

Abstract
AIMS
To develop a population pharmacokinetic–pharmacodynamic model to describe the occurrence and severity of bleeding or bruising as a function of enoxaparin exposure.
METHODS
Data were obtained from a randomized controlled trial (n = 118) that compared conventional dosing of enoxaparin (product label) with an individualized dosing regimen. Anti-Xa concentrations were sampled using a sparse design and the size, location and type of bruising and bleeding event, during enoxaparin therapy, were collected daily. A population pharmacokinetic–pharmacodynamic analysis was performed using nonlinear mixed effects techniques. The final model was used to explore how the probability of events in patients with obesity and/or renal impairment varied under differing dosing strategies.
RESULTS
Three hundred and forty-nine anti-Xa concentrations were available for analysis. A two-compartment first-order absorption and elimination model best fit the data, with lean body weight describing between-subject variability in clearance and central volume of distribution. A three-category proportional-odds model described the occurrence and severity of events as a function of both cumulative enoxaparin AUC (cAUC) and subject age. Simulations showed that individualized dosing decreased the probability of a bleeding or major bruising event when compared with conventional dosing, which was most noticeable in subjects with obesity and renal impairment.
CONCLUSIONS
The occurrence and severity of a bleeding or major bruising event to enoxaparin, administered for the treatment of a thromboembolic disease, can be described as a function of both cAUC and subject age. Individualized dosing of enoxaparin will reduce the probability of an event.
Keywords: anticoagulants, enoxaparin, exposure-response modelling, obesity, renal disease
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Enoxaparin is an effective anticoagulant used to treat thromboembolic diseases.
Dose individualization of enoxaparin based on renal impairment and lean body weight reduces bleeding and bruising end-points for renally compromised and obese subjects.
Scant data exist that quantify the dose–exposure–adverse event relationship for enoxaparin, especially in subjects with renal impairment and/or obesity.
WHAT THIS STUDY ADDS
The occurrence and severity of a bleeding or bruising event is described as a function of both cumulative enoxaparin AUC and subject age.
When compared with conventional (product label) dosing, dose individualization of enoxaparin based on lean body weight and renal function reduces the probability of a bleeding or bruising event.
Introduction
Enoxaparin is a low-molecular-weight heparin that is proven to be at least as effective as unfractionated heparin (UFH) for the treatment of acute coronary syndromes (ACS) [1–4] and other thromboembolic disorders [5, 6]. Enoxaparin has linear pharmacokinetics and a purported predictable dose–exposure–response relationship that negates the need for therapeutic drug monitoring. The cited dose in contemporary clinical guidelines [7–9] and the approved product label is 1 mg kg−1 twice a day or 1.5 mg kg−1 once a day, based on total body weight (Wt), with a dose reduction to 1 mg kg−1 once a day when the subject's estimated creatinine clearance (CLCR) is <30 ml min−1[10–12].
Despite strong data to support enoxaparin's effectiveness, little information exists that quantifies the risk of bleeding events as a function of drug exposure. Observational data collected during the TIMI 11A study [13] demonstrated that an increase in dose from 1 mg kg−1 twice a day to 1.25 mg kg−1 twice a day elevated major bleeding events from 1.9% [95% confidence interval (CI) 0.80, 4.4] to 6.5% (95% CI 4.2, 10.0), respectively, with no improvement in effectiveness. The median (range) trough anti-Xa concentrations, before the third weight-adjusted dose, were reported to be 0.5 IU ml−1 (0.3–0.7) and 0.6 IU ml−1 (0.3–1.0) for the 1 mg kg−1vs. the 1.25 mg kg−1 dose, respectively, with the peak (3–5 h post dose) concentrations reported as 1.0 IU ml−1 (0.9–1.2) and 1.5 IU ml−1 (1.2–1.7), respectively. These trial data were pivotal in supporting the dose label of 1 mg kg−1 based on Wt.
The clearance (CL) of enoxaparin is described by a composite of renal elimination and metabolism [14]. Both of these processes have been shown to be proportional to lean body weight (LBW) [14, 15]. Subjects with renal impairment and/or obesity are therefore at risk of excessive inhibition of factor-Xa and increased bleeding events if doses are not adjusted accordingly [16]. Many clinicians recognize the limitations of the product label and have reduced doses in an effort to reduce the likelihood of an adverse event [16]. This practice is of concern given that subtherapeutic anti-Xa concentrations have been associated with an increase in mortality [17]. Despite these empirical observations and the intuitive link between enoxaparin exposure and bleeding, quantification of the probability of a bleeding event has not been adequately performed.
We aimed to develop a pharmacokinetic–pharmacodynamic (PK–PD) model to describe the occurrence and severity of bleeding and bruising events as a function of drug exposure, then use this model to explore how the probability of events will vary in patients with obesity and/or renal impairment under dosing strategies that were used in a recent randomized controlled trial (RCT). This trial demonstrated that dose individualization, as a function of obesity and renal function, reduced the relative risk of bleeding to 0.12 (P = 0.03) [16].
