

Abstract
A series of novel curcumin analogs, symmetrical dienones, were previously shown to possess cytotoxic, anti-angiogenic and anti-tumor activities. Analogs 1 (EF24) and 2 (EF31) share the dienone scaffold and serve as Michael acceptors. We propose that the anticancer effects of 1 and 2 are mediated in part by redox-mediated induction of apoptosis. In order to support this concept, 1 and 2 were treated with L-glutathione (GSH) and cysteine containing dipeptides under mild conditions to form colorless water-soluble adducts, which were identified by LC/MS. Comparison of the cytotoxic action of 1, 2 and the corresponding conjugates, 1-(GSH)2 and 2-(GSH)2, illustrated that the two classes of compounds exhibit essentially identical cell killing capabilities. Compared with the yellow, somewhat light sensitive and nearly water insoluble compounds 1 and 2, the glutathione conjugates represent a promising new series of stable and soluble anti-tumor pro-drugs.
Curcumin (diferuloylmethane) is a α,β-diketone constituent of turmeric with antioxidant properties. It has been used in traditional medicine for liver disease, indigestion, urinary tract infections, rheumatoid arthritis, and insect bites.1 This phytochemical has also demonstrated both anti-cancer and anti-angiogenic properties.2,3 Among other things, curcumin blocks several important cellular targets such as nuclear factor kappa-B (NF-κB).4,5,6 This interaction, in turn, induces apoptosis and retards the function of protein kinase C, epidermal growth factor receptor tyrosine kinase and human epidermal growth factor receptor-2 (HER-2).7,8 Recent therapeutic efficacy against pancreatic cancer in a phase II clinical trial further supports the use of curcumin as a lead for the development of a new class of anticancer agents.9 Unfortunately, due to the low potency and poor absorption characteristics of curcumin, its clinical potential may prove to be limited.10 Nonetheless, the compound remains an ideal lead compound for design of derivatives with improved water solubility.11
About 100 curcumin analogs have been synthesized in our laboratory and tested for anti-cancer and anti-angiogenesis properties.12 A subset of 10 was further evaluated in the 60 panel NCI cancer cell lines and in several in vitro anti-angiogenesis screens. Analog 1 with ortho-fluoro groups and its 2-pyridine analog 2 exhibit superior cytotoxic activities compared with other members of the series (Figure 1). Analog 1 has been shown to inhibit the growth of cancer cells at a ca. 10-fold lower dose than curcumin,12 induce apoptosis13 and block the growth of human breast tumors in a mouse xenograft model with relatively low toxicity.14 Our most recent study identified I-kappa B kinase (IKKβ) as an effective target for both compound 1 and curcumin, although the latter is less potent. Compound 1 rapidly blocks the nuclear translocation of NF-κB with an IC50 of 1.3 μM compared with curcumin with an IC50 of 13 μM.15 In spite of the higher activity of 1 (EF24) and 2 (EF31) by comparison with curcumin, the low bioavailability and fast metabolism of these analogs still remains a critical problem for further development.
Figure 1.

Structures of curcumin, analogs 1 and 2.
Our earlier study showed that 1 induces cell cycle arrest and apoptosis by means of a redox-dependent process in MDA-MB-231 human breast cancer and DU-145 human prostate cancer cells.13 Compound 1, containing a dienone moiety, serves as a Michael acceptor to deplete L-glutathione (GSH) and GSSG concentrations in both wild type and Bcl-xL overexpressing HT29 human colon cancer cells. Comparable chemistry is utilized by a series of novel tyrosine kinase inhibitors developed by Smaill and coworkers, in which a mild Michael acceptor is appended to a quinazoline ring. The compounds engage in a very specific alkylation of Cys-773 in the ATP pocket of the EGFR. The α,β-unsaturated tyrosine kinase inhibitors are currently in Phase I clinical trial.16,17 In addition, Dimmock and colleagues have long recognized the affinity of dienones for biological thiols,18 and isolated ethanethiol and 1,2-ethanedithiol adducts of selected enones.19 Unexplored, however, are the reversible or partially reversible nature of the compounds, which may offer potential advantages in terms of target suppression and in vivo pharmacokinetics.
