

Abstract
AIMS
This study reports the pharmacokinetics of nelfinavir, its active metabolite, M8, and active moiety (nelfinavir + M8) in volunteers genotyped for CYP2C19 as extensive metabolizer (*1*1; n = 38), heterozygous poor metabolizer (PM) (*1*2; n = 22) and homozygous PM (*2*2; n = 6).
METHODS
Subjects received nelfinavir at normal dose (3.5 days of 1250 mg q12h) or high dose (1250 mg q12h for 3 days and single dose of 3125 mg on day 4). Steady-state plasma samples were analysed by high-performance liquid chromatography/ultraviolet assay to determine pharmacokinetics.
RESULTS
At steady state, the mean Cmax was 42% [95% confidence interval (CI) 19, 69] and 63% (95% CI 20, 122) higher, and mean AUC was 51% (95% CI 24, 83) and 85% (95% CI 32, 159) higher for *1*2 and *2*2 compared with *1*1 subjects, respectively. For M8, the mean Cmax and AUC were 35% (95% CI 6, 55) and 33% (95% CI −3, 56), respectively, lower for *1*2 compared with *1*1 subjects. M8 was not detectable in *2*2 subjects. The mean Cmax and AUC values for the active moiety were higher by 30–35% for the *1*2 and *2*2 compared with *1*1 subjects.
CONCLUSIONS
Mutation in CYP2C19 increased the systemic exposure of nelfinavir and reduced the exposure of M8. No significant differences were noted among the heterozygous (*1*2) and homozygous (*2*2) PMs. These changes are not considered to be clinically relevant and hence the use of nelfinavir does not require prior assessment of CYP2C19 genotype.
Keywords: CYP2C19, M8, nelfinavir, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The true influence of CYP2C19*2 mutation on the pharmacokinetics of nelfinavir and its active metabolite, M8, is not clear.
Often, published studies have combined *2 hetero- and homozygous poor metabolizers (PMs) and/or have very limited data from *2 homozygotes, which contributes to the lack of clarity.
WHAT THIS STUDY ADDS
The pharmacokinetics of nelfinavir was delineated using pharmacogenomic data from 66 healthy subjects.
The exposure of nelfinavir was elevated, whereas that of M8 was reduced, in heterozygous and homozygous PMs in an incremental manner consistent with the loss of functional alleles.
However, the exposure of active moiety was only modestly elevated in hetero- and homozygous PMs.
Introduction
Nelfinavir (Viracept®; Pfizer Inc., New York, NY, USA) is an inhibitor of the human immunodeficiency virus (HIV) protease [1]. The pharmacokinetics of nelfinavir has been studied in both HIV-positive patients and healthy volunteers [2]. Following oral administration, peak plasma concentrations typically occur 3–4 h after administration of single or multiple doses. The oral bioavailability of nelfinavir is increased by the presence of food. Nelfinavir is approximately 98% plasma protein bound, and has an apparent volume of distribution of 150 l. Upon multiple dosing, the terminal plasma elimination half-life of nelfinavir is 2.5–4.2 h. About 87% of the dose is accounted for in the faeces, of which 22% is unchanged drug [1, 2].
Nelfinavir is extensively metabolized in the liver by cytochrome P450 enzymes CYP2C19 and CYP3A4 and -3A5. It is converted to an active metabolite, nelfinavir hydroxy-t-butylamide (M8), along with several inactive, oxidative metabolic products [3, 4]. M8 has been shown to exhibit in vitro antiviral potency similar to that of nelfinavir [4]. The formation of M8 is primarily mediated by CYP2C19 [3], which is known to exhibit genetic polymorphism [5]. The CYP2C19 wild-type genotype (*1*1) is associated with an extensive metabolizer (EM) phenotype. In all racial groups, CYP2C19*2 (G681A point mutation in exon 5) is the most common allele associated with a poor metabolizer (PM) phenotype. However, there are differences among racial groups in the occurrence of this polymorphism. The frequency of PMs is about 2–4% in Whites and Blacks, about 14–21% in Asians, and about 60% in certain Pacific Islanders [6]. Among the PMs, the frequency of heterozygote mutants, carrying one wild-type and one mutant allele, is much greater than homozygote mutants, which carry two mutant alleles (e.g. 15% vs. 2% in general White population). For this study, the following designations were used to describe the polymorphism in CYP2C19: EM associated with *1*1 genotype, heterozygous PM associated with *1*2 genotype, and homozygous PM associated with *2*2 genotype.
There is some inconsistency in published data on the influence of CYP2C19 polymorphism on nelfinavir and/or M8 concentrations. Whereas Haas et al.[7] reported increased nelfinavir exposure in CYP2C19*2 hetero- and homozygote PMs compared with EMs, this was not reported by other groups [8–11]. Several groups also noted reduced production of M8 in CYP2C19 PMs [7, 8, 10]. With one exception [7], published studies have either combined *2 hetero- and homozygote PMs, and/or had very limited data from *2 homozygote PMs, and thus the true influence of this enzyme on nelfinavir pharmacokinetics may not have been fully characterized.
The objective of this analysis was to characterize the influence of CYP2C19 activity on the pharmacokinetics of nelfinavir and M8 in healthy volunteers, based on pharmacokinetic data obtained after intensive plasma sampling following single and multiple dose administration of nelfinavir as part of a thorough QTc evaluation of nelfinavir [12]. As per the E14 guidance for QT studies [13], the normal therapeutic dose (1250 mg) and a supratherapeutic dose (3125 mg) were included in the study.