Methods
Study population
Details of the data used for this analysis have been extensively described elsewhere [16]. Briefly, PK and PD data were collected during a prospective RCT from subjects treated for pulmonary embolism, deep vein thrombosis, ACS or atrial fibrillation who were allocated to either a dose-individualized or conventional dosing arm. Subjects in the conventional arm received labelled dose regimens, as recommended by the prescriber. Subjects in the individualized arm were dosed according to either Wt or LBW: subjects ≥100 kg (considered obese) using LBW (1.5 mg kg−1 twice a day), subjects <100 kg using Wt (1 mg kg−1 twice a day). Subjects with a CLCR < 50 ml min−1 were dose reduced at 48 h of therapy based on renal function.
Subjects were not eligible for inclusion in the trial if their liver enzymes (aspartate aminotransferase, alanine aminotransferase, γ-glutamyl transferase) were greater than twice normal range; were pregnant; were <18 years of age; had been administered warfarin or heparin therapy in the past 7 days; had an international normalized ratio >1.2; had an activated partial thromboplastin time >60 s; had a CLCR < 10 ml min−1, calculated using the Cockcroft–Gault (C-G) equation [18]; or were receiving haemodialysis.
Data collection
Pharmacokinetic data
A total of 118 subjects were available for analysis. Plasma samples were collected during time windows determined using D-optimality to ensure PK parameters could be estimated with good precision [19]. Four samples were collected: one predose and then one at 15–30, 60–120 and 180–300 min post dose. Venous blood was collected in sodium-citrate tubes and the measurement of anti-Xa activity performed using a validated chromogenic assay by the Queensland Health Pathology Service (STA-Rotachrom®; Diagnostica Stago, Asnieres, France), with a lower limit of assay detection of 0.1 IU ml−1 and an interassay precision (% CV) of 5.9%.
Pharmacodynamic data
Bleeding events were classified using the following criteria: a decrease in haemoglobin >30 g l−1 in the absence of overt bleeding, evidence of an internal bleed as defined in Phase III studies [1, 2]; any overt bleed such as haematemesis, epistaxis, haematuria, injection or a venepuncture site bleed. A major bruise was described as a bruise that had a surface area ≥20 cm2, a criterion matched to a post hoc analysis of the ESSENCE study [20]. Minor bruises were described as those ≥1 cm2 and <20 cm2. Bruises <1 cm2 were considered uninformative with the potential to be influenced by factors such as injection technique and were therefore excluded.
Bleeding and bruising events were identified by a dedicated research nurse who assessed the subject's body and medical notes. The subject was reviewed upon initial recruitment and daily thereafter until enoxaparin therapy was ceased. Only new events following enrolment (baseline) were recorded. Additional drug therapy was not influenced by the study investigators.
Population PK–PD modelling
The PK–PD analysis was carried out using the nonlinear mixed-effect modelling program NONMEM (version V.1.1; with a G77 FORTRAN compiler and Wings for NONMEM) [21]. The PK analysis was undertaken using the first order conditional estimation method with the interaction option to estimate PK–PD parameters.
Pharmacokinetic model
Base heterogeneity and residual error model
To identify the best structural model, one-, two- and three-compartmental models were evaluated together with a variety of first and zero order input models. During model development, PK parameters were assumed to vary between individuals according to a log-normal distribution. Proportional, additive and combined error models were evaluated to explain random unexplained variability. Between-occasion variability was not considered in the analysis as each subject provided only one profile of blood samples.
Covariate model
Model development and covariate selection were based on the hydrophilic properties of enoxaparin, prior population PK studies (Table 1) and previous research on body size descriptors by the authors [22–24]. For example, Green et al. conducted a meta-analysis of studies to quantify the relationship between PK parameters and body size descriptors and found that CL of drugs was best described by LBW [22]. This was confirmed by Han et al., who demonstrated that CL of drugs is best described as a function of LBW [24] and not Wt, especially for subjects of varying body compositions and particularly for the obese [23]. It should be noted that at the time of the RCT, LBW was calculated according to the method described in Green et al.[15]; however, the formula by Janmahasatian has been shown to be superior at describing the CL of enoxaparin at extremes of body compositions [24].
Table 1.