The observation of glutathione depletion suggested a means to solubilize the curcumin analogs for evaluation in various cell-based assays. Thus, solubility comparisons indicated that while aqueous dissolution of curcumin is unaffected by GSH, high concentrations of the peptide are capable of drawing 1 into solution. (Figure 2) The adduction with glutathione is illustrated in Scheme 1. A number of studies have demonstrated that depletion of thiol concentrations prior to treatment with various anticancer drugs has increased cell killing compared to the use of drug alone.20,21
Figure 2.
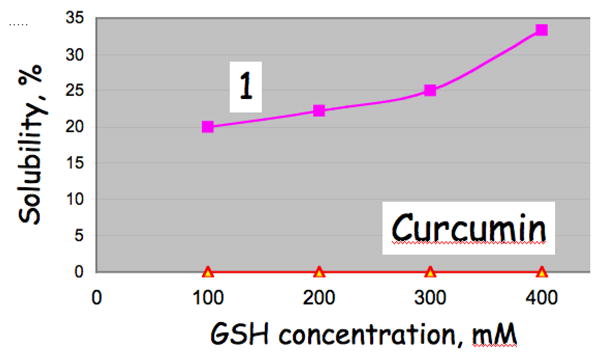
Solubility of 1 and curcumin in GSH-doped aqueous solution. Each point corresponds to 200 mM of compound, and thus a maximum 1:GSH molar ratio of 1:1 and 2:1 at GSH concentrations of 200 and 400 mM, respectively. Powders of the enones and four GSH concentration were mixed and left at room temperature for 30 min. Insoluble 1 and curcumin were collected by centrifugation, and the pellets content (% compound undissolved) were determined by bioassay after dissolution in DMSO. Incomplete solubility of 1-(GSH)2 is due to the short 30 min mixing times employed in this experiment.
Scheme 1.

Combination of 1 and 2 with GSH to give mono-adduct 3 and bis-adduct 4.
Synthesis of GSH conjugates
Compound 1 or 2 dissolved in acetonitrile (CH3CN) was added slowly to 2.1 equivalents of GSH in water at room temperature. The yellow color of the dienone disappears as a result of eliminating the extended chromophore and the simultaneous formation of bis-adducts 1- or 2-(GSH)2. Conjugation of GSH with 2 takes place instantaneously as reflected by the immediate disappearance of the yellow color of the unsaturated ketone, while conjugation with 1 takes several hours to complete. Nevertheless, both reactions provide bis-adducts as the only products along with free GSH based on LC/MS (Figure 3a and b). No mono-adducts were detected under these conditions, although mono-adduct 3 is surely an intermediate to bis-adduct 4. Accordingly, treatment of 2 with 1.0 equivalent of L-glutathione delivers a mixture of mono- and bis-adducts, 2-(GSH) and 2-(GSH)2, with the mono-adduct as the major product by MS. However, due to the similar polarities of 2-(GSH) and 2-(GSH)2, no clear separation by LC was observed. Thus, a broad peak presumed to be overlapping bands of the two conjugates was observed. (Figure 3c)
Figure 3.

LC spectra following separate addition of 1 eq of 1 and 2 to 2.1 eq of GSH; peak mass determined by MS; a) 1-(GSH)2 and GSH; b) 2-(GSH)2 and GSH; c) LC/MS spectra of 1: 1 mixture of 2 and GSH to deliver mono-adduct 2-(GSH) as the major product and bis-product 2-(GSH)2 as the minor product based on the MS.
In order to further investigate the reaction between thiols and curcumin analogs, several cysteine-containing dipeptides were prepared and their reactions with curcumin analogs, examined.