Methods
Study design
Pharmacokinetic data from nelfinavir treatment arms in a thorough QT study were included in this analysis [12]. In brief, this was a multiple-dose, randomized, four-treatment, four-period crossover, positive- and placebo-controlled study in 68 healthy male and female subjects, to study the effect of nelfinavir on QTc interval. There were four treatment periods, each separated by a wash-out period of at least 4 days. Study treatments included nelfinavir 1250 mg q12h on days 1–3 and on day 4 morning (normal dose); nelfinavir 1250 mg q12h on days 1–3 and 3125 mg on day 4 morning (high dose); placebo; and moxifloxacin. Only data from the first two treatments are presented in this analysis. Nelfinavir was administered orally as the 625-mg tablet formulation (Viracept®) with 240 ml water, and following consumption of a standard meal consisting of 500 kcal with 20% fat content.
The study was conducted across four Clinical Research Units: Pfizer Global Research and Development, located in Ann Arbor, MI, USA; New Haven, CT, USA; Brussels, Belgium; and Singapore. The study was conducted in compliance with the ethical principles originating from the Declaration of Helsinki and in compliance with local laws and regulations. The protocol was approved prior to its start by the following Ethics Review Committees: IntegReview Ethical Review Board, Austin, TX, USA; Comite D'Ethique Hopital Erasme, Belgium; and Singapore General Hospital Ethics Committee, Singapore. Written informed consent was obtained prior to the subject entering the study.
Pharmacokinetics
On days 1 and 4 of each treatment period, blood samples (5 ml each) were collected in Vacutainer® tubes containing sodium heparin at the following time points: predose, and at 1, 2, 3, 4, 5, 6, 7, 10 and 12 h after dosing. Plasma was separated within 1 h of blood collection, and samples were stored at −20°C until assay. Plasma samples from normal and high-dose nelfinavir treatments were analysed for nelfinavir and its active metabolite, M8, using a validated high-performance liquid chromatography/ultraviolet assay method reported previously [14]. Calibration standard responses were linear over the range of 0.05–10 µg ml–1 for both nelfinavir and M8. All samples were analysed by PPD Development (Richmond, VA, USA). Pharmacokinetic analyses were carried out with WinNonlin (V.3.2; Pharsight®, Mountain View, CA, USA) using standard noncompartmental methods. Peak plasma concentrations (Cmax) and time to peak plasma concentration (Tmax) were determined based on observed data. Area under the plasma concentration–time profile from time zero to τ (AUC), where τ was defined as 12 h for nelfinavir and M8, was determined by log-linear trapezoidal method. Nelfinavir and M8 concentrations (µg ml–1) were converted to active moiety concentrations (µM l–1) by multiplying each value by 1000/568 and 1000/596, respectively.
Pharmacogenomics
A blood sample (10 ml) was collected prior to Period 1 for pharmacogenomic analysis. Blood sample for pharmacogenomics was not obtained if a subject had previously had the required CYP2C19 genotyping test performed.
Genomic DNA was isolated from whole blood using the QIAamp 96 DNA Blood Kit (Qiagen, Valencia, CA, USA) as per manufacturer's instructions. Genotyping was performed using TaqMan™ (Applied Biosystems, Foster City, CA, USA) allelic discrimination assays for CYP2C19 *2 and *3 (AB Part nos 4312562 and C2786180910, respectively). Polymerase chain reaction conditions were as recommended for the commercially available assays (50°C/2 min, 95°C/10 min, 1 cycle; 92°C/15 s, 60°C/1 min, 35 cycles) and were performed with AB 9700 thermocyclers. A custom TaqMan™ assay was designed for CYP2C19 *4 and performed as previously described by Myrand et al.[15]. Genotyping data were collected on an AB Prism 7900 Sequence Detector System using manual calling.
Statistics
Pharmacokinetic data were summarized by CYP2C19 genotype. Statistical analyses were conducted on day 4 data (which represents steady state) for the normal dose of nelfinavir. The AUC and Cmax values for nelfinavir were log-transformed and analysed using a parametric, one-way anova with genotype as a fixed effect. The AUC and Cmax values were added to 10−5 to handle zero concentrations when log-transformed. Pair-wise comparisons across the three genotypes were also assessed from the anova model, using the Bonferroni method for controlling the significance level. Additionally, as a nonparametric alternative, the Kruskal–Wallis test was performed. Similar tests were also carried out for suitably log-transformed M8 and active moiety AUC and Cmax values. In addition, approximate 95% confidence intervals (CIs) (adjusted for multiplicity using Bonferroni correction) for the ratio of mean Cmax and AUC levels across the three genotypes were also computed by re-transforming the intervals obtained for pair-wise differences (on the log scale) from the anova.