Recent population pharmacokinetic (PK) and pharmacokinetic–pharmacodynamic studies of enoxaparin
| Author | N | Year | Compartments in PK model | Final body size descriptor on Vc | Final covariates on CL | Body size descriptor used in CLCR calculation | BSV (% CV) on CL (final model) | BSV (% CV) Vc (final model) | RUV (IU ml−1‡ or % CV§) |
|---|---|---|---|---|---|---|---|---|---|
| Bruno [31] | 448 | 2002 | 1 | Nil | Wt, CLCR | Wt | 26.9 | 56.1* | 21.5% |
| Hulot [30] | 60 | 2004 | 1 | Wt* | Wt, SCr gender | N/A | 23 | 54* | 0.15 |
| Hulot [40] | 532 | 2005 | 1 | Wt* | Wt, SCr gender | N/A | 27 | 50* | 0.14 |
| Sanchez-Pena [42] | 546 | 2005 | 1 | Wt* | Wt | N/A | 33 | 30* | 0.09 |
| Kane-Gill [43] | 67 | 2005 | 1 | Nil | Wt | N/A | 70 | 57* | N/P |
| Green [15] | 96 | 2002 | 2 | Wt | LBW | N/A | 35.6 | 58 | 0.08 |
| Green [14] | 38 | 2004 | 2 | Wt | CLCR† | IBW | 32.7 | 34.4 | 0.05, 20%¶ |
| Berges [44] | 189 | 2007 | 2 | Wt | Wt, CLCR | MDRD | 26 | 15 | 30% |
| Feng [29] | 48 | 2006 | 2 | Wt | CLCR† | IBW | 40.7 | 29.4 | 0.13, 12%¶**44%†† |
| Barras | 118 | 2009 | 2 | LBW | CLCR† | LBW | 37.8 | 35.6 | 0.09 |
Vc, central volume of distribution; CL, clearance; N, number of subjects in the study; BSV, between-subject variability; RUV, residual unexplained variability; CLCR, creatinine clearance calculated using the Cockcroft–Gault equation [18]; Wt, total body weight; N/A, not applicable; SCr, serum creatinine concentration; LBW, lean body weight; IBW, ideal body weight; MDRD, modification of diet in renal disease formula; N/P, not presented; Year, year of publication.
One-compartment model, therefore results represent total volume of distribution.
Denotes combined renal and nonrenal clearance model.
Additive residual error model.
Proportional residual error model.
Combined additive and proportional residual error model.
General medical subjects.
Intensive care subjects.
Covariates considered included Wt, ideal body weight (IBW), LBW, age, sex and CLCR. CLCR was calculated according to the C-G equation [18] and where Wt, IBW and LBW were included separately into the equation. LBW was calculated using the formula by Janmahasatian [24] where:
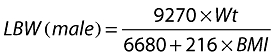 |
(1.1) |
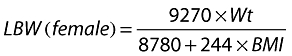 |
(1.2) |
and BMI = body mass index.
Continuous variables were standardized to data-derived median values and included in the model in a proportional, combined renal and nonrenal, exponential and allometrically scaled (exponent fixed to 0.75) design.
Data below the level of quantification
As there were only six samples (1.7%) below the level of quantification (LOQ), they were excluded in the initial model building phase. As per Beal [25], the data were added at 50% LOQ (0.05 IU ml−1) after development of the final model to test for any significant change in parameter estimates.
PK–PD model
Structural model
The PK–PD analysis was conducted in a sequential format by fixing individual PK parameters as described by Zhang [26], with exposure variables of interest computed from the individual empirical Bayes estimates of the parameters. To quantify the exposure–response relationship, a proportional-odds model was constructed to predict the severity of an event. Three levels of severity, S = 1, 2, and 3 (Table 2), were evaluated and the probability (P) of an event less than or equal to an event of grade S expressed as:
Table 2.
Event severity categories for the proportional-odds model
| Category | Event description | N (103) |
|---|---|---|
| S1 | No event (no bruise or a bruise with a surface area <1 cm2) | 36 |
| S2 | Minor bruising event (a bruise with a surface area 1 cm2 ≤ bruise size <20 cm2) | 40 |
| S3 | Bleed or major bruising event (a bleed or bruise with a surface area ≥20 cm2) | 27 |
| (2) |
where N is the level of severity, α and β are parameters to be estimated in the model and × the independent variables.
Model building
Random effects were not included in the proportional odds model due to the small number of multiple events. If multiple events did occur, the event of greatest severity throughout each subject's course of treatment was chosen for analysis. As treatment was ceased after each bleeding event, modelling of repeated measures was not considered.
Multiple exposure measures of enoxaparin were evaluated in the model and included area under the concentration–time curve in the first 24 h (AUC0–24), cumulative under the concentration–time curve (cAUC) from first dose to the time of the event, maximum concentration (Cmax) and minimum concentration (Cmin). The values for Cmax and Cmin were calculated from individual PK model-predicted anti-Xa concentrations using individual PK estimates. The values for AUC0–24 and cAUC were calculated using NONMEM [21], where AUC was allowed to accumulate over time in a hypothetical compartment. The maximum values of Cmax and Cmin during dosing that preceded the event were used in the analysis. Demographic variables such as age, sex, estimated CLCR (using the C-G equation), Wt, IBW and LBW [24] were evaluated.