Synthesis of cysteine-containing di-peptides
The preparation of cysteine-containing dipeptides was initiated with free L-cysteine 5. Due to the sensitivity of the cysteine molecule toward oxidation and elimination, it was necessary to protect the β-sulfhydryl moiety as well as the amino and/or carboxyl group during synthesis.22 Several methodologies for cysteine S-functionalization and protection have been reported previously.23,24,25,26 Dimethyl-thiazolidine (Dmt) has been successfully employed as a sulfhydryl-amino masking group for cysteine in the synthesis of glutathione27 and in the course of natural product syntheses.28,29 The protecting unit can be removed under mild conditions to yield the corresponding aminoethanethiol moiety of cysteine. In the present work, Dmt-cysteine 6 was converted into the Boc acetonide derivative 7. The latter was coupled with the protected amino acid hydrochloride salt 8 using EDCI and HOBt in DMF and further treated with diisopropyl ethyl amine (DIEA) to give Boc-Dmt-cys-amino acid-ester 9 in medium to good yields. Deprotection of the Dmt group with TFA delivered the Cys-Phe-methyl ester TFA salt 12. Hydrolysis of 9 under basic conditions (NaOH) in dioxane delivered the Boc-Dmt-cys-amino acid 10. Removal of the Boc group from 10 under acidic conditions (TFA) and subsequent concentration in the presence of EtOH and H2O (1:1) led to the complete removal of the acetone generated and isolation of free amino and thiol-cysteine-dipeptide TFA salt 11 in excellent yield (95%) (Scheme 2).
Scheme 2.

Preparation of cysteine-containing dipeptides
Reagents: a) acetone, reflux; b)Boc2O, DIEA, MeCN, rt; c) HOBt, EDCI, DMF, then phenylalanine methyl ester. HCl (8), DIEA; d) NaOH (1N), dioxane, then HCl (1N); e) TFA-CH2Cl2(3:1), rt, 3h; then EtOH-H2O (1:1).
Coupling of Cys-dipeptides with 1 and 2
Compounds 1 and 2 were treated with di-peptides Cys-Phe (11) and Cys-Gly (12) respectively. Reaction between 2 and Cys-Gly is instantaneous when the dienone in CH3CN is added dropwise to Cys-Gly in water. The yellow color of 2 disappears immediately with the formation of 2(Cys-Gly)2 (13d), which was confirmed by LC/MS. By comparison, reaction between 1 and Cys-Gly as with GSH is much slower, requiring at least seven hours at room temperature. Both 1 and 2 Cys-Gly conjugates are white, water-soluble powders upon solvent evaporation. Conjugation with higher mass di-peptides, for instance Cys-Phe (11), proceeds much more slowly relative to Cys-Gly. Combination of 2 with Cys-Gly occurs instantly, while conjugation with Cys-Phe takes several hours to complete. Nonetheless, the products, 13a and 13c are also white, water-soluble powders. (Scheme 3)
Scheme 3.
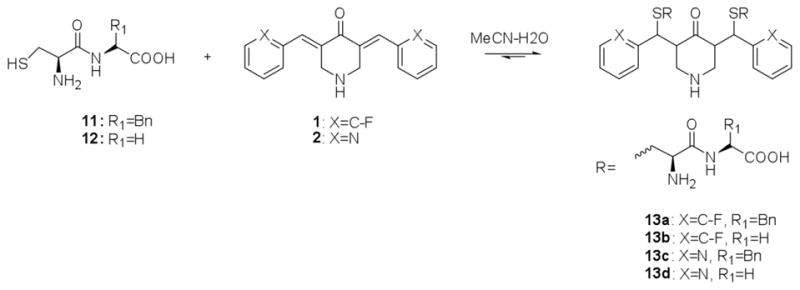
Conjugation of 1 and 2 with cysteine-containing dipeptides.
