

Abstract
Mutations affecting the Na+, K+ ATPase alpha subunit have been implicated in at least two distinct human diseases, rapid-onset dystonia Parkinsonism (RDP), and familial hemiplegic migraine (FHM). Over 40 mutations have been mapped to the human ATP1A2 and ATP1A3 genes and are known to result in RDP, FHM or a variant of FHM with neurological complications. To develop a genetically tractable model system for investigating the role of the Na+, K+ ATPase in neural pathologies we performed genetic screens in Drosophila melanogaster to isolate loss-of-function alleles affecting the Na+, K+ ATPase alpha subunit. Flies heterozygous for these mutations all exhibit reduced respiration, consistent with a loss-of-function in the major ATPase. However, these mutations do not affect all functions of the Na+, K+ ATPase alpha protein since embryos homozygous for these mutations have normal septate junction paracellular barrier function and tracheal morphology. Importantly, all of these mutations cause neurological phenotypes and, akin to the mutations that cause RDP and FHM, these new alleles are missense mutations. All of these alleles exhibit progressive stress-induced locomotor impairment suggesting neuromuscular dysfunction, yet neurodegeneration is observed in an allele-specific manner. Surprisingly, studies of longevity demonstrate that mild hypomorphic mutations in the sodium pump significantly improve longevity, which was verified using the Na+, K+ ATPase antagonist ouabain. The isolation and characterization of a series of new missense alleles of ATPalpha in Drosophila provides the foundation for further studies of these neurological diseases and the role of sodium pump impairment in animal longevity.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-009-0673-2) contains supplementary material, which is available to authorized users.
Introduction
Neurological disorders have a devastating impact on sufferers and their families. For many such diseases, specific disease-causing mutations have been identified; yet the underlying cellular deficits and specific molecular mechanisms remain poorly understood. The use of a tractable genetic organism to model these diseases has become an invaluable method of determining the mechanisms underlying neuropathogenesis of these complex disease states.
ATPalpha (FBgn0002921) encodes the catalytic subunit of the Na+, K+ ATPase (a.k.a. the sodium pump). Mature Na+, K+ ATPases are high molecular weight, integral membrane proteins composed of tetramers of the alpha and beta subunits and may contain auxiliary subunits. These Na+, K+ ATPases are vital for generating and maintaining the electrochemical gradients that drive numerous downstream cellular processes. These proteins are ubiquitously expressed, are highly evolutionarily conserved, and are the predominate users of cellular ATP (Blanco and Mercer 1998; Lingrel et al. 1997; Lopina 2000; Mobasheri et al. 2000; Palmgren and Axelsen 1998). Expression and activity of the Na+, K+ ATPase is exceptionally high within the neuromuscular system, and its activity within the brain accounts for the overwhelming majority of ATP consumption in animals (Attwell and Laughlin 2001; Beal et al. 1993; Erecinska and Dagani 1990; Lees 1993). In addition to maintaining ionic gradients, these proteins are present in large complexes that function in cell adhesion, polarity, signaling and endocytosis (Cai et al. 2008; Cereijido et al. 2004, 2008; Genova and Fehon 2003; Hilgenberg et al. 2006; Paul et al. 2003; Rajasekaran et al. 2005). In many cases, these roles do not require ion transport. For example, in Drosophila, the Na+, K+ ATPase is essential for formation of septate junctions (SJs), which form paracellular diffusion barriers analogous to vertebrate tight junction (Genova and Fehon 2003; Paul et al. 2003). Importantly, the Na+, K+ ATPase appears to have a structural role in SJs since catalytic activity is not required either for SJ barrier function or for the separable function of facilitating lumenal secretion of the protein Verm, which is required for limiting the length of the tubes comprising the tracheal (airway) system (Paul et al. 2007).
The Na+, K+ ATPase has been implicated in two distinct neurological disorders: RDP and FHM (Cannon 2004; de Carvalho Aguiar et al. 2004; Haan et al. 2005; Pietrobon 2007). RDP is a distinct form of dystonia in which patients experience sudden—often stress induced—onset of Parkinsonian symptoms that are unresponsive to standard dopaminergic treatments and not associated with typical Parkinson’s brain pathology. FHM type II is a form of migraine associated with partial paralysis (hemiplegia) that is often accompanied by seizures or symptoms of cognitive dysfunction. These neurological disorders are dominant and result primarily from specific missense mutations. Little is known about pathogenesis of these complex neurological diseases and it is currently unclear whether these chronic diseases might be complicated by degenerative pathology.
We have previously reported that specific alleles of ATPalpha cause neurological dysfunction, impaired locomotion, reduced longevity, and progressive neurodegeneration (Palladino et al. 2002, 2003). To develop Drosophila models of FHM and RDP diseases and enable detailed studies of disease pathogenesis we performed a genetic screen to isolate a series of missense alleles of ATPalpha. We have identified and extensively characterized seven new alleles of ATPalpha. While animals heterozygous for these mutations share some phenotypes with previously described mutants, such as progressive locomotor deficits and mechanical stress-induced paralysis, they also have novel and allele-specific phenotypes. More importantly, we have identified one allele that bears a missense mutation altering the protein at a highly conserved residue in exactly the same manner as is known to result in human FHM.
In addition to further developing Drosophila as a model for investigating neuropathogenic mechanisms resulting from Na+, K+ ATPase alpha dysfunction, our results reveal an unexpected role for the Na+, K+ ATPase in regulating animal longevity. Previous genetic screens for mutants causing progressive neurodegeneration have resulted in alleles of ATPalpha that have significantly reduced longevity (Palladino et al. 2003). The alleles reported here were isolated based upon failure-to-complement null alleles for viability and were thus not biased toward causing neurological phenotypes. One resulting mutation results in a significant reduction in longevity and progressive neuropathology. However, three alleles affecting the Na+, K+ ATPase result in a striking increase in animal longevity. Remarkably, this finding was phenocopied with ouabain, a well-described antagonist of the Na+, K+ ATPase ion-transport activity. These findings demonstrate a role for Na+, K+ ATPase impairment in increased longevity and additional data suggest that this effect is independent of a caloric restriction mechanism.
Materials and methods
Fly stock maintenance and mutagenesis
Fly stocks were maintained on standard cornmeal-molasses agar medium at 22–25°C. Isogenized cn bw; ve e males were mutagenized with ethylmethane sulfonate using standard methods. Following mutagenesis the males were mated to TM6 Tb virgins. Tb F1 males were collected and individually mated to ATPalpha DTS1R1/TM6 Tb or ATPalpha DTS2R3/TM6 Tb virgin females. Putative new alleles were identified as those which failed to complement existing ATPalpha revertant lines for embryonic and early larval lethality, resulting in a lack of the expected 1/3 Tb + progeny. Vials were initially screened without magnification for the presence of non-Tb pupal cases. Strains were outcrossed several times to replace the other mutagenized chromosomes. ATPalpha CJ strains lacking the cn bw chromosome were identified and absence of bw was confirmed using genetics and PCR (data not shown). Hybrid animals bearing Canton S and ve e (mutant or parental control) were used to control for second site recessive mutations. There is a possibility of third chromosome dominant modifiers. Recombination was used to determine whether a lethal mutation was linked to e (which is tightly linked to ATPalpha) and complementation tests were performed between all new alleles and the DTS1R1, DTS2R3, DTS1 or DTS2 alleles using lethality and stress-sensitivity (a.k.a. bang sensitivity) phenotypes. The genetic data suggested that the largest complementation group were new alleles, which were named CJ alleles. Other minor complementation groups failed to complement unlinked sites of either the ATPalpha DTS1R1 or the ATPalpha DTS2R3 chromosome (but not both). One allele was determined to be a complex reciprocal translocation T3:Y and was discarded (data not shown).
CJ allele sequencing
Genomic DNA from each ATPalpha CJ strain was isolated from ten flies (QiaAMP DNA mini kit, Qiagen). ATPalpha amplicons 1.5–2.0 kb in size were amplified with PCR, verified on an agarose gel, purified using Qiaquick PCR, and directly sequenced from ATPalpha CJ/TM6, cn bw; ve e, and TM3/TM6 animals. Heterozygous sequence differences unique to the ATPalpha CJ/TM6 strain were identified within coding regions of the ATPalpha gene with the exception of ATPalpha CJ7. Polymorphisms known to exist between ve e and TM6 have been identified in each amplicon from the ATPalpha CJ7 strain, demonstrating the absence of a large deletion or inversion that would prevent amplification from the mutant chromosome.
Trachea assays
Tracheal morphology and dye exclusion assays were performed as previously described (Paul et al. 2007). The phenotypes of ATPalpha CJ# /ATPalpha null transheterzygotes and ATPalpha CJ# homozygotes were assessed in embryos created by mating ATPalpha CJ# /TM6B GMR-YFP males with ATPalpha null /TM6B GMR-YFP virgin females. TM6B GMR-YFP described in Le et al. (2007).
Lifespan analyses
Longevity testing
Lifespan data were collected and analyzed as previously reported (Celotto et al. 2006a, b). Briefly, 20–25 flies were kept per vial at 25°C, checked daily, and transferred to fresh media every other day to minimize incidental death. Deaths were recorded daily and lifespan plots were generated showing daily percent survivorship over time. Incidental deaths and escapees were noted and removed from the survivorship calculations. Longevity was examined from three independent populations of animals from each genotype, until its median age was reached and animals were killed for histological examination. To control for hybrid vigor, lifespans for mutant and control strains were examined in females heterozygous for w ve and e generated by a single mating between isogenic Canton S or isogenic ve e and the mutant strain, as appropriate. Briefly, Tb + F1 progeny from w;;ATPalpha CJ /TM6 males mated to isogenic Canton S virgin females or w; ve e males mated with ATPalpha DTS1R1 /TM6, ATPalpha DTS1 /TM6, ATPalpha DTS2 /TM6 virgins were examined to maintain heterozygosity of the recessive w, ve, and e mutations.