Model discrimination and evaluation
A series of diagnostics was used to discriminate between competing models. The likelihood ratio test was used with a decrease in the objective function (OF) value (minus twice the log-likelihood of the data) ≥6.61 points (χ2 distribution with one degree of freedom for P < 0.01) required for a nested model to be preferred. Preferred models also had to have a reduction in between-subject variability (BSV). During the covariate analysis, in the event that multiple covariates were included in the model, backward elimination was conducted where a covariate was removed from the model if the increase in OF was <10.83 points (P < 0.001). Goodness of fit plots were used to aid model development, including population-predicted vs. observed concentrations, weighted residual, and unexplained variability in the parameter estimates (ETA) plots [27]. The proportional odds assumption was tested using a score test.
The final PK and PK–PD models were evaluated using a nonparametric bootstrap to quantify uncertainty in the parameter estimates [28]. The median parameter values and nonparametric 95% CI were derived from 1000 bootstraps and compared with the final parameter estimates and asymptotic 95% CI. The final model-based probabilities of a bleeding or major bruising event were compared with the empirical probabilities determined from the observed data.
Visual predictive checks (VPCs) were performed to explore the predictive capability of the final PK model. As the confirmatory RCT [16] investigated subjects with obesity (Wt ≥ 100 kg) and renal impairment (CLCR < 50 ml min−1), these subject groups were examined in separate VPCs. For all VPCs, 500 datasets that mirrored the demographics of subjects recruited in the clinical trial were simulated using the final model parameter estimates. The 10th, 50th and 90th prediction intervals from the simulated concentrations were plotted against time post first dose, with the observed data superimposed.
Demonstration of the inference of the PK–PD model
To apply the final PK–PD model, deterministic PD simulations were performed to compare the individualized and conventional dose strategies from the RCT. The probability of a bleed or major bruising event over time was simulated using the final PK parameter estimates linked to the PD model parameter estimates in three hypothetical subjects, each of whom was dosed using the individualized or conventional dosing strategy: a subject with a CLCR = 30 ml min−1 (at median Wt); a subject with a Wt = 150 kg (at median CLCR); and a subject with both demographic characteristics. A CLCR of 30 ml min−1 was chosen as this matches the CLCR at which a dose reduction occurs in the product label and a Wt of 150 kg was an arbitrary selection to represent an extremely obese subject. As standard duration of enoxaparin therapy is 72 h for ACS [7] and 96 h for other embolic events [9], the probabilities of an event at these times were compared.
Results
PK analysis
Model building
The demographics of subjects recruited are shown in Table 3. Subjects had a median (range) Wt of 77 kg (43–120) and median (range) CLCR of 85 ml min−1 (15–244). A total of 349 anti-Xa concentrations was collected during the study with a mean (range) of three (one to four) samples per subject [16], with sampling primarily occurring in the first 48 h of therapy (93% of concentrations). A two-compartment model with first-order input and linear elimination was the best structural model to fit the data. BSV was included on CL, central volume of distribution (Vc) and absorption rate (Ka) with Vc and CL allowed to co-vary. Residual variability was described by an additive error model. The base model parameter estimates are shown in Table 4.
Table 3.
Characteristics of pharmacokinetic (PK) and pharmacodynamic (PD) study population*
| PK model | PD model | |
|---|---|---|
| Characteristics | n = 118 | n = 103 |
| Demographic variables | ||
| Age, years | 61 (23–91) | 61 (23–91) |
| Sex, no. (%) | ||
| Female | 45 (38) | 36 (35) |
| Male | 73 (62) | 67 (65) |
| Body size descriptor, kg | ||
| Wt | 77 (43–120) | 77 (43–120) |
| LBW† | 55 (30–86) | 57 (34–80) |
| IBW | 64 (39–83) | 66 (46–84) |
| CLCR (Wt), ml min−1 | 85 (15–244) | 95 (18–244). |
| CLCR (LBW)‡, ml min−1 | 70 (10–170) | 73 (17–166) |
| Admission biochemistry | ||
| Platelets, ×109 l−1 | 236 (113–498) | 234 (121–498) |
| Haemoglobin, g l−1 | 141 (92–186) | 141 (85–186) |
| Co-administration of warfarin, no. (%) | 37 (31) | 35 (34) |
| Co-administration of antiplatelet drugs§, no. (%) | 50 (42) | 44 (43) |
| Exposure variables | ||
| Cmin, IU ml−1 | – | 0.45 (0.1–1.33) |
| Cmax, IU ml−1 | – | 0.91 (0.46–3.38) |
| cAUC, h IU−1 ml−1 | – | 23 (4–120) |
| AUC0–24, h IU−1 ml−1 | – | 13.9 (6.9–25.3) |
CLCR, creatinine clearance; Wt, total body weight; SCr, serum creatinine; Cmin, minimum concentration; Cmax, maximum concentration; cAUC, cumulative area under the concentration–time curve; AUC0–24, area under the concentration–time curve in the first 24 h.