Previous studies of the interaction between curcumin and glutathione have suggested the operation of Michael-addition between the α, β-unsaturated chromophore of curcumin and GSH. FAB-MS and MALDI-MS spectra of glutathionated curcumin have been interpreted in terms of mono- and di-glutathionyl-adducts as well as cyclic rearrangement products including feruloyl-methylketone and feruloylaldehyde.30 A recent investigation by Usta and coworkers reports that a combination of glutathione and curcumin leads to the formation of two diastereoisomeric monoglutathionyl curcumin conjugates.31 The structures of both conjugates were identified by LC/MS and one- and two-dimensional 1H NMR analysis. Formation of the two isomers was reported to be reversible, although the compounds appear to be relatively unstable. Compared with the slow association of curcumin and GSH, the reaction of either 1 or 2 with GSH is much faster. In addition, the latter are stable, water-soluble, colorless powders of the bis-adducts 1-(GSH)2 and 2-(GSH)2 with a potential for releasing drugs 1 and 2 by means of a reversible equilibrium (Scheme 1).
Reversible and competitive equilibria of GSH Michael addition to curcumin analogs
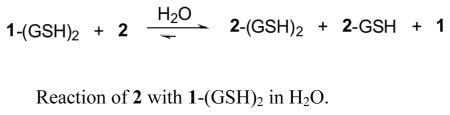
It is common knowledge that Michael additions are reversible reactions,32 yet the literature reveals very few investigations of thiol-mediated equilibria of this type.33 The reversibility of thiol addition to 1 was demonstrated by treatment of 1-(GSH)2 with 2 in water at room temperature. (eq 1)
 |
eq 1 |
The reaction was followed by LC/MS from 5 min to 2 days after mixing 1-(GSH)2 and 2 in a molar ratio of 1:1. Analysis of the reaction mixture by LC/MS after 5 min revealed only starting materials, 2 and 1-(GSH)2. After 8 hr, LC/MS detected 1, 2-GSH and 2-(GSH)2 along with unreacted 2 and 1-(GSH)2. Extending the reaction period to 48 hr gave no evidence of 2, only 1, 2-(GSH)2 and a small amount of unreacted 1-(GSH)2. The Michael addition of the pyridine analog 2 with glutathione was described above as an instantaneous reaction between them, while the corresponding conjugation of 1 proved to be much slower. (Scheme 1). Accordingly, eq. 1 implies the retro-Michael addition of 1-(GSH)2 releasing free 1 and glutathione followed by 2’s capture of GSH to deliver the corresponding mono- and bis-adducts. Thus, the competition confirms that the cross-coupling of 2 with GSH is much faster than 1.
Comparison of the cytotoxic activity of 1, 2 and the corresponding GSH conjugates
Treatment of MDA-MB-435 human breast cancer cells separately with 1, 2, 1-(GSH)2 and 2-(GSH)2 under identical conditions demonstrates that the GSH bis-conjugated analogs are as efficacious in their cell-kill capacity as the parents 1 and 2. Analogs 1 and 1-(GSH)2 exhibit IC50 values of 1.5 μM against MDA-MB-435 breast cancer cells (Figure 4a), while 2 and 2-(GSH)2 likewise show similar IC50 values of 1.0 μM (Figure 4b).
Figure 4.

Dose response curves for the treatment of MDA-MB-435 human breast cancer cells with curcumin analogs and their GSH conjugates. a) 1 and 1-(GSH)2; b) 2 and 2-(GSH)2.
In summary, thiol conjugates such as 1-(GSH)2 and 2-(GSH)2 are easily prepared as stable, water soluble, colorless solids with cytotoxic properties comparable to the unconjugated drugs 1 and 2. The double GSH conjugates can be regarded as a class of pro-drugs capable of releasing the active agents by means of a readily established equilibrium (Scheme 1). While the thiol conjugates of 1 and 2 are in principle capable of existing as a complex mixture of mono- and bis-GSH diastereoisomers in aqueous solution, we expect that certain isomers may well dominate in solution, perhaps as single entities in some cases. Studies underway will explore the relative bioactivity of other pyridone curcumin analogs, the bio-effects of replacing GSH with cysteine-containing peptides, effectiveness against other cell lines, the diastereomeric composition and the potential of the series as anti-cancer pro-drugs.