Ouabain effects on longevity
Lifespans of ouabain-treated flies were performed on three independent populations of an inbred isogenic Canton S strain. Water was used as a vehicle and drug was added at the noted concentrations to a semi-circle piece of filter paper placed on top of the standard fly media (~9 ml) covering ~50% of the surface area. Flies were transferred to fresh media every other day and fresh filter paper with drug was applied.
Behavioral analyses
For behavioral testing, female Tb + F1 progeny from w;;ATPalpha CJ /TM6 males mated to CS virgin females or w;;ve e males mated with ATPalpha DTS1R1/TM6, ATPalpha DTS1/TM6, ATPalpha DTS2/TM6 virgins were examined to maintain heterozygosity of the recessive w, ve, and e mutations.
Conditional locomotor assays
Assays of stress and temperature sensitivity were conducted as described previously (Palladino et al. 2003). For stress-sensitivity testing, time to recovery following 15 s of vortexing was recorded on days 3, 10, 20, and 30 after eclosion. For temperature sensitivity testing, flies were exposed to 38°C for 7 min in groups of 3–5, and time to first signs of sensitivity (bottom-dwelling behavior, brief paralysis) to total paralysis, as well as time to recovery following return to 22°C (first deliberate movement; walking, grooming, flying) were recorded for each fly. Temperature sensitivity was also tested on days 3, 10, 20, and 30.
Locomotor assays
For locomotor activity recording, 9–11-day-old female flies were entrained to a 12:12 L:D schedule for 3–4 days at 25°C. Flies were then inserted into individual locomotor activity tubes supplemented with 5% sucrose, 1% agar media and capped with yarn. Individual fly activity was then recorded utilizing the Drosophila Activity Monitoring (DAM) system monitors and software (Trikinetics, Inc.). Briefly, fly locomotion breaks an infrared beam, and the number of beam breaks in 1-min bins was recorded and analyzed using Insomniac 2.0 software. Sleep, defined as bouts of at least 5 consecutive minutes of zero beam breaks, were identified and from these, total time awake and asleep were calculated. Waking activity levels, a useful measure of hypo/hyperactivity (Wu et al. 2008), were calculated from the total activity levels (total beam breaks/total time awake).
Stimulated locomotor assays
Startle stimulation activity was measured from Zeitgeber time (ZT) 12–14, a time when flies are normally inactive. Monitors were wrapped in aluminum foil just prior to lights off to block light entry, and removed from the incubator. Flies were startled by randomly jarring or moving the monitors, individually or as a group, at least every 3 min. Flies were allowed to recover for the remainder of the night and the foil was removed the following morning.
Behavioral rhythm assays
Flies were monitored using the DAM system as above. Once entrained to a 12:12 (L:D) pattern for 3 days, animals were shifted to a DD pattern and activity was observed for 3 days. Population activity patterns were visualized by binning activity data into 1 h intervals and averaging by genotype. Circadian activity and anticipation behavior were noted.
Respiration assays
Individual metabolic measurements were performed as previously described (Celotto et al. 2006a; Libert et al. 2007; Van Voorhies et al. 2003, 2004). Respiration was measured on male flies that were 5–6 days post-emergent. Briefly, respiration rates were determined by measuring CO2 production from individual flies maintained in a 2.2 ml glass sealed chamber flushed with CO2-free, water-saturated (100% RH) air. Gas samples were directly injected into a 150 ml/min (±1%) STPD, dry, CO2-free air stream controlled with a mass flow meter (Sierra Instruments, Monterrey, CA). A Li-Cor 6251 carbon dioxide gas analyzer (Li-Cor, Lincoln, NE) was used to analyze the samples (sensitivity of <0.1 ppm and an accuracy of <1 ppm). Respiration rates were determined using DATACAN software (Sable Systems International, Henderson, NV). Individual fly mass was determined using a Sartorius M2P microbalance. The CO2 gas analysis system was zeroed daily against CO2-free air, and calibrated against a 51 ppm certified gas standard (Air Products, Long Beach, CA).
Western blots
Eight fly heads were ground by pestle in 50 µl 2× SDS PAGE sample buffer (4% SDS, 4% beta-mercaptoethanol, 130 mM Tris–HCl pH 6.8, 20% glycerol). The proteins were resolved by SDS PAGE and transferred onto nitrocellulose. Following treatment in 1% milk in PBST the blots were treated with anti-TPI (1:5000; rabbit polyclonal FL-249; Santa Cruz Biotechnology) or anti-Na/K ATPase (1:5000; alpha5; Developmental Studies Hybridoma Bank (DSHB)). The blots were washed in PBST, incubated in the appropriate HRP-conjugated secondary antibody, and developed with an ECL kit (Pierce) as previously described (Seigle et al. 2008). Quantification of the scanned films was performed digitally using ImageJ software available from the National Institutes of Health.
Histology
Aged flies were collected at their median age (age of ~50% survivorship) from lifespan experiments. Heads and thoraxes were dissected and fixed for 24 h at room temperature in freshly prepared Carnoy’s fixative, washed in 70% ethanol, processed and embedded in paraffin blocks according to standard histological procedures as previously published (Fergestad et al. 2008; Palladino et al. 2000). Serial 4–5 micron frontal head sections and thoracic cross sections were obtained and stained with hematoxylin and eosin. Incidence and extent of pathology present in neural tissues or flight muscle, were noted (n > 15 animals per genotype). Neuropathology was scored on a scale from 0 to 5 using published methods and criteria (Fergestad et al. 2006).
Feeding assays
Outcrossed male flies were collected from underpopulated vials and feeding assays were performed using 3–5-day-old animals using dyed food consisting of 15% sucrose, 1% agar, and 3% FD&C blue #1 (McCormick). The dye accumulates in the gut and can be used as a quantitative measure of meal size (Edgecomb et al. 1994; Xu et al. 2008). Flies were segregated from nutrients for 1 h and then permitted 2 h to feed. Flies were immediately frozen on dry ice, separated into groups of 30, and decapitated to prevent eye pigment interference. Headless bodies were homogenized in 250 µl of PBS. Samples were centrifuged twice at ~15,000g for 15 min to remove debris. Absorbance of the supernatant (150 µl) was measured at 625 nm using a plate reader. Absorbance from control age-matched flies fed an undyed sucrose–agar mixture was subtracted, and the net absorbance reflected total food ingested. Significance was set at P < 0.01 using a Student’s t test. The positive controls were wild-type animals that were not nutrient deprived prior to the assays and were allowed one-third of the total feeding time, in order to demonstrate the sensitivity of the assay to detect reductions in feeding.
Results
Isolation of novel, dominant alleles of ATPalpha
Mutations affecting the ATPalpha gene in Drosophila have previously been isolated by several labs and cause phenotypes that include stress-sensitivity, temperature-sensitive paralysis, reduced longevity and altered tracheal development (Genova and Fehon 2003; Lebovitz et al. 1989; Palladino et al. 2002, 2003; Paul et al. 2003; Schubiger et al. 1994; Sun et al. 2001; Trotta et al. 2004). Although several ATPalpha mutants have been reported, the vast majority of these are of limited utility for disease modeling and structure–function studies. Recent publications have identified more than 40 missense mutations affecting sodium pump genes are the cause of RDP and FHM diseases. In an effort to identify Drosophila missense mutations affecting ATPalpha that would model these neurological diseases, a genetic screen was performed to isolate novel missense alleles of ATPalpha. Molecular null alleles of ATPalpha that cause homozygous embryonic lethality were previously identified as revertants of dominant temperature sensitive (DTS) mutations (e.g. ATPalpha DTS1R1 or ATPalpha DTS2R3) (Palladino et al. 2003; Paul et al. 2003). As these null alleles fail-to-complement all tested ATPalpha mutations for viability, including the partial loss-of-function mutations, we reasoned that new loss-of-function alleles could be identified as those that fail-to-complement either ATPalpha DTS1R1 or ATPalpha DTS2R3 for viability. Such a screen would avoid the bias toward neural or tracheal phenotypes that have been present in previous screens for ATPase mutations. In total ~12,000 EMS-generated mutant lines were generated and screened for those that failed to complement either ATPalpha DTS1R1 or ATPalpha DTS2R3 null mutations for viability. The screen resulted in eight new alleles named CJ for the student who performed the pilot screen. Recombination mapping of the lethality demonstrated a tight linkage to ebony(e) (<1.75 cM) for the ATPalpha CJ4, ATPalpha CJ6, ATPalpha CJ10, and ATPalpha CJ13 alleles (data not shown). ATPalpha CJ5, ATPalpha CJ7 and ATPalpha CJ12 had recessive lethal mutations that were not linked to e or ATPalpha. ATPalpha CJ3 was found to have a T3:Y reciprocal translocation bearing the ATPalpha region and was discarded from further study.