Results
Demography
Demographic details of study subjects are summarized in Table 1. A total of 68 subjects were enrolled in the study and dosed with study treatments. There were two subjects who discontinued from the study, one due to an adverse event (rash) following completion of the high-dose nelfinavir treatment in Period 1, the other defaulting from the study and not receiving any nelfinavir treatments. Among the subjects completing the study (n = 66), 38 were genotyped as EMs (*1*1), 22 as heterozygous PMs (*1*2), and six subjects as homozygous PMs. One subject was genotyped as *1*3 (same subject who defaulted from the study and did not receive nelfinavir), while another subject was genotyped as *1*4 (omitted from summary statistics). Within each genotype group, most of the subjects were White and about two-thirds of the subjects were male. Age, body weight, and body mass index were similar between the genotype groups (Table 1).
Table 1.
Demographic characteristics of study subjects
| CYP2C19 genotype* | ||||
|---|---|---|---|---|
| Parameter | All subjects | Extensive metabolizer (*1*1) | Heterozygous poor metabolizer (*1*2) | Homozygous poor metabolizer (*2*2) |
| n | 68 | 38 | 22 | 6 |
| Gender, n (%) | ||||
| Male | 44 (64.7) | 25 (65.8) | 13 (59.0) | 4 (66.6) |
| Female | 24 (35.3) | 13 (34.2) | 9 (41.0) | 2 (33.4) |
| Age, years | ||||
| Mean (range) | 32.0 (19.0–55.0) | 31.7 (19.0–54.0) | 32.7 (19.0–54.0) | 25.5 (19.0–37.0) |
| Race, n (%) | ||||
| White | 34 (50.0) | 21 (55.3) | 8 (36.4) | 4 (66.6) |
| Black | 12 (17.6) | 7 (18.4) | 4 (18.2) | 1 (16.7) |
| Asian | 18 (26.5) | 7 (18.4) | 9 (40.9) | 1 (16.7) |
| Other | 4 (5.9) | 3 (7.9) | 1 (4.5) | 0 (0.0) |
| Weight, kg | ||||
| Mean (range) | 72.9 (52.0–96.0) | 73.5 (53.0–92.0) | 72.2 (52.0–96.0) | 70.3 (59.0–82.0) |
| BMI, kg m–2 | ||||
| Mean (range) | 24.6 (18.7–29.8) | 25.0 (18.7–29.8) | 24.5 (20.7–29.0) | 23.2 (21.7–26.6) |
Two subjects were classified as having the *1*3 and *1*4 genotype, and were excluded.
Pharmacokinetics of nelfinavir
The mean plasma concentration–time profiles of nelfinavir on days 1 and 4 after administration of the normal dose are shown in Figure 1. Mean concentrations for EMs (*1*1 subjects) were lower than those observed for heterozygous PMs (*1*2 subjects) and homozygous PMs (*2*2 subjects). On day 4, the differences in mean concentrations between the groups were much more pronounced, and a clear trend in the mean levels was observed as follows: *2*2 > *1*2 > *1*1. Upon administration of the high dose of nelfinavir, similar trends in mean concentration–time profiles for nelfinavir were seen (data not shown).
Figure 1.
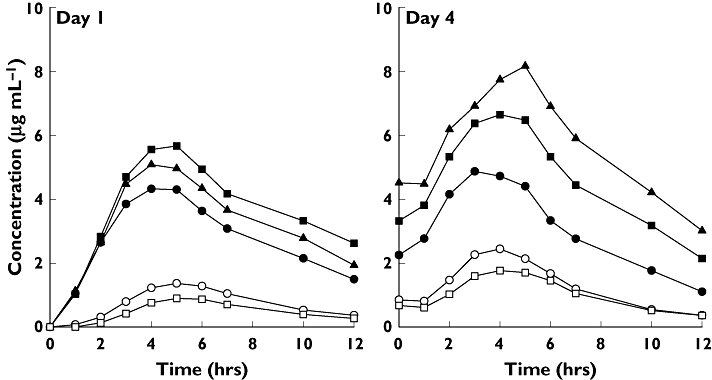
Mean concentration–time profiles of nelfinavir and M8 in CYP2C19 extensive metabolizers (*1*1), heterozygous poor metabolizers (*1*2), and homozygous poor metabolizers (*2*2) following single- and multiple-dose administration of normal dose of nelfinavir. nelfinavir, *1*1 (—•—); nelfinavir, *1*2 ( ); nelfinavir, *2*2 (—▴—); M8, *1*1 (—○—); M8, *1*2 (—□—)
); nelfinavir, *2*2 (—▴—); M8, *1*1 (—○—); M8, *1*2 (—□—)
Pharmacokinetic parameters for nelfinavir are summarized in Table 2. Nelfinavir was absorbed slowly with a median Tmax of 3.0–5.0 h across dose levels, days, and genotype groups (Table 2). Following administration of the normal dose on day 1, the mean Cmax and AUC values of nelfinavir were marginally higher for *1*2 and *2*2 subjects compared with *1*1 subjects. Following administration of the normal dose of nelfinavir on day 4, the mean Cmax was 42% (P < 0.0001) (95% CI 19, 69) and 63% (P = 0.0002) (95% CI 20, 122) higher, and mean AUC was 51% (P < 0.0001) (95% CI 24, 83) and 85% (P < 0.0001) (95% CI 32, 159) higher for *1*2 and *2*2 subjects compared with *1*1 subjects, respectively. The Cmax (P = 0.16) and AUC (P = 0.29) values for *1*2 subjects were not significantly different from *2*2 subjects. The point estimates, CIs and P-values for the ratio of mean Cmax and AUC were obtained by re-transforming the results on the log scale from the anova analysis, and may differ slightly from ratios computed using descriptive statistics as presented in Table 2. Similar trends in the pharmacokinetic parameters were observed across the different genotype subgroups for the high dose of nelfinavir; for example, the mean Cmax values on day 4 were 50 and 68% higher and mean AUC values were 56 and 91% higher for the *1*2 and *2*2 subjects compared with *1*1 subjects, respectively (Table 2).