Acknowledgments
This work was financially supported by the National Institutes of Health (NIH) grants R21 CA82995-01A1 and U.S. Department of Defense, the Division of U.S. Army DAMD17-00-1-0241 (to M. Shoji) and 1 U54 HG003918 (to J.P. Snyder).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Goel A, Kunnumakkara AB, Bharat BA. Biochemical Pharmacology. 2008;75:787–809. doi: 10.1016/j.bcp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 2.Bemis DL, Katz AE, Buttyan R. Expert Opin Invest Drugs. 2006;15:1191–1200. doi: 10.1517/13543784.15.10.1191. [DOI] [PubMed] [Google Scholar]
- 3.Nonn L, Duong D, Peehl DM. Carcinogenesis. 2006;28:1188–1196. doi: 10.1093/carcin/bgl241. [DOI] [PubMed] [Google Scholar]
- 4.Tomita M, Kawakami H, Uchihara JN, Okudaira T, Masuda M, Takasu N, Matsuda T, Ohta T, Tanaka Y, Ohshiro K, Mori N. Int J Cancer. 2006;118:765–772. doi: 10.1002/ijc.21389. [DOI] [PubMed] [Google Scholar]
- 5.Jobin C, Bradham CA, Russo MP, Juma B, Narula AS, Brenner DA, Sartor RB. J Immunol. 1999;163:3474–3483. [PubMed] [Google Scholar]
- 6.Shishodia S, Potdar P, Gariola CG, Aggarwal BB. Carcinogenesis. 2003;24:1269–1279. doi: 10.1093/carcin/bgg078. [DOI] [PubMed] [Google Scholar]
- 7.Aggarwal BB, Shishodia S. Biochem Pharmacol. 2006;71:1397–1421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Sharma RA, Gescher AJ, Steward WP. Eur J Cancer. 2005;41:1955–1968. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Dhillon N, Wolff RA, Abbruzzese JL, Hong DS, Camacho LH, Li L, Braiteh FS, Kurzrock R. J Clin Oncol. 2006;24:14151. (Abstract) [Google Scholar]
- 10.Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS. Planta Med. 1998;64:353–356. doi: 10.1055/s-2006-957450. [DOI] [PubMed] [Google Scholar]
- 11.Safavy A, Raisch KP, Mantena S, Sanford LL, Sham SW, Krishna NR, Bonner JA. J Med Chem. 2007;50:6284–6288. doi: 10.1021/jm700988f. [DOI] [PubMed] [Google Scholar]
- 12.Adams BK, Ferstl EM, Davis MC, Herold M, Kurtkaya S, Camalier RF, Hollingshead MG, Kaur G, Sausville EA, Rickles FR, Snyder JP, Liotta DC, Shoji M. Bioorg & Med Chem. 2004;12:3871–3883. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Adams BK, Cai J, Armstrong J, Herold M, Lu Y, Sun A, Snyder JP, Liotta DC, Jones DP, Shoji M. Anticancer Drugs. 2005;16:263–275. doi: 10.1097/00001813-200503000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Shoji M, et al. Unpublished results. [Google Scholar]
- 15.Kasinski AL, Du Y, Thomas SL, Zhao J, Sun Shi-Y, Khuri FR, Wang C, Shoji M, Sun A, Snyder JP, Liotta DC, Fu H. Molecular Pharmacology. 2008;74:654–661. doi: 10.1124/mol.108.046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, Smaill JB, Trumpp-Kallmeyer S, Dobrusin EM. Proceedings of the National Academy of Sciences. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klutchko SR, Zhou H, Winters RT, Tran TP, Bridges AJ, Althaus IW, Amato DM, Elliott WL, Ellis PA, Meade MA, Roberts BJ, Fry DW, Gonzales AJ, Harvey PJ, Nelson JM, Sherwood V, Han HK, Pace G, Smaill JB, Denny WA, Hollis SHD. J Med Chem. 2006;49:1475–1485. doi: 10.1021/jm050936o. [DOI] [PubMed] [Google Scholar]