To further test the allelic nature of the novel ATPalpha strains, complementation tests for recessive lethality were performed for all pair-wise combinations (Table 1). All but ATPalpha CJ7 failed to complement both loss-of-function alleles ATPalpha DTS1R1 and ATPalpha DTS2R3 for lethality. ATPalpha CJ4, ATPalpha CJ5, ATPalpha CJ6, ATPalpha CJ10, and ATPalpha CJ13 failed to complement each other as well, resulting in lethality. ATPalpha CJ12 was lethal in combination with ATPalpha CJ10 and ATPalpha CJ13. ATPalpha CJ7 failed to complement ATPalpha CJ4, ATPalpha CJ6 and ATPalpha CJ13 and produced fewer than the Mendelian expected progeny with three other alleles, all in a temperature-dependent manner (Table 1). Animals that complemented for lethality were tested for stress-sensitive paralysis. The genetic analyses suggested that this complementation group represents novel ATPalpha alleles. The genetic analysis also demonstrates that four alleles are not lethal with the entire complementation group but exhibit markedly reduced viability or conditional lethality with certain other alleles suggesting that ATPalpha CJ4, ATPalpha CJ6, ATPalpha CJ7 and ATPalpha CJ12 are likely mild hypomorphic alleles. The data could indicate that there is some degree of intragenic complementation within ATPalpha CJ4/CJ12 and ATPalpha CJ6/CJ12 animals. Further data will be needed to resolve these possibilities.
Table 1.
Viability of existing ATPalpha alleles with new missense mutants
| DTS1R1 | DTS2R3 | CJ 4 | CJ 5 | CJ 6 | CJ 7 | CJ 10 | CJ 12 | CJ 13 | |
|---|---|---|---|---|---|---|---|---|---|
| + | BS | BS | BS | BS | BS | BS | BS, TS | BS | BS |
| DTS1R1 | L | L | L | L | L | BS | L | L | L |
| DTS2R3 | L | L | L | L | BS | L | L | L | |
| CJ 4 | L | L | L | BS**, L29 | L | BS**, L29 | L | ||
| CJ 5 | L | L | BS**29 | L | L | L | |||
| CJ 6 | L | BS, L29 | L | BS | L | ||||
| CJ 7 | L | BS**29 | BS*,**29 | BS, L29 | |||||
| CJ 10 | L | L | L | ||||||
| CJ 12 | L | L**29 | |||||||
| CJ 13 | L |
Viability to adulthood of each genotype combination at 25°C. Complementation was also examined at 29°C, differences from 25°C data are noted. L is developmental lethal. An asterisk indicates adult viable but a significant reduction from the Mendelian expected one-third of total adults (P < 0.05). Two asterisks indicate a more severe reduction and only rare escapers were observed. BS, bang or stress-sensitive paralysis
TS Temperature-sensitive paralysis (38°C)
CJ alleles have molecular lesions in ATPalpha
The above genetic data strongly suggest that several novel ATPalpha alleles have been isolated. To confirm this, we directly sequenced PCR amplicons covering the major coding regions of ATPalpha from each mutant and two control strains. We discovered mutations in six of the seven strains and, in all cases, these were missense mutations (Fig. 1; Supplemental Fig. 1). These mutations did not correspond with any polymorphisms found between the two control sequences (ve e and TM6) and each alters a highly conserved amino acid among species from hydra to human (Supplemental Fig. 2). We did not find a mutation within the coding sequences of the CJ7 allele. It is possible that this mutation affects non-coding sequences or a different gene that interacts with ATPalpha, possibly by reducing its expression (see Fig. 2b).
Fig. 1.
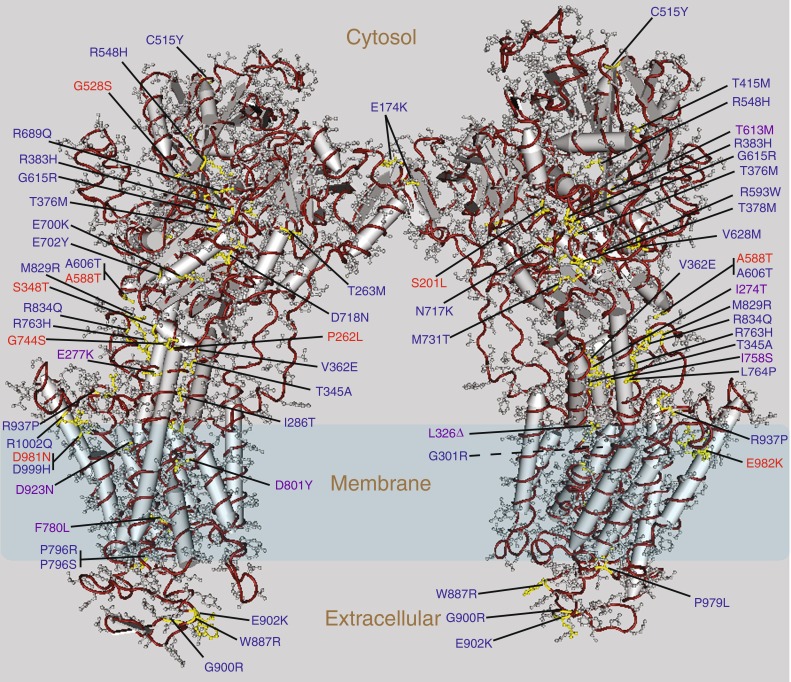
Pathogenic mutations affecting Na, K ATPase alpha subunits. A 3.5 Å crystal structure of the Na, K ATPase as a dimer is available (Morth et al. 2007). Numbered amino acids indicate those known to be affected by pathogenic mutations in the human ATP1A2 (FHM), human ATP1A3 (RDP), or Drosophila ATPalpha genes. These residues have been mapped onto a common structure for clarity. Citations for human ATP1A2 mutations: Ambrosini et al. (2005), Castro et al. (2008), De Fusco et al. (2003), Fernandez et al. (2008), Gallanti et al. (2008), Jen et al. (2007), Jurkat-Rott et al. (2004), Kaunisto et al. (2004), Koenderink et al. (2005), Pierelli et al. (2006), Riant et al. (2005), Segall et al. (2004, 2005), Spadaro et al. (2004), Swoboda et al. (2004), Todt et al. (2005), Vanmolkot et al. (2003, 2006a, b, 2007). Citations for human ATP1A3 mutations: Brashear et al. (2007), de Carvalho Aguiar et al. (2004), Kamm et al. (2008), Zanotti-Fregonara et al. (2008)
Fig. 2.

Adult ATPalpha protein expression in animals heterozygous for dominant alleles. a Previously described ATPalpha mutants demonstrate a significant decrease in expression in the revertant lines examined but not in the temperature-sensitive alleles, compared to wild-type controls. ATPalpha DTS2R2 and ATPalpha DTS2R3 express significantly more protein than the 50% expected for a heterozygous null (P < 0.05) suggesting the possibility of a compensatory mechanism. b Newly isolated missense mutants ATPalpha CJ6, ATPalpha CJ7, and ATPalpha CJ13 have significantly reduced ATPalpha protein relative to wild-type controls, whereas the remaining CJ alleles do not have significantly altered expression. In all cases: n = 3, ATPalpha protein was normalized to the internal control protein TPI. Error shown is SEM. Student t test reduction from control: * P < 0.05, ** P < 0.01, *** P < 0.001
The molecular and complementation data provide evidence that the CJ alleles affect ATPalpha and that at least six of the new alleles directly alter the encoded protein. Consistent with the EMS method of mutagenesis, these are all point mutations that result in a single amino acid substitution in the ATPalpha protein. Known Na+, K+ ATPases alpha disease-causing mutations are dominant and appear to confer a loss-of-function resulting either from haploinsufficiency or a mild hypomorphic impairment in pump function (de Carvalho Aguiar et al. 2004; Pietrobon 2007). To examine the range of impairment caused by these newly isolated alleles, we assessed the effects of these mutations in five functional tests and compared the new mutations with representative previously isolated alleles.
Adult ATPalpha protein expression in mutant alleles
We examined ATPalpha protein expression from ATPalpha DTS mutants, their revertants and control animals. All mutations are homozygous lethal and protein levels were examined as heterozygotes. Consistent with ATPalpha DTS mutants resulting in a dominant gain-of-function, these alleles have normal expression by western blot analysis (Fig. 2a). The revertant strains all have expression reduced by 30–50% compared to wild type. These results are in accordance with those found previously by immunohistochemistry for ATPalpha DTS1 and ATPalpha DTS1R1 (Fergestad et al. 2006). While one might expect a 50% reduction in protein levels in animals heterozygous for null ATPase mutations, only ATPalpha DTS1R1 shows this degree of reduction, suggesting the presence of a compensatory mechanism that either stabilizes the protein or increases its level of expression in the other revertants. ATPalpha DTS2R2 and ATPalpha DTS2R3 do exhibit significantly higher expression than the expected 50% of wild-type ATPalpha protein (P < 0.05), whereas, ATPalpha DTS1R1 and ATPalpha DTS1R2 do not deviate significantly from this expected value (P > 0.05). Interestingly, the ATPalpha DTS1R1 mutation is a four nucleotide deletion causing a frameshift that introduces a premature stop codon in an earlier exon, suggesting the possibility that non-sense mediated decay (NMD) might actively degrade ATPalpha DTS1R1 mRNAs. Consistent with NMD mRNA targeting we did not observe a truncated protein product by western blot.