Table 2.
Mean (SD) pharmacokinetic parameters of nelfinavir by CYP2C19 genotype following single (day 1) and multiple (day 4) dose administration of nelfinavir in healthy subjects
| Normal dose* | High dose* | ||||||
|---|---|---|---|---|---|---|---|
| Parameter (units) | Study day | *1*1†[n = 38] | *1*2†[n = 22] | *2*2†[n = 6] | *1*1 [n = 38] | *1*2 [n = 22] | *2*2 [n = 6] |
| Cmax (µg ml–1) | Day 1 | 4.5 (1.4) | 6.0 (1.1) | 5.1 (1.3) | 4.8 (1.5) | 5.4 (1.8) | 5.8 (1.9) |
| Day 4 | 5.0 (1.4) | 7.1 (1.9) | 8.3 (2.8) | 6.5 (1.8) | 9.8 (2.2) | 10.9 (2.1) | |
| Tmax (h) | Day 1 | 4.0 (2.0, 6.0) | 4.0 (3.0, 6.0) | 4.0 (4.0, 5.0) | 4.0 (2.0, 5.0) | 5.0 (3.0, 6.0) | 4.0 (3.0, 5.0) |
| Day 4 | 3.0 (2.0, 5.0) | 4.0 (2.0, 5.0) | 5.0 (3.0, 5.0) | 4.0 (2.0, 6.0) | 4.0 (2.0, 5.0) | 4.0 (3.0, 5.0) | |
| AUC (µg h–1 ml–1) | Day 1 | 32.5 (10.9) | 43.5 (8.2) | 38.5 (12.8) | 34.1 (10.8) | 39.9 (14.4) | 43.4 (16.1) |
| Day 4 | 36.3 (11.2) | 54.2 (15.1) | 67.9 (24.4) | 48.0 (14.1) | 74.9 (17.6) | 91.9 (20.3) | |
Normal dose was 1250 mg nelfinavir q12h on days 1–3 and the morning dose of 1250 mg on day 4. High dose was 1250 mg nelfinavir q12h on days 1–3 and the morning dose of 3125 mg on day 4.
*1*1, *1*2 and *2*2 genotypes correspond to extensive metabolizer, heterozygous poor metabolizer, and homozygous poor metabolizer phenotypes, respectively.
Nelfinavir accumulated upon multiple dosing. Based on ratio of day 4 to day 1 mean AUC values for the normal dose, the accumulation ratio was 1.12, 1.25 and 1.76 for the *1*1, *1*2 and *2*2 groups, respectively (Table 2). Accumulation ratio could not be determined for the high dose due to difference in the dose on days 1 and 4 (1250 mg q12h on days 1–3 and 3125 mg on day 4). Comparison of AUC values on day 4 for the high dose vs. the normal dose indicated less than dose proportional increase in exposure of nelfinavir. For a 2.5-fold increase in the dose, the mean AUC values were 1.32-, 1.38- and 1.35-fold higher for the *1*1, *1*2 and *2*2 groups, respectively (Table 2).
Pharmacokinetics of M8
The mean plasma concentration–time profiles of M8 on days 1 and 4 after administration of the normal dose are shown in Figure 1. Levels of M8 were markedly lower than nelfinavir, and a clear trend toward reduced mean levels was observed in *1*2 compared with *1*1 subjects. M8 levels were undetectable in all pharmacokinetic samples from all *2*2 subjects. Upon administration of the high dose of nelfinavir, similar trends in mean concentration–time profiles for nelfinavir and M8 were seen (data not shown).
Pharmacokinetic parameters for M8 are summarized in Table 3. M8 had median Tmax values ranging from 4.0 to 5.0 h across dose levels, days, and genotype groups (Table 3). Cmax and AUC values of M8, which is directly formed from nelfinavir via CYP2C19-mediated enzymatic reaction, were significantly reduced in *1*2 subjects compared with *1*1 on days 1 and 4 and for both dose levels. For example, following administration of the normal dose, the mean Cmax and mean AUC value was 35% (P = 0.005) (95% CI 6, 55) and 33% (P = 0.02) (95% CI −3, 56) lower on day 4, respectively, for *1*2 subjects compared with *1*1 subjects. Consistent with the undetectable levels of M8 in *2*2 subjects, the Cmax and AUC values for this group were significantly different from *1*1 subjects. The point estimates, CIs and P-values for the ratio of mean Cmax and AUC were obtained by re-transforming the results on the log scale from the anova analysis and may differ slightly from ratios computed using descriptive statistics as presented in Table 3. M8 also accumulated upon multiple dosing; the accumulation ratio for the normal dose, based on mean AUC value on day 4 vs. day 1, was 1.79 and 2.22 for *1*1 and *1*2 subjects, respectively.
Table 3.