Data presented as median (range) or number (%).
LBW formulae as used by Janmahasatian [24].
CLCR is calculated using the Cockcroft–Gault equation [18] and substituting LBW as the body size descriptor. LBW formula as used by Janmahasatian [24].
Antiplatelet drugs include aspirin and clopidogrel.
Table 4.
Parameter estimates for pharmacokinetic (PK) and pharmacodynamic (PD) population parameters, corresponding bootstrap and relative percentage (%) standard errors (SE) of the estimates of the final model
| Parameter estimate | Base model (95% CI) | Covariate model (95% CI)* | Bootstrap (95% CI)† | Relative % SE of the parameter estimate‡ |
|---|---|---|---|---|
| PK model | ||||
| CL (l h−1) | 0.69 | 0.3 renal (0.14, 0.45) | 0.29 (0.13, 0.46) | 27.4 |
| 0.42 non-renal (0.24, 0.61) | 0.42 (0.25, 0.60) | 22.3 | ||
| Vc (l) | 3.46 | 3.43 (2.20, 4.66) | 3.42 (2.07, 5.17) | 18.3 |
| Ka (h) | 0.25 | 0.26 (0.17, 0.35) | 0.26 (0.16, 0.40) | 18.3 |
| Vp (l) | 6.14 | 5.77 (0.75, 10.8) | 6.42 (3.0, 17.8) | 44.3 |
| Q (l h−1) | 0.3 | 0.31 (0.15, 0.47) | 0.28 (0.1, 0.44) | 27.0 |
| ωCL (% CV) | 48.0 | 37.8 (22.3, 48.0) | 37.4 (28.3, 50.9) | 32.2 |
| ωVc (% CV) | 42.5 | 35.6 (7, 51.0) | 34.6 (5.4, 52.9) | 52.9 |
| ωKa (CV%) | 32.7 | 30.3 (5.3, 42.5) | 29.8 (1.7, 49.8) | 49.6 |
| ε (IU ml−1) | 0.1 | 0.09 (0.086, 0.1) | 0.09 (0.083, 0.1) | 14.5 |
| OF | −859.9 | −909.16 | – | – |
| Proportional-odds model (PD) | ||||
| θ1 | 2.83 | 2.83 (1.26, 4.40) | 2.87 (1.26, 4.69) | 28.3 |
| θ2*Age/61 | −2.75 | −2.75 (−4.31, −1.19) | −2.8 (−4.51, −1.13) | 29.0 |
| θ3*cAUC/23 | −0.54 | −0.54 (−0.84, −0.24) | −0.54 (−0.86, −0.21) | 28.7 |
| θ4 | 2.05 | 2.05 (1.47, 2.63) | 2.1 (1.55, 2.80) | 14.4 |
CL, clearance; Vc, central volume of distribution; Ka, absorption rate constant; Vp, peripheral volume of distribution; Q, intercompartmental clearance; ω, between-subject variability; ε, additive residual error; OF, objective function; cAUC, cumulative area under the concentration–time curve; Age (years).
Confidence intervals (CI) presented as mean ± standard error ×1.96.
Denotes 95% bootstrapped CI (2.5 and 97.5 %ile values).
Calculated as standard error divided by the mean (×100).
LBW, calculated using the formula by Janmahasatian [24], was the most significant body size descriptor to describe BSV in both CL and Vc, reducing the OF by 21 and 11 points from the base model, respectively (P < 0.001). As enoxaparin is extensively renally eliminated, the influence of CLCR, where LBW was used in the C-G equation, on CL was also investigated, which resulted in a further reduction in OF of 31 points. To reflect nonrenal elimination pathways [14], total CL was parameterized as a composite of nonrenal and renal CL incorporating LBW into both pathways. This resulted in a reduction in OF of 50 points and was the best covariate model to fit the data. Other covariate models including allometrically scaled covariates were of no additional benefit. Addition of data below the LOQ at half the LOQ did not affect the parameter estimates, which are shown in Table 4. BSV on CL, Vc and Ka in the final covariate model decreased by 21.3%, 16.3% and 7.4%, respectively, from the base model. Plots of the unexplained variability (ETA) of CL and Vcvs. LBW and CLCR between the base and final model show the reduction in variability after these covariates were added to the model (Figure 1).
Figure 1.