Protein expression was also examined in the novel ATPalpha CJ alleles by western blot. Several alleles, namely ATPalpha CJ6, ATPalpha CJ7, and ATPalpha CJ13, exhibit a significant reduction in ATPalpha protein (Fig. 2b). In contrast, the ATPalpha CJ4, ATPalpha CJ5, ATPalpha CJ10, and ATPalpha CJ12 mutants do not have significantly altered ATPalpha protein levels.
Conditional temperature-dependent paralysis
ATPalpha DTS mutants were originally isolated based upon their temperature-dependent paralysis phenotype. Null mutations were isolated as revertants of this temperature-sensitive paralysis, suggesting that the DTS mutations cause a dominant gain-of-function. To examine whether any of the ATPalpha CJ alleles exhibit temperature-sensitivity akin to other gain-of-function alleles we examined their locomotor function when the ambient temperature was acutely elevated. Only ATPalpha CJ10 exhibited temperature-sensitive paralysis. This phenotype was not observed in young ATPalpha CJ10 flies and manifested in a progressive manner: first evident at approximately 20 days post-eclosion and becoming highly penetrant by day 30. The average time to paralysis of day 20 ATPalpha CJ10 flies when acutely shifted to the non-permissive temperature (38°C) was 334 ± 17.3 s. Wild-type flies and the remaining ATPalpha CJ mutants did not paralyze in the 7-min test period. The paralysis in ATPalpha CJ10 flies was reversible: recovery of normal locomotion in ATPalpha CJ10 took an average of 209 ± 35 s at the permissive temperature (room temperature ~22°C).
Circadian rhythms and locomotor function in ATPalpha alleles
We examined whether circadian rhythm or locomotor function in ATPalpha CJ mutants were aberrant using the DAM system. ATPalpha CJ mutants, revertant animals, and age-matched controls were examined at day 10–12 post-eclosion. All of the ATPalpha mutant lines examined, except ATPalpha CJ12, display normal waking activity levels: a measure of total activity/total time active (Fig. 3a). The ATPalpha CJ12 strain is more active than wild type in this measure. In contrast, waking locomotor activity of ATPalpha CJ mutants in response to startle stimulation was reduced from that of age-matched control animals (Fig. 3b). It was noted that all ATPalpha CJ mutants did exhibit an increased activity level upon startle stimulation but this increase was significantly less than that observed in wild-type controls. Additionally, total activity levels and total time active were significantly reduced in ATPalpha CJ mutants from that of controls, yet a normal circadian pattern to their behavior is evident (Supplemental Fig. 3). Thus, ATPalpha CJ mutants are less active but exhibit normal waking locomotor function at permissive temperatures. These mutants exhibit circadian behavior and are also capable of responding to a startle stimulus; however, their startle response locomotion is significantly reduced, suggesting a decrease in their maximal locomotion capacity.
Fig. 3.

Locomotor impairment in ATPalpha alleles. Waking activity levels (measured in total activity counts/total minutes active) were determined for daytime baseline locomotor activity (a) and a 2-h period of repeated startle stimulation during a period of inactivity (b) for heterozygous populations at 25°C (see “Materials and methods”). At the age of testing (12–15 days) no gross locomotor deficits were observed in any line, and baseline waking activity levels were not significantly different from ATPalpha DTS1R1 or wild type (ve e) except for ATPalpha CJ12, which appeared to be significantly hyperactive (** P < 0.005). Upon stimulation, however, all ATPalpha mutant strains display significantly lower activity levels than wild type controls (* P < 0.05)
Neuromuscular pathology in ATPalpha alleles
Familial hemiplegic migraine and RDP are two distinct neurological diseases that manifest largely from missense mutations in the genes encoding the alpha subunit of Na+, K+ ATPases. It remains controversial whether pathology is associated with chronic RDP or FHM diseases, especially FHM associated with epilepsy or convulsions. We examined the integrity of the neuromuscular system of aged ATPalpha mutants and control animals and discovered allele-specific pathology (Fig. 4). Some ATPalpha CJ alleles, namely ATPalpha CJ7, ATPalpha CJ10, ATPalpha CJ13, exhibit marked vacuolar pathology throughout the brains of these animals (Fig. 4; Table 2). ATPalpha CJ5, ATPalpha CJ6, and ATPalpha DTS1R1 have mild neuropathology, whereas, ATPalpha CJ4 and ATPalpha CJ12 have minimal neuropathology, similar to that commonly seen in aged wild-type control animals (Fig. 4; Table 2, and data not shown). Interestingly, ATPalpha CJ10 and ATPalpha CJ13 exhibit large clustering vacuolar pathology similar in appearance to that seen in ATPalpha DTS mutants (Palladino et al. 2003). In contrast, the neuropathology in ATPalpha CJ7 is a small non-clustering vacuolar pathology similar to that reported in the TPI sugarkill mutant strain (Celotto et al. 2006b). Histology was also performed on flight muscles from the ATPalpha CJ mutants and only the ATPalpha CJ12 exhibit significant myopathology (Fig. 4i).
Fig. 4.
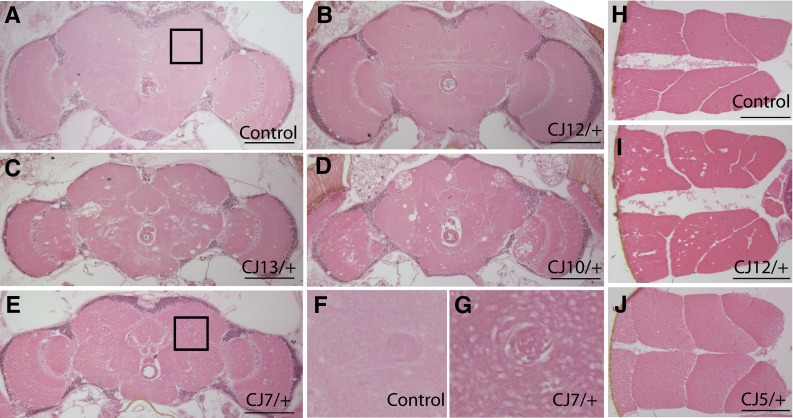
Histopathology from ATPalpha alleles. a–g Brain histology from aged ATPalpha CJ alleles and control tissues. b ATPalpha CJ12 animals exhibit modest neuropathology akin to that observed in aged wild-type control brains (a). ATPalpha CJ13 and ATPalpha CJ10 exhibit marked neuropathology as is evident by vacuolar and spongiform-like neuropath throughout the neuropil and optic lobes (c and d, respectively). e ATPalpha CJ7 exhibits a fine vacuolar pathology that is more clearly observed at higher magnification as seen in g (compare to panel f from wild type). h–j Muscle histology from aged animals. Wild-type and ATPalpha CJ5 mutants do not exhibit significant myopathology in aged specimens (h and j, respectively). I ATPalpha CJ12 do exhibit pathology. Bars are all 100 µm. Histopathology was obtained from animals at the median age for their genotype
Table 2.
Summary of ATPalpha allele phenotypes
| Genotype | TS para | BS para | Respir. | Protein | Neurodeg. | Myodeg. | Longevity |
|---|---|---|---|---|---|---|---|
| Wild type | No | No | → | → | 0–1 | No | → |
| DTS1R1 | No | Yes | ↓ | ↓ | 2 | No | ↑ |
| DTS1 | Yesψ | Mildψ | ↓ | → | 4ψ | – | ↓↓ψ |
| DTS2 | Yesψ | Mildψ | – | → | 4ψ | – | ↓ψ |
| CJ 4 | No | Yes | ↓↓ | → | 0–1 | No | → |
| CJ 5 | No | Yes | ↓↓ | → | 2 | No | → |
| CJ 6 | No | Yes | ↓↓ | ↓ | 1–2 | No | ↑↑ |
| CJ 7 | No | Yes | ↓ | ↓ | 2–3 | - | → |
| CJ 10 | Yes | Yes | ↓↓ | → | 2–3 | No | ↓ |
| CJ 12 | No | Yes | ↓ | → | 1 | Yes | ↑ |
| CJ 13 | No | Yes | ↓↓ | ↓ | 3 | No | → |
Neurodeg. Neurodegeneration and is scored on a scale of 0–5 where 5 is marked neuropathology, Myodeg. myodegeneration. Protein is expression levels. Respir. respiration
ψ indicates previously published data (Palladino et al. 2003), – indicates not tested
ATPalphaCJ alleles have decreased metabolic rates
The Na+, K+ ATPase is known to be broadly expressed, exhibit high expression in the neuromuscular system and be a major consumer of cellular energy. To test the hypothesis that these ATPalpha mutants result in a loss of normal ATPase function, we examined whole animal respiration using a sensitive, well-established single fly assay (Celotto et al. 2006a; Van Voorhies et al. 2003, 2004). In all mutants examined there is a significant reduction in the rate of animal respiration versus age-matched control animals (Fig. 5). Surprisingly, heterozygosity for the ATPalpha DTS1 mutation resulted in an ~10% decrease in respiration, the revertant strains produced an ~15% reduction, and the ATPalpha CJ mutants produced a 16–27% decrease in total animal respiration. These data are consistent with the ATPalpha CJ mutants representing a range of loss-of-function phenotypes from mild hypomorph to strong loss-of-function.
Fig. 5.