Mean (SD) pharmacokinetic parameters of M8 by CYP2C19 genotype following single (day 1) and multiple (day 4) dose administration of nelfinavir in healthy subjects
| Normal dose* | High dose* | ||||||
|---|---|---|---|---|---|---|---|
| Parameter (units) | Study day | *1*1†[n = 38] | *1*2†[n = 22] | *2*2†[n = 6] | *1*1 [n = 38] | *1*2 [n = 22] | *2*2 [n = 6] |
| Cmax (µg ml–1) | Day 1 | 1.4 (0.8) | 0.9 (0.4) | 0.0 (0.0)‡ | 1.5 (0.7) | 0.8 (0.5) | 0.0 (0.0)‡ |
| Day 4 | 2.5 (0.9) | 1.8 (0.9) | 0.0 (0.0)‡ | 3.4 (1.2) | 2.8 (1.3) | 0.0 (0.0)‡ | |
| Tmax (h) | Day 1 | 5.0 (3.0, 7.0) | 5.0 (4.0, 7.0) | –‡ | 5.0 (3.0, 6.0) | 5.0 (4.0, 6.0) | –‡ |
| Day 4 | 4.0 (3.0, 5.0) | 4.0 (3.0, 6.0) | –‡ | 4.0 (3.0, 6.0) | 5.0 (3.0, 5.0) | –‡ | |
| AUC (µg h–1 ml–1) | Day 1 | 8.5 (5.6) | 5.3 (2.6) | 0.0 (0.0)‡ | 9.0 (4.5) | 4.8 (3.0) | 0.0 (0.0)‡ |
| Day 4 | 15.2 (6.3) | 11.8 (5.8) | 0.0 (0.0)‡ | 21.2 (7.8) | 18.2 (8.4) | 0.0 (0.0)‡ | |
Normal dose was 1250 mg nelfinavir q12h on days 1–3 and the morning dose of 1250 mg on day 4. High dose was 1250 mg nelfinavir q12h on days 1–3 and the morning dose of 3125 mg on day 4.
*1*1, *1*2 and *2*2 genotypes correspond to extensive metabolizer, heterozygous poor metabolizer and homozygous poor metabolizer phenotypes, respectively.
No quantifiable concentration was observed in any *2*2 subjects, hence Cmax and AUC values are zero and Tmax could not be determined.
Metabolic ratios were determined based on the AUC of metabolite vs. the AUC of parent drug. The metabolic ratio for the normal dose was 0.42 and 0.21 for the EMs and heterozygote PMs; similar ratios were seen with the high-dose group of nelfinavir.
Pharmacokinetics of active moiety
Cmax and AUC values for the active moiety are summarized in Table 4. The mean AUC values on day 4 for both dose levels were higher by 29% and 34% for the *1*2 (P = 0.001) and *2*2 (P = 0.04) subjects compared with *1*1 subjects. Of note, the mean AUC values for the active moiety were similar between the heterozygous PMs (*1*2) and the homozygous PMs (*2*2) (P = 0.92) (Table 4).
Table 4.
Mean (SD) pharmacokinetic parameters of active moiety by CYP2C19 genotype following single (day 1) and multiple (day 4) dose administration of nelfinavir in healthy subjects
| Normal dose* | High dose* | ||||||
|---|---|---|---|---|---|---|---|
| Parameter (units) | Study day | *1*1†[n = 38] | *1*2†[n = 22] | *2*2†[n = 6] | *1*1 [n = 38] | *1*2 [n = 22] | *2*2 [n = 6] |
| Cmax (µg ml–1) | Day 1 | 10.4 (3.4) | 12.0 (2.2) | 9.0 (2.2) | 11.1 (3.3) | 10.8 (3.4) | 10.1 (3.4) |
| Day 4 | 13.0 (3.3) | 15.6 (4.2) | 14.5 (4.9) | 17.3 (4.4) | 21.9 (5.2) | 19.3 (3.6) | |
| AUC (µg h–1 ml–1) | Day 1 | 71.5 (26.3) | 85.5 (15.3) | 67.8 (22.5) | 75.1 (23.6) | 78.3 (27.4) | 76.5 (28.4) |
| Day 4 | 89.2 (26.6) | 115.2 (30.9) | 119.5 (43.0) | 120.0 (32.5) | 162.4 (35.8) | 161.9 (35.8) | |
Normal dose was 1250 mg nelfinavir q12h on days 1–3 and the morning dose of 1250 mg on day 4. High dose was 1250 mg nelfinavir q12h on days 1–3 and the morning dose of 3125 mg on day 4.
*1*1, *1*2 and *2*2 genotypes correspond to extensive metabolizer, heterozygous poor metabolizer, and homozygous poor metabolizer phenotypes, respectively.
A scatter plot of the individual subject AUC values for nelfinavir, M8 and active moiety following administration of the normal dose on day 4 is presented in Figure 2. These data suggested that the hetero- and homozygous PMs had incrementally higher exposure of nelfinavir and, conversely, lower exposure to M8 compared with EMs. The exposure to active moiety was modestly higher in PMs compared with EMs. However, there appeared to be no difference between the hetero- and homozygous PMs in their exposure to the active moiety.
Figure 2.