- 18.a) Dimmock JR, Padmanilayam MP, Puthucode RN, Nazarali AJ, Motaganahalli NL, Zello GA, Quail JW, Oloo EO, Kraatz HB, Prisciak JS, Allen TM, Santos CL, Balzarini J, De Clercq E, Manavathu EK. J Med Chem. 2001;44:586–593. doi: 10.1021/jm0002580. [DOI] [PubMed] [Google Scholar]; b) Pati HN, Das U, Quail JW, Kawase M, Sakagami H, Dimmock JR. Eur J Med Chem. 2008;43:1–7. doi: 10.1016/j.ejmech.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dimmock JR, Smith LM, Smith PJ. Can J Chem. 1980;58:984–991. [Google Scholar]
- 20.Balendiran GK, Dabur R, Fraser D. Cell Biochem Funct. 2004;22:343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Ruiz C, Mari M, Morals A, Colell A, Ardite E, Fernandez-Checa JC. Hepatology. 2000;32:56–65. doi: 10.1053/jhep.2000.8267. [DOI] [PubMed] [Google Scholar]
- 22.Alves D, Esser D, Broadbridge R, Beevers A, Chapman C, Winsor C, Betley J. J Pep Science. 2003;9:221–228. doi: 10.1002/psc.448. [DOI] [PubMed] [Google Scholar]
- 23.a) Brois J, Pilot J, Barnum H. J Am Chem Soc. 1970;92:7629–7631. [Google Scholar]; b) Hiskey R, Li C, Vunnam R. J Org Chem. 1975;40:3697–3703. doi: 10.1021/jo00913a017. [DOI] [PubMed] [Google Scholar]; c) Kamber B. Helv Chim Acta. 1973;56:1370–1381. doi: 10.1002/hlca.19730560420. [DOI] [PubMed] [Google Scholar]; d) Fotouhi N, Galakatos N, Kemp D. J Org Chem. 1989;54:2803–2817. [Google Scholar]
- 24.a) Ruiz-Gayo M, Albericio F, Pedroso E, Giralt E. J Chem Soc Chem Commun. 1986:1501–1502. [Google Scholar]; b) Corey EJ, Gin DY, Kania RS. J Am Chem Soc. 1996;118:9202–9203. [Google Scholar]; c) Ponsati B, Giralt E, Andreu D. Tetrahedron. 1990;46:8255–8266. [Google Scholar]
- 25.West C, Estiarte M, Rich R. Org Lett. 2001;3:1205–1208. doi: 10.1021/ol015678d. [DOI] [PubMed] [Google Scholar]
- 26.Katritzky A, Angrish P, Hür D, Suzuki K. Synthesis. 2005;3:397–402. [Google Scholar]
- 27.a) King F, Clark-Lewis J, Wade R. J Chem Soc. 1957:880. [Google Scholar]; b) Sheehan J, Yang DD. J Am Chem Soc. 1958;80:1154–1158. [Google Scholar]
- 28.Nicolaou KC, Nevalainen M, Safina B, Zak M, Bulat S. Angew Chem Int Ed. 2002;41:1941–1945. [PubMed] [Google Scholar]
- 29.West C, Estiarte M, Rich R. Org Lett. 2001;3:1205–1208. doi: 10.1021/ol015678d. [DOI] [PubMed] [Google Scholar]
- 30.Awasthi S, Pandya U, Singhal SS, Lin JT, Thiviyanathan V, Seifert WE, Jr, Yogesh C, Awasthi YC, Ansari GA. Chem Biol Interact. 2000;128:19–38. doi: 10.1016/s0009-2797(00)00185-x. [DOI] [PubMed] [Google Scholar]
- 31.Usta M, Wortelboer HM, Vervoort J, Boersma MG, Rietjens IMCM, van Bladeren PJ, Cnubben NHP. Chem Res Toxicol. 2007;20:1895–1902. doi: 10.1021/tx7002245. [DOI] [PubMed] [Google Scholar]
- 32.(a) March J. Advanced Organic Chemistry. 4. Wiley; New York: 1992. p. 796. [Google Scholar]; (b) Bergmann ED, Ginsburg D, Pappo R. Org React. 1959;10:179. [Google Scholar]
- 33.Brickley J. Rubber Age (New York) 1943;53:331–334. [Google Scholar]