Animals heterozygous for ATPalpha alleles have reduced respiration. Respiration was measured from the emergent carbon dioxide from individual ATPalpha mutants and age-matched wild-type controls. ATPalpha mutants uniformly have significantly lower metabolic rates than controls. Error is SEM, n = 4–10, per genotype. Animals were 5–6 days old adults. Statistical significance was determined by Student’s t test (* P < 0.05, ** P < 0.01, and *** P < 0.001)
ATPalphaCJalleles maintain normal septate junction barrier and tracheal tube-size control function
In addition to its essential function as an ion pump, we have previously shown that the Drosophila Na+, K+ ATPase also has critical pump-independent structural or scaffolding functions in septate junctions, which, like the vertebrate tight junction, provide paracellular diffusion barriers (Genova and Fehon 2003; Paul et al. 2003, 2007). To determine whether the ATPalpha CJ mutations affect Na+, K+ ATPase septate junction function (ion-transport independent) we examined paracellular barrier function and tracheal tube morphogenesis in the new mutants using established methods (Paul et al. 2007). ATPalpha CJ alleles and two known null alleles, ATPalpha DTS1R1 or ATPalpha DTS1R2, were tested as homozygotes derived from heterozygous parents. In addition, two ATPalpha CJ were tested in trans with the null alleles (see “Materials and methods”). We found that paracellular barriers formed in all ATPalpha CJ mutants (Table S1). Further, all ATPalpha CJ alleles examined also supported normal tracheal formation, indicating that apical secretion of the Verm protein was normal (Fig. 6; Table S1). In contrast, ATPalpha DTS1R2 homozygous animals cannot form paracellular barriers and show elongated dorsal trunks (Fig. 6e) and gaps in the ganglionic branches (Fig. 6f) that are not present in ATPalpha CJ mutants (Fig. 6c, d; Table S1) or the heterozygous control animals (Fig. 6a, b). These data demonstrate that none of the tested ATPalpha CJ alleles are null alleles because they retain significant and perhaps normal tracheal size control and barrier function activity. Rather, these alleles appear to specifically affect ion-transport functions of the Na+, K+ ATPase.
Fig. 6.

ATPalpha CJ alleles have normal septate junction function and tracheal morphogenesis. a–b Heterozygous null alleles have normal dorsal trunks and ganglionic branches. c–d ATPalpha CJ alleles in trans with null alleles are also normal. e–f Homozygous null alleles demonstrate a lengthened dorsal trunk and incomplete ganglionic branches. Scale bars are 15 µm
ATPalpha mutants display progressive, stress-sensitive paralysis
Numerous mutations affecting ATPalpha have been shown to result in varying degrees of conditional paralysis resulting from mechanical stress (Lebovitz et al. 1989; Palladino et al. 2003; Schubiger et al. 1994; Sun et al. 2001; Trotta et al. 2004). However, observations of young flies in our ATPalpha CJ mutants revealed no overt locomotor defects—qualitatively all flies displayed robust geotaxic responses and appeared to walk, climb, and fly normally. We used a quantitative measure of stress-sensitive paralysis to examine the locomotor function of ATPalpha mutants as they age to determine whether this phenotype was progressive. Young ATPalpha CJ flies (3 days post-eclosion) were tested for mechanical stress-induced paralysis and none of the lines revealed a striking defect. However, ATPalpha CJ5 and ATPalpha CJ10 did recover more slowly from the stress than age-matched control animals (Fig. 7a). As the ATPalpha CJ mutant strains aged the progressive nature of this locomotor defect became evident with all lines exhibiting marked paralysis that resulted in a significantly longer recovery time than age-matched controls and with young animals of the same genotype (Fig. 7a). There was some variability in severity of the stress-sensitive paralysis and in the age of onset between the ATPalpha CJ strains. All ATPalpha CJ mutants exhibited 100% penetrance of this phenotype once they were aged 15–30 days post-eclosion.
Fig. 7.

Longevity and stress-sensitive locomotor impairment in ATPalpha alleles. a Progressive locomotor impairment in heterozygous ATPalpha mutants. Recovery from mechanical stress-induced paralysis (seconds) was measured in ATPalpha mutants and aged-matched wild type controls at 25°C. Paralysis was never observed in wild type controls, but all mutant lines showed some degree of progressive impairment by 20–30 days post-eclosion. Asterisks indicate significant differences from age-matched wild type controls using a Student’s t test (* P < 0.001). Time points examined are days 3, 10 and 30 post-eclosion. b Lifespans were performed on heterozygous ATPalpha mutant and control animals. Median lifespan was used to compare longevity between the genotypes. Asterisks indicate significant differences from wild type controls using a Student’s t test (* P < 0.05, ** P < 0.01, and *** P < 0.001). Error is SEM
ATPalpha CJ alleles have different effects on lifespan
Previously, gain-of-function alleles of ATPalpha were shown to have significantly reduced longevity from that of control animals (Palladino et al. 2003). The severity of degenerative pathology observed in some ATPalpha CJ mutants suggested that some of these alleles might also have reduced longevity. We measured lifespans in all of our new alleles as well as a cytologically normal revertant strain ATPalpha DTS1R1. Only the ATPalpha CJ10 mutant exhibited a reduction in longevity (data not shown). This result is consistent with the severity of the stress-sensitive locomotor impairment and marked neurodegenerative pathology observed in this strain. Surprisingly, several strains had significantly increased longevity (data not shown). To properly control for hybrid vigor and strain effects, we replicated these experiments using single outcrossed mutant and controls lacking balancer chromosomes and maintaining heterozygosity of w, ve, and e in all mutants and in the control populations (see “Materials and methods” for details). The increase observed in longevity associated with the ATPalpha CJ6, ATPalpha CJ12 and in ATPalpha DTS1R1 mutations was reproduced (Fig. 7b). The ATPalpha CJ6 strain demonstrated a highly reproducible 22% increase longevity over the control strain. Importantly, not all of the ATPalpha CJ mutants exhibit increased longevity and the independently isolated ATPalpha DTS1R1 strain also has increased longevity, ruling out numerous trivial explanations for this finding. The allele specificity of the finding and the fact that the ATPalpha DTS1R1 strain has increased longevity suggest that a specific level of ATPalpha impairment might be advantageous to animal longevity.
Pharmacological phenocopy with ouabain
The Na+, K+ ATPase has numerous diverse functions including ion transport and non-pumping functions. Activity of the Na+, K+ ATPase is known to serve the essential roles of maintaining ion gradients in numerous tissues, including the neuromuscular system where high membrane potentials are required for signaling and in numerous other tissues where they are tied to various cellular homeostatic processes. To confirm that dose-dependent loss-of-function of the Na+, K+ ATPase is responsible for modulating longevity, we utilized the well-characterized pharmacological antagonist ouabain. These experiments allowed us to examine the effect of varied Na+, K+ ATPase impairment within a standardized control genotype (isogenic Canton S) to completely control for genetic background. These experiments revealed a dose-dependent increase in longevity, where low doses exhibited the most striking increase in lifespan (25%) and high doses exhibited toxicity, as predicted (Fig. 8). The finding that genetic and pharmacologic impairment of the Na+, K+ ATPase each exhibit a similar 22–25% increase in longevity suggests an important role for this protein in regulating organism lifespan. ATPalpha mutant longevity and other phenotypes are summarized in Table 2.
Fig. 8.

Ouabain improves longevity in wild type animals. Longevity assays were performed on wild-type animals administrated one of four test doses of ouabain or vehicle. a The presence of ouabain altered survivorship and resulted in a right or left shift to the survival curve relative to the control, representing an increase or decrease, respectively. b Median lifespan was used to compare the survival curves. Asterisks indicate significant differences from wild-type controls using a Student’s t test (* P < 0.05, ** P < 0.01)
Caloric restriction mechanism of increased longevity?
Several Caenorhabditis elegans eat mutants were found to have increased longevity, including eat-6 that encodes the Na+, K+ ATPase alpha subunit (Hamilton et al. 2005; Lakowski and Hekimi 1998). It was proposed that the increase in longevity was due to the dramatic decrease in their feeding rates thus inducing a state of caloric restriction—a well-known method of increasing animal longevity (Lakowski and Hekimi 1998). We examined the mass of the ATPalpha CJ animals and age- and gender-matched controls to determine whether the animals were malnourished akin to the eat-6 mutants. There was no reduction in the mass of ATPalpha CJ mutant animals as is observed in eat-6 mutants (Supplemental Fig. 4). To more directly assess nutrient intake in ATPalpha CJ mutants we utilized an established Drosophila feeding assay (Edgecomb et al. 1994; Xu et al. 2008). Animals were acutely nutrient deprived and then provided media containing a dye that can be quantified using a spectrophotometer. All of the ATPalpha CJ mutants demonstrated robust ingestion of the test media (Fig. 9). Although there was variability in consumption noted between strains, none of the mutants examined showed a marked reduction in feeding (Fig. 9). There was no correlation between feeding and animal longevity (Pearson r = 0.383) or between mass and longevity (Pearson r = −0.308). Although the feeding assay media is distinct from the standard Drosophila media used in longevity assay, the feeding assay results clearly demonstrate the animals are all capable of normal levels of nutrient consumption. Thus, the data argue that the increase in longevity observed from loss of Na+, K+ ATPase function in Drosophila is likely distinct from that reported for eat-6 mutants.
Fig. 9.