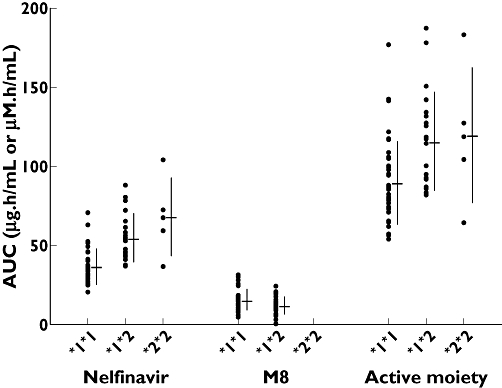
AUC values of nelfinavir, M8 and active moiety by CYP2C19 genotype following administration of normal dose of nelfinavir on day 4. CYP2C19 genotypes are denoted as follows: extensive metabolizer (*1*1), heterozygous poor metabolizer (*1*2) and homozygous poor metabolizer (*2*2). Points represent individual subject value and adjacent line indicates mean (horizontal line) and standard deviation (vertical line)
Discussion
The main findings of the present study were as follows: (i) the exposure of nelfinavir was elevated in heterozygous PMs (*1*2) and homozygous PMs (*2*2) in an incremental manner consistent with the loss of functional alleles; (ii) the exposure of active metabolite, M8, was reduced in heterozygous PMs, whereas M8 was undetectable in homozygous PMs; and (iii) the exposure of active moiety was only modestly (30–35%) elevated in PMs. These findings were similar across different doses of nelfinavir. Statistical analysis using a parametric, one-way anova showed significant differences in Cmax and AUC values across the three genotypes. The evidence was similar using a nonparametric Kruskal–Wallis test. In addition, pair-wise comparisons from the anova model revealed significant differences in pharmacokinetics between *1*1 vs.*1*2 and *2*2 subjects, but showed that the pharmacokinetics were not significantly different between *1*2 and *2*2 for nelfinavir and the active moiety.
Studies characterizing the effect of CYP2C19 genotype on nelfinavir pharmacokinetics have done so in terms of nelfinavir and M8 exposures or with metabolite/parent ratio. However, there has been a lack of clarity on this topic for two reasons. Some studies have combined data from heterozygous (*1*2) and homozygous (*2*2) PMs rather than presenting them separately [10, 11]. Others have arbitrarily set M8 concentration values that were below the assay limit of quantification (LOQ) as a proportion of the LOQ, for the purpose of calculating metabolite/parent ratio [7, 10]. Our data, and those from others [7], show that homozygous PMs do not have detectable M8 concentrations. Instead of calculating metabolite/parent ratio based on arbitrarily determined M8 levels, a more appropriate approach may be to calculate active moiety (parent plus metabolite, expressed as molar units). Nelfinavir and M8 are both highly (>99%) protein bound, and therefore no adjustments were made for protein binding for calculating the active moiety concentration [16]. Furthermore, it is clear that heterozygous PMs do produce M8, although at lower levels than EMs [7, 8, 10]. Based on this difference (reduced vs. no M8 production), it may not be appropriate to combine homozygous and heterozygous PMs [10, 11] when analysing the effects of genotype on nelfinavir pharmacokinetics.
To compare the results from the present study with those reported previously, the key differences observed in the pharmacokinetics of nelfinavir and M8 across genotypes among various studies are summarized in Table 5. The influence of CYP2C19 polymorphism on the pharmacokinetics of nelfinavir was not consistent across studies (Table 5). Nelfinavir exposures were increased in the present study and the Haas et al. study [7] due to mutations in CYP2C19 compared with EMs. In contrast, the other studies that enrolled smaller cohort of subjects (Burger et al.[8] and Fellay et al.[9]) were unable to differentiate any changes due to *1*2 mutation, or had too few *2*2 subjects to draw meaningful comparison with *1*1 subjects regarding the pharmacokinetics of nelfinavir. Differences in nelfinavir exposure were also not detected by Hirt et al.[10] and Saitoh et al.[11], although these two studies grouped heterozygous and homozygous PMs together. It is likely that the moderate increases in nelfinavir exposure or the unchanged levels of nelfinavir in heterozygous PMs may indicate increased clearance via alternative metabolic pathways (e.g. CYP3A4 and -5), as suggested by Hirt et al.[10]. The differences between studies in detecting changes in nelfinavir levels due to genetic variations in CYP2C19 may be dependent on several factors such as the use of healthy subjects vs. HIV-infected patients, and sparse sampling with population pharmacokinetic analysis vs. intensive sampling with noncompartmental analyses. Other factors that could influence the variability in nelfinavir pharmacokinetics, thus making it difficult to detect differences, are concomitant medications (especially in HIV patients) and caloric/fat content of the food. Smaller sample sizes, especially for the *2*2 subjects, also limit the interpretation of nelfinavir data. In contrast to nelfinavir, the influence of CYP2C19 polymorphism on the pharmacokinetics of M8 was consistently observed across studies (Table 5). M8 was undetectable in *2*2 subjects, and detectable but lower exposures were observed in *1*2 subjects. The M8 data are consistent with previous reports that CYP2C19 is mainly responsible for conversion of nelfinavir to M8 [3]. Therefore, *1*2 subjects with one active allele produce reduced amounts of M8 compared with *1*1 subjects, whereas *2*2 subjects with no active alleles produce no M8.
Table 5.
