

Abstract
TAR DNA-binding protein-43 (TDP-43) proteinopathies are classified based upon the extent of modified TDP-43 inclusions and include a growing number of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration with ubiquitin immunoreactive, tau negative inclusions (FTLD-U) and FTLD with motor neuron disease (FTLD-MND). In addition, TDP-43 inclusions have also been identified in a number of other neurodegenerative disorders including Alzheimer's disease, corticobasal degeneration, Lewy body related diseases and Pick's disease. Current understanding suggests that in these diseases, TDP-43 is relocated from the nucleus to the cytoplasm and sequestered into inclusions that contain modified TDP-43. Major modifications of TDP-43 have been identified as being hyperphosphorylation and proteolytic cleavage by caspases. In this review a summary of the major findings regarding the proteolytic modification of TDP-43 will be discussed as well as potential toxic-gain mechanisms these fragments may cause including cytoskeletal disruptions.
Keywords: Pick's disease, Pick bodies, Caspases, TDP-43, Hirano Bodies, Tau, Review, Alzheimer's disease, FTLD-U, ALS, actin
Biological function of TDP-43
The TAR DNA-binding protein-43 (TDP-43) is a highly conserved 414 amino acid protein with an apparent molecular weight of approximately 43 kDa. TDP-43 was initially identified by localization within the nucleus and its ability to bind the HIV-1 TAR DNA element and in this role may act as a transcriptional repressor (Ou et al., 1995). More recently, studies have suggested TDP-43 may in fact function in RNA transcription and splicing of specific genes (Buratti & Baralle, 2008). Structurally, TDP-43 is composed of two RNA recognition regions followed by a glycine-rich C-terminal domain. The RNA recognition regions are thought to mediate RNA binding as well as protein-protein interactions (Buratti & Baralle, 2001). In contrast, the C-terminal domain of TDP-43 does not bind RNA but appears necessary for modulation of splicing (Buratti et al., 2005). In addition, previous studies have shown that the C-terminal region of TDP-43 is critical for the correct nuclear distribution of this protein (Ayala et al., 2008). This may be of particular importance in neurodegenerative TDP-43 proteinopathies in which a critical feature is the presence of cytoplasmic C-terminal fragments of TDP-43 that have the ability to aggregate (Igaz et al., 2008, Winton et al., 2008). Therefore, although TDP-43 originally was thought to function in the nucleus in relative anonymity, research in the last few years has indicated a direct link between certain neurodegenerative disorders and the presence of cytoplasmic inclusions of TDP-43.
Cytoplasmic inclusions of TDP-43 in various neurodegenerative diseases: cause or effect?
In a seminal study by Neumann et al., TDP-43 was linked to FTLD-U and ALS following an immunological approach to isolate the protein (Neumann et al., 2006). Their rationale was based on part of other insoluble proteins that have a propensity to aggregate as what occurs for beta-amyloid in AD. Isolating high-molecular weight proteins from FTLD-U and ALS subjects followed by immunization, resulted in the generation of antibodies that were then used to further screen brain extracts from affected subjects. Following an exhaustive analysis, TDP-43 was identified in inclusions in all 53 cases of FTLD-U tested as well as in 18 cases of ALS (Neumann et al., 2006). Further confirmation that TDP-43 is a major player in these newly classified TDP-43 proteinopathies was the discovery of mutations of TDP-43 in familial and sporadic forms of ALS (Kabashi et al., 2008, Rutherford et al., 2008, Sreedharan et al., 2008). Interestingly, all of the TDP-43 mutations to date are dominant, suggesting that mutant TDP-43 causes neurodegeneration through a toxic gain of function rather than a loss of normal protein function.
Post-translational modifications of TDP-43 in neurodegenerative disorders
A key feature of TDP-43 proteinopathies is the presence of modified TDP-43 located predominantly within the cytoplasm of neurons (Arai et al., 2006, Neumann et al., 2006). Three major modifications of TDP-43 have been identified: ubiquitination, hyperphosphorylation, and proteolytic processing (Arai et al., 2006, Neumann et al., 2006, Hasegawa et al., 2008, Inukai et al., 2008) For example, analysis of brain extracts from FTLD-U and ALS brains indicated N-terminal truncation of TDP-43 into 25 and 35 kDa fragments specifically within insoluble fractions (Arai et al., 2006, Neumann et al., 2006, Igaz et al., 2008). A similar 25 kDa-truncated fragment of TDP-43 has also been identified in the Alzheimer's disease (AD) brain (Amador-Ortiz et al., 2007). Thus, in several neurodegenerative disorders, TDP-43 redistributes from the nucleus to the cytoplasm where it is sequestered as truncated, hyperphosphorylated insoluble aggregates. It is possible that truncation of TDP-43 interferes with its ability to interact with cellular proteins and thereby leads to altered trafficking of TDP-43 between the nucleus and cytoplasm. Recently, utilizing a yeast TDP-43 proteinopathy model, Johnson et al. demonstrated that cytoplasmic aggregation of TDP-43 is a critical factor in promoting toxicity (Johnson et al., 2008). Further, their results supported the hypothesis that only aggregating forms of TDP-43 was toxic (Johnson et al., 2008). The question remains, however, on what is the molecular link between aberrant TDP-43 trafficking, aggregation and protein misfolding? A clue to this puzzle was the finding by the same authors that a C-terminal fragment of TDP-43 was necessary for cellular toxicity (Johnson et al., 2008), suggesting the involvement of a specific protease in promoting TDP-43 toxicity.