Drosophila feeding assays. Feeding was measured as 625 nm absorption owing to dye included in the test media. The ATPalpha CJ6 strain consumed significantly more media than controls and the positive control strain consumed significantly less. n = 2–3 groups per genotype, representing 60–90 animals. Error is standard deviation. * P < 0.01 and ** P < 0.001 (Student’s t test). All genotypes consumed a significant amount of media and their absorption was in the linear range of detection. Genotypes tested were young age-matched adults (days 3–5)
Discussion
Sodium potassium pumps were first theorized almost 70 years ago (Dean 1941) and first demonstrated biochemically over 50 years ago (Skou 1957), an achievement that led to the shared Nobel prize in chemistry many years later in 1997 (Skou 1998). For decades it was widely hypothesized that mutations affecting sodium pump genes might cause various heritable neurological diseases; however, many attempts to link such diseases to mutations in these genes were not successful. The extremely high degree of evolutionary conservation of Na+, K+ ATPase alpha subunits and their indispensable role in numerous essential processes led some researchers to wonder whether these genes could be targets of genetic disease. Reports of Drosophila sodium pump mutants with stress-sensitive paralysis and ouabain sensitivity (Lebovitz et al. 1989), and later those with temperature-dependent paralysis, reduced longevity and neural degeneration (Palladino et al. 2003), suggested that sodium pump loci were capable of mutations that could result in neurological disease-like states, at least in invertebrates. Later in 2003 the first human disease, FHM, was mapped to the ATP1A2 locus (De Fusco et al. 2003) and a year later several RDP mutations were mapped to the ATP1A3 locus (de Carvalho Aguiar et al. 2004). Currently more than 40 human disease-causing sodium pump mutations are known to be associated with one of these two neurological conditions or a variant thereof. In accordance with the high degree of evolutionary conservation of these proteins and their numerous essential cellular functions, disease-causing mutations are dominantly inherited, missense mutations thought to confer a mild hypomorphic loss-of-function condition.
Several Drosophila ATPalpha mutations have previously been reported; however, most of these are transposon-induced or inversion breaks within the locus and it was not evident that these would accurately model RDP and FHM diseases resulting from numerous distinct missense mutations. There is compelling evidence that dominant, loss-of-function, missense mutations result in the RDP and FHM diseases (de Carvalho Aguiar et al. 2004; De Fusco et al. 2003). We sought to perform a genetic screen to identify novel EMS induced alleles of the ATPalpha gene, in the hopes that an allelic series of loss-of-function mutations would emerge to serve as animal models of these neurological diseases and enable structure–function studies in a tractable genetic system. The screen produced seven new useful alleles affecting the Na+, K+ ATPase, six of which were missense mutations. These ATPalpha CJ mutants all exhibit reduced respiration consistent with each resulting in a loss of ATPase ion-transport function. Several alleles exhibit reduced ATPalpha protein by western blot, also consistent with these being loss-of-function alleles. All of the ATPalpha CJ mutants have normal or near normal septate junction barrier and tracheal morphogenesis function, demonstrating that they are not null mutations and are likely hypomorphic alleles affecting other protein functions. Because of the tetramer organization of the mature Na+, K+ ATPase, hypomorphic loss-of-function mutations have the potential to be more severe than protein null mutations due to possible dominant-negative effects. Such a mutation may exert a dominant-negative effect functionally on the mature ATPase or by altering protein assembly, trafficking, or stability. Dependent upon the mechanism of action and the efficiency of protein degradation, protein abundance may not be significantly altered as determined by western blot. Further studies will be needed to more definitively establish the nature of these mutations; however, these mutations provide important tools for studying the multiple ion-transport dependent and independent functions of the Na+, K+ ATPase.
We have extensively characterized locomotor function and behavior in these mutants. All of the novel mutants exhibit circadian behavior and are capable of normal waking locomotor function. Total activity of ATPalpha CJ mutants is reduced. The Drosophila startle response causes increased locomotor activity in wild-type strains, and in ATPalpha CJ mutants, however to a lesser extent in these mutants. These data suggest that ATPalpha CJ mutants have a specific defect affecting intensive or maximal activity. Gain-of-function alleles have previously been isolated that exhibit temperature-dependent paralysis. Of these new alleles, only the ATPalpha CJ10 mutant exhibits temperature-sensitive paralysis. One perplexing but characteristic feature of RDP is that while the disease is progressive, its onset is rapid and often follows a physical stress. We examined stress-dependent locomotor function in ATPalpha CJ mutants and found that all of the ATPalpha CJ mutants exhibit this feature and the dysfunction that manifests increases markedly with time after the initial onset.
We examined ATPalpha CJ mutants for pathology within the neuromuscular system. We discovered neuropathology associated with specific missense mutations. Neuropathology was most severe in ATPalpha CJ7, ATPalpha CJ10 and ATPalpha CJ13. We had previously reported temperature-sensitive alleles that exhibit marked neuropathology and ATPalpha CJ10 also exhibits temperature-sensitive paralysis. These results suggest that the specific gain-of-function that confers temperature-sensitivity is neurotoxic: understanding pathogenicity of this gain-of-function will require additional study. Interestingly, neuropathology exhibits allelic variation and many of the ATPalpha CJ mutants do not exhibit neuropathology or the pathology observed is mild, whereas other mutants exhibit marked neuropathology, including ATPalpha CJ7, ATPalpha CJ10, ATPalpha CJ13, ATPalpha DTS1, and ATPalpha DTS2. The ATPalpha CJ13 missense mutation results in the identical amino acid substitution to the protein (A588T) as is known to cause FHM (A606T in ATP1A2). This mutant will serve as a valuable model to elucidate the specific dysfunction associated with FHM pathogenesis. ATPalpha DTS2 affects the aspartic acid amino acid 981. This amino acid is also known to be affected in FHM patients bearing the D999H mutation. There is limited data regarding long-term pathology in RDP and FHM patients, the data presented here suggest that specific missense alterations affecting the ATP1A proteins are capable of being neuropathogenic, which should be investigated further.
It was surprising to find that specific alleles of ATPalpha increase longevity by as much as 25% in the absence of a decrease in animal weight or a defect in feeding as was reported for C. elegans eat-6 mutants (Lakowski and Hekimi 1998). Na+, K+ ATPases have been implicated in numerous essential cellular homeostatic processes, making it surprising that mild hypomorphic loss-of-function mutations affecting this protein would result in increased longevity. Whether genetic or pharmacological, impairment of the Na+, K+ ATPase was found to increase the median lifespan by ~20–25%. The sodium pump is actively involved in numerous cellular processes in the brain as well as in other organ systems. Thus, the increase in longevity could derive from altered metabolic activity, stress-response, insulin signaling, or decreased sensory input (or capacity) similar to results in other C. elegans and Drosophila aging models (Antebi 2007; Giannakou and Partridge 2007; Libert et al. 2007; Samuelson et al. 2007; Tatar 2007; Tatar et al. 2001; Vermeulen and Loeschcke 2007). It is also possible that a novel mechanism of action or combination of mechanisms underlies the observed increase in longevity.