Summary of changes in nelfinavir and M8 pharmacokinetics in heterozygous (*1*2) and homozygous (*2*2) poor metabolizers relative to extensive metabolizers (*1*1) in the present study and in published studies
| Change* in NFV levels relative to *1 *1 | Change in M8 levels relative to *1*1 | |||
|---|---|---|---|---|
| Study [reference no.] | *1*2 | *2*2 | *1*2 | *2*2 |
| Present study | ↑.556% (n = 22) | ↑.591% (n = 6) | ↓ | M8 was undetectable |
| Burger et al. 2006 [8] | ↔ (n = 8) | Not enrolled | ↓ | Not enrolled |
| Fellay et al. 2002 [9] | ↔ (n = 11) | ↔ (n = 2) | Not studied | Not studied |
| Haas et al. 2005 [7] | ↑.525% (n = 85) | ↑.536% (n = 10) | ↓ | M8 was undetectable |
| Hirt et al. 2008 [10] | ↔ (n = 13)† | ↓ | ||
| Saitoh et al. 2005 [11] | ↔ (n = 22)‡ | Not studied | ||
Change in nelfinavir (NFV) levels was assessed based on AUC or apparent oral clearance at steady-state. ↔, similar; ↑, increased; ↓, decreased.
Data from *1*2 (n = 11) and *2*2 (n = 2) were combined.
Data from *1*2 (n = 21) and *2*2 (n = 1) were combined.
Since M8 is an active metabolite, and has in vitro activity similar to that of nelfinavir, we examined the change in the active moiety. Interestingly, these data indicated that genetic polymorphism in the CYP2C19 enzyme had modest (30–35%), albeit statistically significant, influence in the systemic exposure of the active moiety compared with the marked changes in nelfinavir and M8. In other words, the increased level of nelfinavir in CYP2C19 variants was counterbalanced by decreased formation of the active metabolite, M8. The increase in nelfinavir exposure in subjects with one or no active alleles is unlikely to be clinically meaningful. Wyles and Gerber [17] have noted that studies to date showing increased exposure of nelfinavir in HIV patients co-infected with hepatitis C virus or having chronic liver disease [18, 19] have not reported increased systemic toxicity or decreased clinical tolerance. This finding may be secondary to the fact that the adverse effects of nelfinavir are mainly localized within the gastrointestinal tract, and that increasing systemic plasma concentrations may not be associated with increased toxicity.
Other findings from this study include confirmation of the nonlinear pharmacokinetics at higher doses, in which systemic exposure increases in a less than dose-proportional manner. The mechanistic basis for this is probably due to solubility-limited absorption of orally administered nelfinavir [14]. Supporting this mechanism are the pharmacokinetic data indicating that genetic mutation in CYP2C19 did not influence the nonlinearity, as the exposure increment was 32–38% across the various genotypes for a 2.5-fold increase in dose. Despite the nonlinear pharmacokinetics, the observed changes in nelfinavir exposure due to polymorphism in CYP2C19 were consistent between high and normal dose levels. An interesting observation from this study is that the accumulation of nelfinavir after multiple dosing may be dependent on CYP2C19 mutation. Whereas *1*1 and *1*2 subjects had minimal accumulation (11 and 25%, respectively), *2*2 subjects had considerably higher nelfinavir exposures (76%) after multiple doses compared with the first dose. Given the small number of *2*2 subjects, this requires confirmation in a larger study.
In summary, the results from this study suggest that whereas nelfinavir levels were increased and M8 levels were decreased due to mutation in CYP2C19, exposure to activity moiety was only modestly affected by CYP2C19 polymorphism. No significant differences were noted among the heterozygous and homozygous PMs. These changes are not considered to be clinically meaningful, and hence the use of nelfinavir does not require prior assessment of CYP2C19 genotype.
Competing interests
All authors are employees of Pfizer Inc., and hold stock in Pfizer Inc. This study was sponsored by Pfizer Inc.
REFERENCES
- 1.Viracept® (nelfinavir mesylate) US Package Insert, Pfizer Inc., July 2007.