Caspase-cleavage of TDP-43: providing a mechanistic link between aggregation of TDP-43 and cellular pathology
A candidate protease responsible for generating truncated fragments of TDP-43 has recently been identified as belonging to the caspase family. Caspases are indispensable for the execution of apoptosis, being responsible for the phenotypic characteristics of apoptosis following the cleavage of critical cellular proteins (Riedl & Shi, 2004), and are highly specific, cleaving only after aspartic residues (Fuentes-Prior & Salvesen, 2004). In a report by Zhang et al., the authors eloquently demonstrated a role for executioner caspases including caspase-3 and caspase-7 in cleaving TDP-43 using both cell-free assay systems as well as in vitro models of apoptosis (Zhang et al., 2007). First, the authors were able to show the cleavage of recombinant TDP-43 by caspase-3 and -7 but not caspase-8 into 35 and 25 kDa fragments (Zhang et al., 2007). Second, by down-regulating progranulin using progranulin siRNA, they were able to show this was sufficient to stimulate caspase-3 activity and subsequent cleavage of TDP-43. Interestingly, the 25 and 35 kDa fragments from cell lysates following such an experiment displayed the same molecular weight as those found from brains of FTLD-U. Third, and probably most importantly, the authors demonstrated that redistribution of TDP-43 from the nucleus to the cytoplasm is critically dependent upon the caspase-cleavage of TDP-43: in mutant caspase-resistant TDP-43 cell lines, the addition of the classical apoptotic insult, staurosporine, had no effect in promoting the cellular redistribution of TDP-43 as compared with wild-type TDP-43 (Fig. 1). Taken together, these results strongly suggest that caspase activation and cleavage of TDP-43 is a key molecular step linking cellular redistribution and toxicity to the neurodegeneration observed in TDP-43 proteinopathies.
Figure 1. Redistribution of TDP-43 is caspase-dependent.

Wild-type (WT) TDP-43 redistributes from the nucleus to the cytoplasm following treatment of H4 cells with the apoptotic insult, staurosporine (Stspn). However, under similar conditions when a caspase-resistant mutant of TDP-43 was expressed, sub-cellular redistribution to the cytoplasm did not occur (top right and bottom of Panel). These experiments were carried out using a mutant form of TDP-43 in which the consensus motif for caspase cleavage at two different sites was mutated. Reproduced with permission from Zhang et al. (Zhang et al., 2007).
Further evidence that caspase-cleavage of TDP-43 may occur in vivo, were results obtained in post-mortem brain samples from AD following the synthesis and application of a site-directed caspase-cleavage antibody to TDP-43 (Rohn, 2008). The rationale for synthesis of this antibody was the identification of a putative caspase-cleavage consensus site, DVMD219, that would generate an approximate 25-kDa fragment following cleavage by caspases (Zhang et al., 2007). Following verification that this antibody (termed TDP caspase-cleavage product (ccp) antibody) was specific for caspase-cleaved TDP-43, it was determined whether immunoreactivity could be detected with this antibody in the AD brain. Application of the TDPccp antibody revealed strong immunoreactivity in tangles, plaques, and most interestingly within Hirano bodies of area CA1 of the hippocampus (Rohn, 2008). Staining of Hirano bodies with the TDPccp antibody was restricted to area CA1 of the hippocampus in the AD brain and was not found in age-matched control brains (Fig. 2A). In addition, the TDPccp antibody co-localized with an anti-ubiquitin antibody in Hirano bodies of the AD brain (Rohn, 2008). Previous studies have demonstrated that pathological TDP-43 consists of not only truncated fragments, but is also heavily ubiquitinated (Kwong et al., 2007).
Figure 2. Caspase-cleaved TDP-43 within Hirano bodies in Alzheimer's and Pick's disease.