A plethora of human sodium pump mutations have been identified recently: more than 40 described mutations are known that affect the ATP1A2 and ATP1A3 proteins. Why are there so many distinct mutations in the ATP1A genes? The genes are large, ~28 kb genetic loci with ~3.6 kb mRNAs containing ~1,000 codons, but the size of the genes does not seem to fully explain the observed incidence. Numerous processes are known to determine the frequency of disease mutations, namely: mutation rate, selection, genetic drift, and founder effects and sufficient data does not currently exist to determine the role, if any, of these factors. Systematic and detailed studies will be needed to understand whether mutation rates of ATP1A genes are increased. It is possible that the large number of mutations, especially those causing FHM, exist simply due to the dominant loss-of-function nature of mutations that cause this disease. The lack of any obvious decrease in fitness is also relevant. The high degree of evolutionary conservation of Na+, K+ ATPases suggests that there is significant functional constraint as well. Although speculative, it is possible that certain ATP1A mutations exhibit antagonistic pleiotropy and confer an advantage to the affected individuals or at least do so under certain situations and that this contributes to their preponderance. The finding that several mild hypomorphic mutations do not cause pathology and confer an increase in longevity suggests the existence of some significant benefit associated with mutation in ATP1A genes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank the National Institutes of Health NIA AG025046 (MJP), NCI U54CA132383 (WVV), GMS GM069540 (GJB), the Lung Biology Training Grant 5 T32 HL076139-03 (SMP), The University of Pittsburgh Department of Pharmacology and Chemical Biology, and The University of Pittsburgh School of Medicine for financial support; Colette Johnston, Bob Kreber, and Barry Ganetzky for assistance with the pilot genetic screen (supported by R01NS15390-29); Sunil Iyer and Mark Langhans for assistance with the molecular characterization of ATPalpha alleles; Felix Akinrinola for assistance with stress-sensitivity testing; Dr. Alicia Celotto, Dr. Charleen Chu, and Dr. Al Fisher for helpful comments; and the Bloomington Stock Center for fly strains.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Ambrosini A, D’Onofrio M, Grieco GS, Di Mambro A, Montagna G, Fortini D, Nicoletti F, Nappi G, Sances G, Schoenen J, Buzzi MG, Santorelli FM, Pierelli F. Familial basilar migraine associated with a new mutation in the ATP1A2 gene. Neurology. 2005;65:1826–1828. doi: 10.1212/01.wnl.0000187072.71931.c0. [DOI] [PubMed] [Google Scholar]
- Antebi A. Genetics of aging in Caenorhabditis elegans. PLoS Genet. 2007;3:1565–1571. doi: 10.1371/journal.pgen.0030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Beal MF, Hyman BT, Koroshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci. 1993;16:125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- Brashear A, Dobyns WB, De Carvalho Aguiar P, Borg M, Frijns CJ, Gollamudi S, Green A, Guimaraes J, Haake BC, Klein C, Linazasoro G, Munchau A, Raymond D, Riley D, Saunders-Pullman R, Tijssen MA, Webb D, Zaremba J, Bressman SB, Ozelius LJ. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain. 2007;130:828–835. doi: 10.1093/brain/awl340. [DOI] [PubMed] [Google Scholar]
- Cai T, Wang H, Chen Y, Liu L, Gunning WT, Quintas LE, Xie ZJ. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol. 2008;182:1153–1169. doi: 10.1083/jcb.200712022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC. Paying the price at the pump: dystonia from mutations in a Na+/K+-ATPase. Neuron. 2004;43:153–154. doi: 10.1016/j.neuron.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Castro MJ, Nunes B, de Vries B, Lemos C, Vanmolkot KR, van den Heuvel JJ, Temudo T, Barros J, Sequeiros J, Frants RR, Koenderink JB, Pereira-Monteiro JM, van den Maagdenberg AM. Two novel functional mutations in the Na+, K+-ATPase alpha2-subunit ATP1A2 gene in patients with familial hemiplegic migraine and associated neurological phenotypes. Clin Genet. 2008;73:37–43. doi: 10.1111/j.1399-0004.2007.00918.x. [DOI] [PubMed] [Google Scholar]
- Celotto AM, Frank AC, McGrath SW, Fergestad T, Van Voorhies WA, Buttle KF, Mannella CA, Palladino MJ. Mitochondrial encephalomyopathy in Drosophila. J Neurosci. 2006;26:810–820. doi: 10.1523/JNEUROSCI.4162-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celotto AM, Frank AC, Seigle JL, Palladino MJ. Drosophila model of human inherited triosephosphate isomerase deficiency glycolytic enzymopathy. Genetics. 2006;174:1237–1246. doi: 10.1534/genetics.106.063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereijido M, Contreras RG, Shoshani L. Cell adhesion, polarity, and epithelia in the dawn of metazoans. Physiol Rev. 2004;84:1229–1262. doi: 10.1152/physrev.00001.2004. [DOI] [PubMed] [Google Scholar]
- Cereijido M, Contreras RG, Shoshani L, Flores-Benitez D, Larre I. Tight junction and polarity interaction in the transporting epithelial phenotype. Biochim Biophys Acta. 2008;1778:770–793. doi: 10.1016/j.bbamem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, Linazasoro G, Borg M, Tijssen MA, Bressman SB, Dobyns WB, Brashear A, Ozelius LJ. Mutations in the Na+/K+-ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–175. doi: 10.1016/j.neuron.2004.06.028. [DOI] [PubMed] [Google Scholar]
- De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G. Haploinsufficiency of ATP1A2 encoding the Na+/K+pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192–196. doi: 10.1038/ng1081. [DOI] [PubMed] [Google Scholar]
- Dean RB. Theories of electrolyte equilibrium in muscle. Biol Symp. 1941;3:331–348. [Google Scholar]
- Edgecomb RS, Harth CE, Schneiderman AM. Regulation of feeding behavior in adult Drosophila melanogaster varies with feeding regime and nutritional state. J Exp Biol. 1994;197:215–235. doi: 10.1242/jeb.197.1.215. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Dagani F. Relationships between the neuronal sodium/potassium pump and energy metabolism. Effects of K+, Na+, and adenosine triphosphate in isolated brain synaptosomes. J Gen Physiol. 1990;95:591–616. doi: 10.1085/jgp.95.4.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergestad T, Bostwick B, Ganetzky B. Metabolic disruption in Drosophila bang-sensitive seizure mutants. Genetics. 2006;173:1357–1364. doi: 10.1534/genetics.106.057463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergestad T, Olson L, Patel KP, Miller R, Palladino MJ, Ganetzky B. Neuropathology in Drosophila mutants with increased seizure susceptibility. Genetics. 2008;178:947–956. doi: 10.1534/genetics.107.082115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez DM, Hand CK, Sweeney BJ, Parfrey NA. A novel ATP1A2 gene mutation in an Irish familial hemiplegic migraine kindred. Headache. 2008;48:101–108. doi: 10.1111/j.1526-4610.2007.00848.x. [DOI] [PubMed] [Google Scholar]
- Gallanti A, Tonelli A, Cardin V, Bussone G, Bresolin N, Bassi MT. A novel de novo nonsense mutation in ATP1A2 associated with sporadic hemiplegic migraine and epileptic seizures. J Neurol Sci. 2008;273:123–126. doi: 10.1016/j.jns.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Genova JL, Fehon RG. Neuroglian, Gliotactin, and the Na+/K+ATPase are essential for septate junction function in Drosophila. J Cell Biol. 2003;161:979–989. doi: 10.1083/jcb.200212054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakou ME, Partridge L. Role of insulin-like signalling in Drosophila lifespan. Trends Biochem Sci. 2007;32:180–188. doi: 10.1016/j.tibs.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Haan J, Kors EE, Vanmolkot KR, van den Maagdenberg AM, Frants RR, Ferrari MD. Migraine genetics: an update. Curr Pain Headache Rep. 2005;9:213–220. doi: 10.1007/s11916-005-0065-9. [DOI] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenberg LG, Su H, Gu H, O’Dowd DK, Smith MA. Alpha3Na+/K+-ATPase is a neuronal receptor for agrin. Cell. 2006;125:359–369. doi: 10.1016/j.cell.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Jen JC, Klein A, Boltshauser E, Cartwright MS, Roach ES, Mamsa H, Baloh RW. Prolonged hemiplegic episodes in children due to mutations in ATP1A2. J Neurol Neurosurg Psychiatry. 2007;78:523–526. doi: 10.1136/jnnp.2006.103267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat-Rott K, Freilinger T, Dreier JP, Herzog J, Gobel H, Petzold GC, Montagna P, Gasser T, Lehmann-Horn F, Dichgans M. Variability of familial hemiplegic migraine with novel A1A2 Na+/K+-ATPase variants. Neurology. 2004;62:1857–1861. doi: 10.1212/01.wnl.0000127310.11526.fd. [DOI] [PubMed] [Google Scholar]
- Kamm C, Fogel W, Wachter T, Schweitzer K, Berg D, Kruger R, Freudenstein D, Gasser T. Novel ATP1A3 mutation in a sporadic RDP patient with minimal benefit from deep brain stimulation. Neurology. 2008;70:1501–1503. doi: 10.1212/01.wnl.0000310431.41036.e0. [DOI] [PubMed] [Google Scholar]
- Kaunisto MA, Harno H, Vanmolkot KR, Gargus JJ, Sun G, Hamalainen E, Liukkonen E, Kallela M, van den Maagdenberg AM, Frants RR, Farkkila M, Palotie A, Wessman M. A novel missense ATP1A2 mutation in a Finnish family with familial hemiplegic migraine type 2. Neurogenetics. 2004;5:141–146. doi: 10.1007/s10048-004-0178-z. [DOI] [PubMed] [Google Scholar]