- 2.Perry CM, Frampton JE, McCormack PL, Siddiqui MA, Cvetkovic RS. Nelfinavir: a review of its use in the management of HIV infection. Drugs. 2005;65:2209–44. doi: 10.2165/00003495-200565150-00015. [DOI] [PubMed] [Google Scholar]
- 3.Hirani VN, Raucy JL, Lasker JM. Conversion of the HIV protease inhibitor nelfinavir to a bioactive metabolite by human liver CYP2C19. Drug Metab Dispos. 2004;32:1462–67. doi: 10.1124/dmd.104.001743. [DOI] [PubMed] [Google Scholar]
- 4.Zhang KE, Wu E, Patrick AK, Kerr B, Zorbas M, Lankford A, Kobayashi T, Maeda Y, Shetty B, Webber S. Circulating metabolites of the human immunodeficiency virus protease inhibitor nelfinavir in humans: structural identification, levels in plasma, and antiviral activities. Antimicrob Agents Chemother. 2001;45:1086–93. doi: 10.1128/AAC.45.4.1086-1093.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desta Z, Zhao X, Shin J-G, Flockhart DA. Clinical significance of the cytochrome P4502C19 genetic polymorphism. Clin Pharmacokinet. 2002;41:913–58. doi: 10.2165/00003088-200241120-00002. [DOI] [PubMed] [Google Scholar]
- 6.Wedlund PJ. The CYP2C19 enzyme polymorphism. Pharmacology. 2000;61:174–83. doi: 10.1159/000028398. [DOI] [PubMed] [Google Scholar]
- 7.Haas DW, Smeaton LM, Shafer RW, Robbins GK, Morse GD, Labbe L, Wilkinson GR, Clifford DB, D'Aquila RT, De Gruttola V, Pollard RB, Merigan TC, Hirsch MS, George AL, Jr, Donahue JP, Kim RB. Pharmacogenetics of long-term responses to antiretroviral regimens containing efavirenz and/or nelfinavir: an Adult AIDS Clinical Trials Group Study. J Infect Dis. 2005;192:1931–42. doi: 10.1086/497610. [DOI] [PubMed] [Google Scholar]
- 8.Burger DM, Schwietert HR, Colbers EP, Becker M. The effect of the CYP2C19*2 heterozygote genotype on the pharmacokinetics of nelfinavir. Br J Clin Pharmacol. 2006;62:250–2. doi: 10.1111/j.1365-2125.2006.02635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fellay J, Marzolini C, Meaden ER, Back DJ, Buclin T, Chave JP, Decosterd LA, Furrer H, Opravil M, Pantaleo G, Retelska D, Ruiz L, Schinkel AH, Vernazza P, Eap CB, Telenti A, for the Swiss HIV Cohort Study Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet. 2002;359:30–6. doi: 10.1016/S0140-6736(02)07276-8. [DOI] [PubMed] [Google Scholar]
- 10.Hirt D, Mentre F, Tran A, Rey E, Auleley S, Salmon D, Duval X, Treluyer JM COPHAR2-ANRS Study Group. Effect of CYP2C19 polymorphism on nelfinavir to M8 biotransformation in HIV patients. Br J Clin Pharmacol. 2008;65:548–57. doi: 10.1111/j.1365-2125.2007.03039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saitoh A, Singh KK, Powell CA, Fenton T, Fletcher CV, Brundage R, Starr S, Spector SA. An MDR1-3435 variant is associated with higher plasma nelfinavir levels and more rapid virologic response in HIV-1 infected children. AIDS. 2005;19:371–80. doi: 10.1097/01.aids.0000161766.13782.2f. [DOI] [PubMed] [Google Scholar]
- 12.Damle B, Fosser C, Ito K, Tran A, Clax P, Uderman H, Glue P. Effects of standard and supratherapeutic doses of nelfinavir on cardiac repolarization: a thorough QT study. J Clin Pharmacol. 2009;49:291–300. doi: 10.1177/0091270008329551. [DOI] [PubMed] [Google Scholar]
- 13.E14 Implementation Working Group Questions & Answers. June 4, 2008. Available at http://www.ich.org/LOB/media/MEDIA4719.pdf (last accessed 4 August 2009.
- 14.Fang AF, Damle BD, LaBadie RR, Crownover PH, Hewlett D, Jr, Glue PW. Significant decrease in nelfinavir systemic exposure after omeprazole coadministration in healthy subjects. Pharmacotheraypy. 2008;28:42–50. doi: 10.1592/phco.28.1.42. [DOI] [PubMed] [Google Scholar]
- 15.Myrand SP, Sekiguchi K, Man MZ, Lin X, Tzeng RY, Teng CH, Hee B, Garrett M, Kikkawa H, Lin CY, Eddy SM, Dostalik J, Mount J, Azuma J, Fujio Y, Jang IJ, Shin IG, Bleavins MR, Williams JA, Paulauskis JD, Wilner K. Pharmacokinetics/ genotype associations for major cytochrome P450 enzymes in native and first- and third-generation Japanese populations: comparison with Korean, Chinese and Caucasian populations. Clin Pharmacol Ther. 2008;84:347–61. doi: 10.1038/sj.clpt.6100482. [DOI] [PubMed] [Google Scholar]
- 16.Aweeka F, Motoya T, Thevanayagam L, Blaschke T, Stone J, Jayewardene A, Chi J. Nelfinavir unbound drug interactions and protein binding characteristics. 9th Conference on Retrovirous and Opportunistic Infections. Seattle WA. 2002. [Abstract 448-W.
- 17.Wyles DL, Gerber JG. Antiretroviral drug pharmacokinetics in hepatitis with hepatic dysfunction. Clin Infect Dis. 2005;40:174–81. doi: 10.1086/426021. [DOI] [PubMed] [Google Scholar]
- 18.Regazzi M, Maserati R, Villani P, Cusato M, Zucchi P, Briganti E, Roda R, Sacchelli L, Gatti F, Foglie PD, Nardini G, Fabris P, Mori F, Castelli P, Testa L. Clinical pharmacokinetics of nelfinavir and its metabolite M8 in human immunodeficiency virus (HIV)-positive and HIV-hepatitis C virus-coinfected subjects. Antimicrob Agents Chemother. 2005;49:643–9. doi: 10.1128/AAC.49.2.643-649.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peytavin G, Landman R, Lamotte C, Trylesinski A, Legac S, Mentre F, Yeni P. Therapeutic drug monitoring of nelfinavir in a prospective study (LIVIR IMEA 014) in HIV-HCV co-infected patients with chronic liver disease) 1st International AIDS Society Conference on HIV Pathogenesis and Treatment; Buenos Aires, Argentina. 2001 [Abstract 349.