo M, Guzman M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6:313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- Glaser ST, Kaczocha M, Deutsch DG. Anandamide transport: A critical review. Life Sci. 2005;77:1584–1604. doi: 10.1016/j.lfs.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Grimaldi C, Pisanti S, Laezza C, Malfitano AM, Santoro A, Vitale M, Caruso MG, Notarnicola M, Iacuzzo I, Portella G, Di Marzo V, Bifulco M. Anandamide inhibits adhesion and migration of breast cancer cells. Exp Cell Res. 2006;312:363–373. doi: 10.1016/j.yexcr.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Hart S, Fischer OM, Ullrich A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 2004;64:1943–1950. doi: 10.1158/0008-5472.can-03-3720. [DOI] [PubMed] [Google Scholar]
- Hinz B, Ramer R, Eichele K, Weinzierl U, Brune K. R(+)-methanandamide-induced cyclooxygenase-2 expression in H4 human neuroglioma cells: Possible involvement of membrane lipid rafts. Biochem Biophys Res Commun. 2004a;324:621–626. doi: 10.1016/j.bbrc.2004.09.095. [DOI] [PubMed] [Google Scholar]
- Hinz B, Ramer R, Eichele K, Weinzierl U, Brune K. Up-regulation of cyclooxygenase-2 expression is involved in R(+)-methanandamide-induced apoptotic death of human neuroglioma cells. Mol Pharmacol. 2004b;66:1643–1651. doi: 10.1124/mol.104.002618. [DOI] [PubMed] [Google Scholar]
- Hsu SS, Huang CJ, Cheng HH, Chou CT, Lee HY, Wang JL, Chen IS, Liu SI, Lu YC, Chang HT, Huang JK, Chen JS, et al. Anandamide-induced Ca2+ elevation leading to p38 MAPK phosphorylation and subsequent cell death via apoptosis in human osteosarcoma cells. Toxicology. 2007;231:21–29. doi: 10.1016/j.tox.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Islam TC, Asplund AC, Lindvall JM, Nygren L, Liden J, Kimby E, Christensson B, Smith CI, Sander B. High level of cannabinoid receptor 1, absence of regulator of G protein signalling 13 and differential expression of Cyclin D1 in mantle cell lymphoma. Leukemia. 2003;17:1880–1890. doi: 10.1038/sj.leu.2403057. [DOI] [PubMed] [Google Scholar]
- Jacobsson SO, Wallin T, Fowler CJ. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J Pharmacol Exp Ther. 2001;299:951–959. [PubMed] [Google Scholar]
- Joseph J, Niggemann B, Zaenker KS, Entschladen F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol Immunother. 2004;53:723–728. doi: 10.1007/s00262-004-0509-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokha R, Waterhouse P, Yagel S, Lala PK, Overall CM, Norton G, Denhardt DT. Antisense RNA-induced reduction in murine TIMP levels confers oncogenicity on Swiss 3T3 cells. Science. 1989;243:947–950. doi: 10.1126/science.2465572. [DOI] [PubMed] [Google Scholar]
- Laezza C, Pisanti S, Crescenzi E, Bifulco M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006;580:6076–6082. doi: 10.1016/j.febslet.2006.09.074. [DOI] [PubMed] [Google Scholar]
- Lambert S, Vind-Kezunovic D, Karvinen S, Gniadecki R. Ligand-independent activation of the EGFR by lipid raft disruption. J Invest Dermatol. 2006;126:954–962. doi: 10.1038/sj.jid.5700168. [DOI] [PubMed] [Google Scholar]
- Ligresti A, Bisogno T, Matias I, De Petrocellis L, Cascio MG, Cosenza V, D'Argenio G, Scaglione G, Bifulco M, Sorrentini I, Di Marzo V. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology. 2003;125:677–687. doi: 10.1016/s0016-5085(03)00881-3. [DOI] [PubMed] [Google Scholar]
- Manzanares J, Corchero J, Romero J, Fernandez-Ruiz JJ, Ramos JA, Fuentes JA. Pharmacological and biochemical interactions between opioids and cannabinoids. Trends Pharmacol Sci. 1999;20:287–294. doi: 10.1016/s0165-6147(99)01339-5. [DOI] [PubMed] [Google Scholar]
- Mathay C, Giltaire S, Minner F, Bera E, Herin M, Poumay Y. Heparin-binding EGF-like growth factor is induced by disruption of lipid rafts and oxidative stress in keratinocytes and participates in the epidermal response to cutaneous wounds. J Invest Dermatol. 2008;128:717–727. doi: 10.1038/sj.jid.5701069. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- McKallip RJ, Lombard C, Fisher M, Martin BR, Ryu S, Grant S, Nagarkatti PS, Nagarkatti M. Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. Blood. 2002;100:627–634. doi: 10.1182/blood-2002-01-0098. [DOI] [PubMed] [Google Scholar]
- McKallip RJ, Nagarkatti M, Nagarkatti PS. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J Immunol. 2005;174:3281–3289. doi: 10.4049/jimmunol.174.6.3281. [DOI] [PubMed] [Google Scholar]
- Melck D, Rueda D, Galve-Roperh I, De Petrocellis L, Guzman M, Di Marzo V. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999;463:235–240. doi: 10.1016/s0014-5793(99)01639-7. [DOI] [PubMed] [Google Scholar]
- Melck D, De Petrocellis L, Orlando P, Bisogno T, Laezza C, Bifulco M, Di Marzo V. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology. 2000;141:118–126. doi: 10.1210/endo.141.1.7239. [DOI] [PubMed] [Google Scholar]