- Koenderink JB, Zifarelli G, Qiu LY, Schwarz W, De Pont JJ, Bamberg E, Friedrich T. Na, K-ATPase mutations in familial hemiplegic migraine lead to functional inactivation. Biochim Biophys Acta. 2005;1669:61–68. doi: 10.1016/j.bbamem.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T, Yu M, Williams B, Goel S, Paul SM, Beitel GJ. CaSpeR5, a family of Drosophila transgenesis and shuttle vectors with improved multiple cloning sites. Biotechniques. 2007;42:164–166. doi: 10.2144/000112386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Takeyasu K, Fambrough DM. Molecular characterization and expression of the (Na++K+)-ATPase alpha-subunit in Drosophila melanogaster. Embo J. 1989;8:193–202. doi: 10.1002/j.1460-2075.1989.tb03364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees GJ. Contributory mechanisms in the causation of neurodegenerative disorders. Neuroscience. 1993;54:287–322. doi: 10.1016/0306-4522(93)90254-D. [DOI] [PubMed] [Google Scholar]
- Libert S, Zwiener J, Chu X, Vanvoorhies W, Roman G, Pletcher SD. Regulation of Drosophila life span by olfaction and food-derived odors. Science. 2007;315:1133–1137. doi: 10.1126/science.1136610. [DOI] [PubMed] [Google Scholar]
- Lingrel JB, Arguello JM, Van Huysse J, Kuntzweiler TA. Cation and cardiac glycoside binding sites of the Na, K-ATPase. Ann N Y Acad Sci. 1997;834:194–206. doi: 10.1111/j.1749-6632.1997.tb52251.x. [DOI] [PubMed] [Google Scholar]
- Lopina OD. Na+, K+-ATPase: structure, mechanism, and regulation. Membr Cell Biol. 2000;13:721–744. [PubMed] [Google Scholar]
- Mobasheri A, Avila J, Cozar-Castellano I, Brownleader MD, Trevan M, Francis MJ, Lamb JF, Martin-Vasallo P. Na+, K+-ATPase isozyme diversity; comparative biochemistry and physiological implications of novel functional interactions. Biosci Rep. 2000;20:51–91. doi: 10.1023/A:1005580332144. [DOI] [PubMed] [Google Scholar]
- Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TL, Petersen J, Andersen JP, Vilsen B, Nissen P (2007) Crystal structure of the sodium‐potassium pump. Nature 450:1043–1049 [DOI] [PubMed]
- Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000;102:437–449. doi: 10.1016/S0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- Palladino MJ, Hadley TJ, Ganetzky B. Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila. Genetics. 2002;161:1197–1208. doi: 10.1093/genetics/161.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Bower JE, Kreber R, Ganetzky B. Neural dysfunction and neurodegeneration in Drosophila Na+/K+ATPase alpha subunit mutants. J Neurosci. 2003;23:1276–1286. doi: 10.1523/JNEUROSCI.23-04-01276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmgren MG, Axelsen KB. Evolution of P-type ATPases. Biochim Biophys Acta. 1998;1365:37–45. doi: 10.1016/S0005-2728(98)00041-3. [DOI] [PubMed] [Google Scholar]
- Paul SM, Ternet M, Salvaterra PM, Beitel GJ. The Na+/K+ ATPase is required for septate junction function and epithelial tube-size control in the Drosophila tracheal system. Development. 2003;130:4963–4974. doi: 10.1242/dev.00691. [DOI] [PubMed] [Google Scholar]
- Paul SM, Palladino MJ, Beitel GJ. A pump-independent function of the Na, K-ATPase is required for epithelial junction function and tracheal tube-size control. Development. 2007;134:147–155. doi: 10.1242/dev.02710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierelli F, Grieco GS, Pauri F, Pirro C, Fiermonte G, Ambrosini A, Costa A, Buzzi MG, Valoppi M, Caltagirone C, Nappi G, Santorelli FM. A novel ATP1A2 mutation in a family with FHM type II. Cephalalgia. 2006;26:324–328. doi: 10.1111/j.1468-2982.2006.01002.x. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. Familial hemiplegic migraine. Neurotherapeutics. 2007;4:274–284. doi: 10.1016/j.nurt.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Rajasekaran SA, Barwe SP, Rajasekaran AK. Multiple functions of Na, K-ATPase in epithelial cells. Semin Nephrol. 2005;25:328–334. doi: 10.1016/j.semnephrol.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Riant F, De Fusco M, Aridon P, Ducros A, Ploton C, Marchelli F, Maciazek J, Bousser MG, Casari G, Tournier-Lasserve E. ATP1A2 mutations in 11 families with familial hemiplegic migraine. Hum Mutat. 2005;26:281. doi: 10.1002/humu.9361. [DOI] [PubMed] [Google Scholar]
- Samuelson AV, Carr CE, Ruvkun G. Gene activities that mediate increased life span of C. elegans insulin-like signaling mutants. Genes Dev. 2007;21:2976–2994. doi: 10.1101/gad.1588907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubiger M, Feng Y, Fambrough DM, Palka J. A mutation of the Drosophila sodium pump alpha subunit gene results in bang-sensitive paralysis. Neuron. 1994;12:373–381. doi: 10.1016/0896-6273(94)90278-X. [DOI] [PubMed] [Google Scholar]
- Segall L, Scanzano R, Kaunisto MA, Wessman M, Palotie A, Gargus JJ, Blostein R. Kinetic alterations due to a missense mutation in the Na, K-ATPase alpha2 subunit cause familial hemiplegic migraine type 2. J Biol Chem. 2004;279:43692–43696. doi: 10.1074/jbc.M407471200. [DOI] [PubMed] [Google Scholar]
- Segall L, Mezzetti A, Scanzano R, Gargus JJ, Purisima E, Blostein R. Alterations in the alpha2 isoform of Na, K-ATPase associated with familial hemiplegic migraine type 2. Proc Natl Acad Sci USA. 2005;102:11106–11111. doi: 10.1073/pnas.0504323102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigle JL, Celotto AM, Palladino MJ. Degradation of functional triose phosphate isomerase protein underlies sugarkill pathology. Genetics. 2008;179:855–862. doi: 10.1534/genetics.108.087551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta. 1957;23:394–401. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- Skou JC. Nobel Lecture. The identification of the sodium pump. Biosci Rep. 1998;18:155–169. doi: 10.1023/A:1020196612909. [DOI] [PubMed] [Google Scholar]
- Spadaro M, Ursu S, Lehmann-Horn F, Veneziano L, Antonini G, Giunti P, Frontali M, Jurkat-Rott K. A G301R Na+/K+-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics. 2004;5:177–185. doi: 10.1007/s10048-004-0183-2. [DOI] [PubMed] [Google Scholar]
- Sun B, Xu P, Wang W, Salvaterra PM. In vivo modification of Na(+), K(+)-ATPase activity in Drosophila. Comp Biochem Physiol B Biochem Mol Biol. 2001;130:521–536. doi: 10.1016/S1096-4959(01)00470-5. [DOI] [PubMed] [Google Scholar]
- Swoboda KJ, Kanavakis E, Xaidara A, Johnson JE, Leppert MF, Schlesinger-Massart MB, Ptacek LJ, Silver K, Youroukos S. Alternating hemiplegia of childhood or familial hemiplegic migraine? A novel ATP1A2 mutation. Ann Neurol. 2004;55:884–887. doi: 10.1002/ana.20134. [DOI] [PubMed] [Google Scholar]
- Tatar M. Diet restriction in Drosophila melanogaster. Design and analysis. Interdiscip Top Gerontol. 2007;35:115–136. doi: 10.1159/000096559. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Todt U, Dichgans M, Jurkat-Rott K, Heinze A, Zifarelli G, Koenderink JB, Goebel I, Zumbroich V, Stiller A, Ramirez A, Friedrich T, Gobel H, Kubisch C. Rare missense variants in ATP1A2 in families with clustering of common forms of migraine. Hum Mutat. 2005;26:315–321. doi: 10.1002/humu.20229. [DOI] [PubMed] [Google Scholar]
- Trotta N, Rodesch CK, Fergestad T, Broadie K. Cellular bases of activity-dependent paralysis in Drosophila stress-sensitive mutants. J Neurobiol. 2004;60:328–347. doi: 10.1002/neu.20017. [DOI] [PubMed] [Google Scholar]
- Van Voorhies WA, Khazaeli AA, Curtsinger JW. Selected contribution: long-lived Drosophila melanogaster lines exhibit normal metabolic rates. J Appl Physiol. 2003;95:2605–2613. doi: 10.1152/japplphysiol.00448.2003. [DOI] [PubMed] [Google Scholar]
- Van Voorhies WA, Khazaeli AA, Curtsinger JW. Testing the “rate of living” model: further evidence that longevity and metabolic rate are not inversely correlated in Drosophila melanogaster. J Appl Physiol. 2004;97:1915–1922. doi: 10.1152/japplphysiol.00505.2004. [DOI] [PubMed] [Google Scholar]
- Vanmolkot KR, Kors EE, Hottenga JJ, Terwindt GM, Haan J, Hoefnagels WA, Black DF, Sandkuijl LA, Frants RR, Ferrari MD, van den Maagdenberg AM. Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol. 2003;54:360–366. doi: 10.1002/ana.10674. [DOI] [PubMed] [Google Scholar]
- Vanmolkot KR, Kors EE, Turk U, Turkdogan D, Keyser A, Broos LA, Kia SK, van den Heuvel JJ, Black DF, Haan J, Frants RR, Barone V, Ferrari MD, Casari G, Koenderink JB, van den Maagdenberg AM. Two de novo mutations in the Na, K-ATPase gene ATP1A2 associated with pure familial hemiplegic migraine. Eur J Hum Genet. 2006;14:555–560. doi: 10.1038/sj.ejhg.5201607. [DOI] [PubMed] [Google Scholar]
- Vanmolkot KR, Stroink H, Koenderink JB, Kors EE, van den Heuvel JJ, van den Boogerd EH, Stam AH, Haan J, De Vries BB, Terwindt GM, Frants RR, Ferrari MD, van den Maagdenberg AM. Severe episodic neurological deficits and permanent mental retardation in a child with a novel FHM2 ATP1A2 mutation. Ann Neurol. 2006;59:310–314. doi: 10.1002/ana.20760. [DOI] [PubMed] [Google Scholar]
- Vanmolkot KR, Stam AH, Raman A, Koenderink JB, de Vries B, van den Boogerd EH, van Vark J, van den Heuvel JJ, Bajaj N, Terwindt GM, Haan J, Frants RR, Ferrari MD, van den Maagdenberg AM. First case of compound heterozygosity in Na, K-ATPase gene ATP1A2 in familial hemiplegic migraine. Eur J Hum Genet. 2007;15:884–888. doi: 10.1038/sj.ejhg.5201841. [DOI] [PubMed] [Google Scholar]
- Vermeulen CJ, Loeschcke V. Longevity and the stress response in Drosophila. Exp Gerontol. 2007;42:153–159. doi: 10.1016/j.exger.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Wu MN, Koh K, Yue Z, Joiner WJ, Sehgal A. A genetic screen for sleep and circadian mutants reveals mechanisms underlying regulation of sleep in Drosophila. Sleep. 2008;31:465–472. doi: 10.1093/sleep/31.4.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zheng X, Sehgal A. Regulation of feeding and metabolism by neuronal and peripheral clocks in Drosophila. Cell Metab. 2008;8:289–300. doi: 10.1016/j.cmet.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti-Fregonara P, Vidailhet M, Kas A, Ozelius LJ, Clot F, Hindie E, Ravasi L, Devaux JY, Roze E. [123I]-FP-CIT and [99mTc]-HMPAO single photon emission computed tomography in a new sporadic case of rapid-onset dystonia-parkinsonism. J Neurol Sci. 2008;273:148–151. doi: 10.1016/j.jns.2008.06.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.