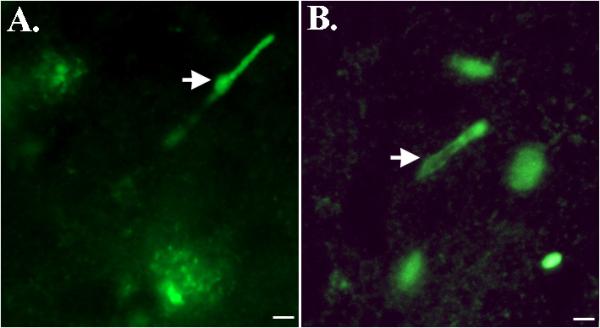
Both panels depict immunofluorescence labeling utilizing a caspase-cleavage site-directed antibody to TDP-43 (Rohn, 2008). Panel A reflects representative labeling in an Alzheimer's case and along with staining of a Hirano body (arrow), labeling within plaques is also observed. A similar staining profile was observed in Pick's disease (B), and in additional to labeling of Hirano bodies, staining of Pick bodies was also evident. All scale bars represent 10 μm.
Recently, we have extended these findings to an additional tauopathy, Pick's disease, by demonstrating the presence of caspase-cleaved TDP-43 within Hirano bodies (Rohn & Kokoulina, 2009) (Fig. 2B). Similar to what we observed in AD, labeled Hirano bodies was a predominant finding, along with labeling within Pick bodies and in addition, was regionally restricted largely within area CA1 of the hippocampus (Rohn & Kokoulina, 2009). Thus, the finding of caspase-cleaved TDP-43 within Hirano bodies of the AD and Pick's disease brain suggests this might be a common feature of tauopathies.
Role of Hirano bodies and the link to TDP-43 in neurodegenerative diseases
The consistent finding of caspase-cleaved TDP-43 within Hirano bodies in various tauopathies including AD and Pick's diseases, raises a number of interesting questions. First, are Hirano bodies causative in promoting neurodegeneration in neurodegenerative diseases? Second, what role are truncated fragments of TDP-43 playing within Hirano bodies? Hirano bodies were first described in 1965 and are characterized as rod-shaped, paracrystalline structures in the neurons of the central nervous system (Hirano, 1994). Hirano bodies have been identified in a number of different neurodegenerative disorders including AD (Gibson & Tomlinson, 1977), Creutzfeldt-Jacob disease (Cartier et al., 1985), Pick's disease (Hirano, 1994) and Parkinson's (Hirano et al., 1968). Whether or not these structures contribute to the neurodegeneration observed in these disorders is unknown. Additional characteristics of Hirano bodies are that they are generally restricted to the CA1 region of the hippocampus and are rich in cytoskeletal proteins including tau and actin (Galloway, Perry & Gambetti, 1987, Galloway, Perry, Kosik et al., 1987). Interestingly, a previous study by Rossiter et al., has demonstrated the presence of caspase-cleaved actin within Hirano bodies of the CA1 region in the AD brain (Rossiter et al., 2000). These results parallel our findings of caspase-cleaved TDP-43 within Hirano bodies, and support a general role for caspase activation and cleavage of proteins within these eosinophilic structures. Moreover, the fact that Hirano bodies are often found in the vicinity of pyramidal cells containing cytoplasmic granulovacoular degeneration (Maciver & Harrington, 1995) provides further support that these structures may promote neurodegeneration. What role may TDP-43 be playing within Hirano bodies? One possibility is that TDP-43 may be involved in the regulation of the cytoskeleton. This idea is supported by a study by Wang et al. who demonstrated the presence of TDP-43 within the somatodendrites of hippocampal neurons where it co-localized with beta-actin mRNA (Wang et al., 2008). Based on these findings, it is possible that TDP-43 plays some normal role in the regulation of the cytoskeleton, a role that may be disrupted following the activation of caspases and cleavage of TDP-43. In certain neurodegenerative diseases, including AD and Pick's disease, the cleavage of TDP-43 could lead to a dysregulation of the cytoskeleton network, leading to the formation of Hirano bodies. However, further studies are needed to determine whether TDP-43 regulates the cytoskeleton and also what role pathological TDP-43 may play in the formation of Hirano bodies.