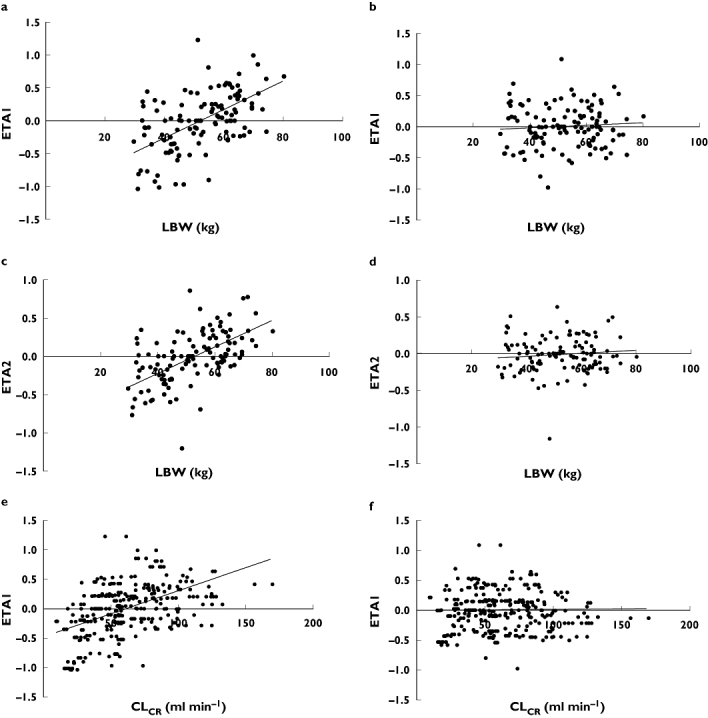
Plots of unexplained variability of clearance (ETA1) and central volume of distribution (ETA2) vs. lean body weight (LBW) and creatinine clearance (CLCR) for the base model and the final covariate model. (a) ETA1 vs. LBW (base model). (b) ETA1 vs. LBW (final covariate model). (c) ETA2 vs. LBW (base model). (d) ETA2 vs. LBW (final covariate model). (e) ETA1 vs. CLCR (base model). (f) ETA1 vs. CLCR (final covariate model)
The final covariate model was described as: CL (l h−1) = 0.3 × CLCR/70 (ml min−1) + 0.42 × LBW/55 (kg)
| (3) |
where CLCR is calculated using LBW [24] in the C-G equation.
Model evaluation
Of the 1000 bootstrap datasets, 873 runs successfully minimized, which were used to compute the median PK parameter estimates and nonparametric CIs shown in Table 4. These results were comparable to the final model, indicating good precision and little bias in the parameter estimates. Figure 2 shows the VPCs for the three populations investigated. All plots indicate the model was suitably predictive with approximately 18% of the data lying outside the 10th and 90th percentile in all subjects (Figure 2a), 23% in subjects with renal impairment (Figure 2b) and 19% in obese subjects (Figure 2c).
Figure 2.
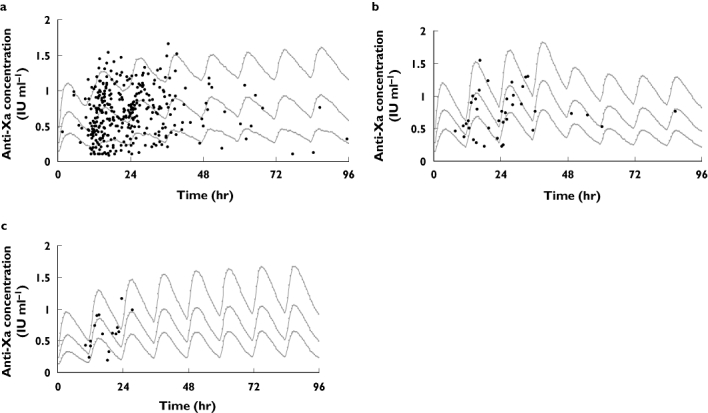
Visual predictive checks for the pharmacokinetic model. The 10th, 50th and 90th prediction intervals from the simulated concentrations are plotted against time post first dose, with the observed data superimposed. (a) All subjects. (b) Subjects with a CLCR < 50 ml min−1[considered to have renal impairment in the randomized controlled trial (RCT)]. Note that subjects in the individualized arm of the RCT that had a CLCR < 50 ml min−1 were dose reduced at 48 h of therapy based on renal function. (c) Subjects with a Wt ≥ 100 kg (considered obese in the RCT)
PK–PD analysis
A total of 103 subjects had a bleeding and bruising assessment beyond baseline and were used to develop the PD model. Relevant subject characteristics are detailed in Table 3. The 15 subjects who did not receive a bruising assessment beyond baseline were either transferred to UFH or the initial diagnosis was rejected after a subsequent radiological scan that resulted in enoxaparin cessation. Of the 103 subjects included in the PD analysis, 27 had a major event (S = 3), 40 had a minor event (S = 2) and 36 had no event (S = 1), as shown in Table 2.