- Mimeault M, Pommery N, Wattez N, Bailly C, Henichart JP. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: Implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate. 2003;56:1–12. doi: 10.1002/pros.10190. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Pagotto U, Marsicano G, Fezza F, Theodoropoulou M, Grubler Y, Stalla J, Arzberger T, Milone A, Losa M, Di Marzo V, Lutz B, Stalla GK. Normal human pituitary gland and pituitary adenomas express cannabinoid receptor type 1 and synthesize endogenous cannabinoids: First evidence for a direct role of cannabinoids on hormone modulation at the human pituitary level. J Clin Endocrinol Metab. 2001;86:2687–2696. doi: 10.1210/jcem.86.6.7565. [DOI] [PubMed] [Google Scholar]
- Patsos HA, Hicks DJ, Dobson RR, Greenhough A, Woodman N, Lane JD, Williams AC, Paraskeva C. The endogenous cannabinoid, anandamide, induces cell death in colorectal carcinoma cells: A possible role for cyclooxygenase 2. Gut. 2005;54:1741–1750. doi: 10.1136/gut.2005.073403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen G, Moesgaard B, Schmid PC, Schmid HH, Broholm H, Kosteljanetz M, Hansen HS. Endocannabinoid metabolism in human glioblastomas and meningiomas compared to human non-tumour brain tissue. J Neurochem. 2005;93:299–309. doi: 10.1111/j.1471-4159.2005.03013.x. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Giuffrida A, Calignano A, Rodriguez de Fonseca F. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- Pisanti S, Borselli C, Oliviero O, Laezza C, Gazzerro P, Bifulco M. Antiangiogenic activity of the endocannabinoid anandamide: Correlation to its tumor-suppressor efficacy. J Cell Physiol. 2007;211:495–503. doi: 10.1002/jcp.20954. [DOI] [PubMed] [Google Scholar]
- Ramer R, Hinz B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J Natl Cancer Inst. 2008;100:59–69. doi: 10.1093/jnci/djm268. [DOI] [PubMed] [Google Scholar]
- Ramer R, Brune K, Pahl A, Hinz B. R(+)-methanandamide induces cyclooxygenase-2 expression in human neuroglioma cells via a non-cannabinoid receptor-mediated mechanism. Biochem Biophys Res Commun. 2001;286:1144–1152. doi: 10.1006/bbrc.2001.5518. [DOI] [PubMed] [Google Scholar]
- Ruiz L, Miguel A, Diaz-Laviada I. Delta9-tetrahydrocannabinol induces apoptosis in human prostate PC-3 cells via a receptor-independent mechanism. FEBS Lett. 1999;458:400–404. doi: 10.1016/s0014-5793(99)01073-x. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Galve-Roperh I, Canova C, Brachet P, Guzman M. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998;436:6–10. doi: 10.1016/s0014-5793(98)01085-0. [DOI] [PubMed] [Google Scholar]
- Sanchez C, de Ceballos ML, del Pulgar TG, Rueda D, Corbacho C, Velasco G, Galve-Roperh I, Huffman JW, Ramon y Cajal S, Guzman M. Inhibition of glioma growth in vivo by selective activation of the CB(2) cannabinoid receptor. Cancer Res. 2001;61:5784–5789. [PubMed] [Google Scholar]
- Sanchez MG, Sanchez AM, Ruiz-Llorente L, Diaz-Laviada I. Enhancement of androgen receptor expression induced by (R)-methanandamide in prostate LNCaP cells. FEBS Lett. 2003;555:561–566. doi: 10.1016/s0014-5793(03)01349-8. [DOI] [PubMed] [Google Scholar]
- Sarfaraz S, Afaq F, Adhami VM, Mukhtar H. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005;65:1635–1641. doi: 10.1158/0008-5472.CAN-04-3410. [DOI] [PubMed] [Google Scholar]
- Schmid PC, Wold LE, Krebsbach RJ, Berdyshev EV, Schmid HH. Anandamide and other N-acylethanolamines in human tumors. Lipids. 2002;37:907–912. doi: 10.1007/s11745-002-0978-z. [DOI] [PubMed] [Google Scholar]
- Shi Y, Zou M, Baitei EY, Alzahrani AS, Parhar RS, Al-Makhalafi Z, Al-Mohanna FA. Cannabinoid 2 receptor induction by IL-12 and its potential as a therapeutic target for the treatment of anaplastic thyroid carcinoma. Cancer Gene Ther. 2008;15:101–107. doi: 10.1038/sj.cgt.7701101. [DOI] [PubMed] [Google Scholar]
- Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol. 2000;10:415–433. doi: 10.1006/scbi.2000.0379. [DOI] [PubMed] [Google Scholar]
- Takeda S, Yamaori S, Motoya E, Matsunaga T, Kimura T, Yamamoto I, Watanabe K. Delta(9)-Tetrahydrocannabinol enhances MCF-7 cell proliferation via cannabinoid receptor-independent signaling. Toxicology. 2008;245:141–146. doi: 10.1016/j.tox.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Voth EA, Schwartz RH. Medicinal applications of delta-9-tetrahydrocannabinol and marijuana. Ann Intern Med. 1997;126:791–798. doi: 10.7326/0003-4819-126-10-199705150-00008. [DOI] [PubMed] [Google Scholar]
- Zacchigna S, Zentilin L, Morini M, Dell'Eva R, Noonan DM, Albini A, Giacca M. AAV-mediated gene transfer of tissue inhibitor of metalloproteinases-1 inhibits vascular tumor growth and angiogenesis in vivo. Cancer Gene Ther. 2004;11:73–80. doi: 10.1038/sj.cgt.7700657. [DOI] [PubMed] [Google Scholar]