Concluding remarks
TDP-43 represents a rather ordinary protein with mundane functions in the every day life of a cell, however, upon posttranslational modification, TDP-43 is transformed into a protein that may lead to cellular dysfunction, neurodegeneration, and disease. In particular, proteolytic truncation by caspases appears to be a key step leading to aggregation and cellular redistribution of TDP-43 to the cytoplasm. This is supported by biochemical studies of TDP-43 demonstrating the presence of ubiquitinated high-molecular weight species and C-terminal fragments of TDP-43 in FTLD-U, ALS, and the AD brain (Neumann et al., 2006, Amador-Ortiz et al., 2007). Moreover, expression of mutant TDP-43, in vitro, leads to disruption of TDP-43 trafficking between the nucleus and cytoplasm and leads to aggregate formation (Winton et al., 2008). Therefore, it appears TDP-43 can be added to the list of other proteins whose modification leads to potential aggregation and cellular toxicity including alpha-synuclein (Liu et al., 2005, Dufty et al., 2007) , tau (Gamblin et al., 2003, Rissman et al., 2004), beta-amyloid (Checler & Vincent, 2002), and the prion protein (Checler & Vincent, 2002). Although current understanding of TDP-43 supports this scenario, there is no direct evidence indicating that caspase-cleavage of TDP-43 facilitates the formation of aggregated species that are neurotoxic and therefore, additional studies are warranted. However, it is likely that TDP-43 pathology will share common disease mechanisms with other neurodegenerative disorders including tauopathies and α-synucleinopathies. Future studies should address this question as well as to the mechanism of cellular toxicity and neurodegeneration caused by modified TDP-43. Many of these questions should be answered by the development of an animal model of TDP-43, and with the recent identification of familial cases of ALS caused by mutations in TDP-43 (Kabashi et al., 2008, Rutherford et al., 2008, Sreedharan et al., 2008), a transgenic animal model overexpressing mutant forms of TDP-43 is no doubt forthcoming.
Figure 3. Proposed pathway of TDP-43 redistribution and proteolytic modification.

1) Under normal conditions, TDP-43 resides in the nucleus and serves in the capacity of regulating transcription. 2) In this hypothesis, some unknown stimulus activates apoptosis and executioner caspases, such as caspase-3 or -7. Following caspase activation and cleavage, TDP-43 fragments are sequestered in the cytoplasm. In the cytoplasm, the truncated forms of TDP-43 may promote aggregation, and cellular toxicity perhaps by disrupting the cytoskeleton. 3) Caspase-cleaved TDP-43 is shown to be localized within Hirano bodies, which may protect the cell from the toxic actions of TDP-43. 4) The mechanism of cellular toxicity is unknown, but based on current understanding most likely represents a toxic-gain of function for modified TDP-43.
Acknowledgments
Funded by NIH/NCRR grant #P20RR016454 and a grant from the American Health Assistance Foundation (AHAF).
References Cited
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- Cartier L, Galvez S, Gajdusek DC. Familial clustering of the ataxic form of Creutzfeldt-Jakob disease with Hirano bodies. J Neurol Neurosurg Psychiatry. 1985;48:234–238. doi: 10.1136/jnnp.48.3.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checler F, Vincent B. Alzheimer's and prion diseases: distinct pathologies, common proteolytic denominators. Trends Neurosci. 2002;25:616–620. doi: 10.1016/s0166-2236(02)02263-4. [DOI] [PubMed] [Google Scholar]
- Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007;170:1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway PG, Perry G, Gambetti P. Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol. 1987;46:185–199. doi: 10.1097/00005072-198703000-00006. [DOI] [PubMed] [Google Scholar]
- Galloway PG, Perry G, Kosik KS, Gambetti P. Hirano bodies contain tau protein. Brain Res. 1987;403:337–340. doi: 10.1016/0006-8993(87)90071-0. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson PH, Tomlinson BE. Numbers of Hirano bodies in the hippocampus of normal and demented people with Alzheimer's disease. J Neurol Sci. 1977;33:199–206. doi: 10.1016/0022-510x(77)90193-9. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A. Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol. 1994;20:3–11. doi: 10.1111/j.1365-2990.1994.tb00951.x. [DOI] [PubMed] [Google Scholar]
- Hirano A, Dembitzer HM, Kurland LT, Zimmerman HM. The fine structure of some intraganglionic alterations. Neurofibrillary tangles, granulovacuolar bodies and “rod-like” structures as seen in Guam amyotrophic lateral sclerosis and parkinsonismdementia complex. J Neuropathol Exp Neurol. 1968;27:167–182. [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle FE, Akiyama H, Hisanaga S, Hasegawa M. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008;582:2899–2904. doi: 10.1016/j.febslet.2008.07.027. [DOI] [PubMed] [Google Scholar]
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2008;105:6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114:63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- Liu CW, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson's disease. J Biol Chem. 2005 doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Harrington CR. Two actin binding proteins, actin depolymerizing factor and cofilin, are associated with Hirano bodies. Neuroreport. 1995;6:1985–1988. doi: 10.1097/00001756-199510010-00008. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT. Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer's disease. Brain Res. 2008;1228:189–198. doi: 10.1016/j.brainres.2008.06.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Kokoulina P. Caspase-cleaved TAR DNA-binding protein-43 in Pick's disease. Int J Physio Pathophysio Pharmacol. 2009;1:24–31. [PMC free article] [PubMed] [Google Scholar]
- Rossiter JP, Anderson LL, Yang F, Cole GM. Caspase-cleaved actin (fractin) immunolabelling of Hirano bodies. Neuropathol Appl Neurobiol. 2000;26:342–346. doi: 10.1046/j.1365-2990.2000.00252.x. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L, Rademakers R. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]