Model building
A summary of physiological variables tested during model building is shown in Table 3. During the univariate analysis, both minimum concentration (Cmin) and CLCR (where LBW was included in the C-G equation) were significant covariates (reduction in OF > 6.61 points), although the best single physiological and exposure measures to describe events were subject age and cumulative AUC (cAUC), which reduced the OF by 14.9 and 15.2 points, respectively. Other variables such as AUC0–24, Cmax and descriptors of body size did not significantly reduce the OF. During the multivariate analysis subject age and cAUC, when combined in a linear model, resulted in a further reduction of the OF by 28.3 points. The addition of further covariates, the use of a maximal effect (Emax) model or a power model did not improve the fit of the model to the data. Colinearity between the two variables was negligible (variance inflation factor = 1.01). The test of proportionality of odds across the response categories (score test) was not significant; χ2 = 0.49, P = 0.78. The final parameter values for the covariate model are shown in Table 4 with the final proportional-odds model represented as:
 |
(4) |
Model evaluation
Figure 3 shows the model-predicted probability of a bleed or major bruise, which increases with cAUC and age. The model-predicted and observed empirical probabilities were closely aligned, suggesting good agreement between the estimated and observed frequency of events, for all severity levels. The difference between the model-predicted and observed probability at higher values of cAUC (120–160 bin) can be partly explained by the small number of subjects in this bin (n = 3). All parameter values and their corresponding nonparametric bootstrap values were comparable (Table 4).
Figure 3.
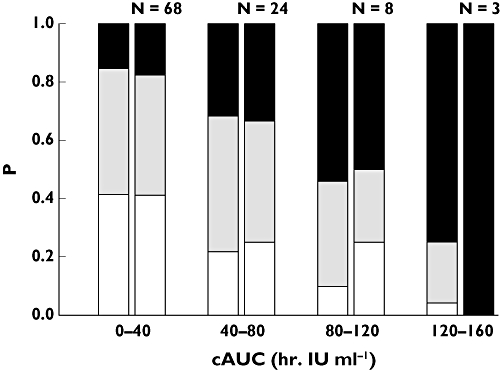
Plot of empirical (right column) and model-predicted (left column) probability (P) of event severity vs. cumulative area under the concentration–time curve (cAUC) at median age. N, number of subjects in each bin; white segment, category S1; grey segment, category S2; black segment, category S3
The probability vs. time plots, at median, 10th and 90th percentiles for age, from the deterministic PD simulations are shown in Figure 4a–c. The model-predicted probability of a bleed or major bruising event increases in both dosing groups over time but there is a clear divergence between groups for all three populations tested and it is most pronounced in subjects who are obese and have renal impairment (Figure 4c). In subjects with both renal impairment and obesity, the divergence begins within the first 24 h of therapy and is particularly evident at 72 h, where the median probability of an event is 0.41 in the conventional group compared with 0.25 in the individualized group (39% decrease), and at 96 h, where the probability is 0.58 and 0.30, respectively (48% decrease). It should be noted that the probability of an event for a patient with renal impairment is the same in both dosing groups for the first 48 h (Figure 4a), as the dose strategies are identical during this time [16]. It is only after 48 h that the dose is individualized and the difference in probability between dosing groups becomes evident. The simulations demonstrate that the probability of an event increases as a function of drug exposure and that dose individualization reduces the probability of an event over time when compared with conventional dosing.
Figure 4.
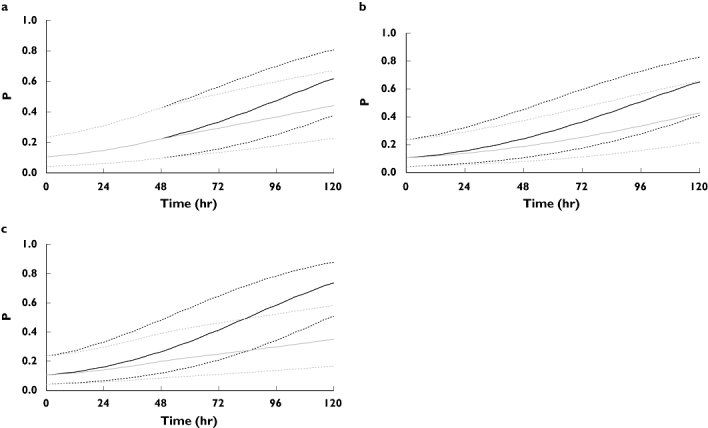
Plots of the model-predicted probability (P) of a bleeding or major bruising event vs. time for individualized (grey) and conventional dosing strategies (black) with 10th and 90th percentiles of age from the study population (dot). (a) Subject with a CLCR = 30 ml min−1 (selected example of a subject with renal impairment that matches the CLCR at which a dose adjustment occurs in the product label). (b) Subject with a Wt = 150 kg (arbitrary selection of a subject who is obese). (c) Subject with a CLCR = 30 ml min−1 and Wt = 150 kg
To assess the PD model performance, the PD dataset was bootstrapped 500 times using each variable, age and cAUC, independently in the model. For each bootstrap, the observed data were divided into four bins, each with the same number of subjects per bin. The empirical probability for each bin was computed and plotted against the midpoint of each bin, which was overlaid with the 10th, 50th and 90th predicted probabilities for each unique value of age and cAUC. Figure 5 shows these plots for age (Figure 5a) and cAUC (Figure 5b) and, as seen, the central tendency is generally captured in both plots, as the median model-predicted probability dissects the empirical bootstrapped observations. These graphs, while separately validating the two variables as predictors of an event, show that age is a better predictor of an event than cAUC.
Figure 5.
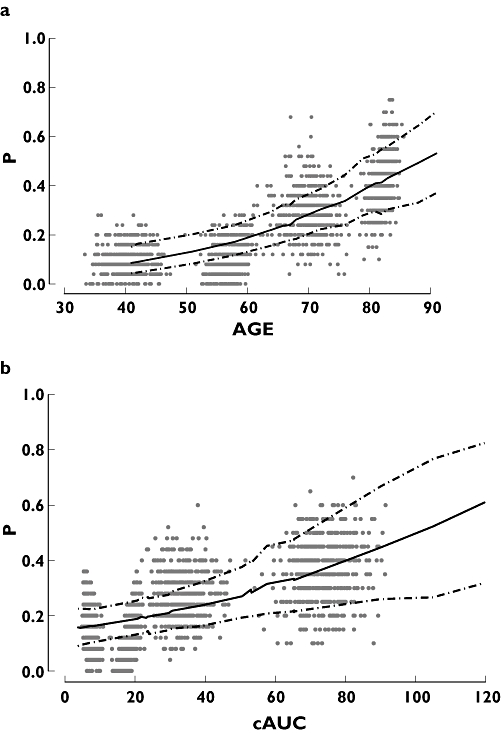
Plots of the bootstrapped empirical probabilities of a bleed or major bruising event vs. variable, superimposed on the median model-predicted probability of an event, with 90% confidence intervals. (a) Probability of an event vs. age (years). (b) Probability of an event vs. cAUC (h IU−1 ml−1)
Discussion
Enoxaparin significantly reduces major ischaemic events when compared with UFH in the treatment of thromboembolic diseases [1, 2, 5, 6]. However, limited information is available that quantifies the probability of bleeding events as a function of drug exposure, especially for patients who are obese and/or have renal impairment.
Pharmacokinetics
The PK of enoxaparin in this study was best described by a two-compartment model with first-order input and linear elimination and was comparable to previously published PK models [14, 15, 29]. One-compartment models have also been used to describe the disposition of enoxaparin (Table 1), although the inability to estimate a two-compartment system may in part be explained by non-optimal sparse sampling or use of observational clinical data that were not intended to be used for PK model development [30, 31].
Enoxaparin is a hydrophilic molecule that is distributed in plasma and lean tissue and predominantly renally eliminated. It was of no surprise that the two most influential covariates on CL were LBW and CLCR, which is comparable to previously published models for enoxaparin where CL was described by a combined renal and nonrenal model [14, 29]. Both these prior and current models used measures of ‘lean mass’ as a substitute for Wt in the C-G equation to calculate CLCR, with LBW [24] preferred in the model presented in this study. This finding complements a recent report that demonstrated LBW was the descriptor of choice when estimating CLCR[32]. This nuance is of minor importance for subjects of normal weight, but becomes extremely important when estimating CLCR in the obese. Creatinine is a by-product of muscle metabolism, and the excess body mass seen in the obese is mostly adipose tissue as opposed to muscle. Therefore, when no adjustment is made for the increase in body mass such as standardization with LBW, CLCR will be routinely overestimated, which can equate to significant clinical ramifications on enoxaparin dose selection.
Prior data suggest approximately 30% of enoxaparin CL is nonrenal [14], with the model presented in this paper also incorporating a nonrenal component of CL. The nonrenal component in our model was represented as a function of LBW, which has recently been supported in a commentary by Han et al.[23]. It is not surprising that the nonrenal estimate of CL for the model presented in this study was significantly greater than the 30% previously published [14]. We propose that this result occurred as a function of the experimental design as subjects with severe renal impairment (CLCR < 30 ml min−1) represented only 7% of the study population and contributed only 5% of all anti-Xa concentrations. This hypothesis was supported by Kim et al., who demonstrated that the most reliable estimate of the renal effect on the CL of enoxaparin was gained if subjects were recruited into groups stratified on renal function: normal (CLCR ≥ 80 ml min−1); mild (70–79.9 ml min−1); moderate (30–49.9 ml min−1); severe (<30 ml min−1) [33]. Unfortunately, recruitment of subjects with severe renal impairment was limited in the study hospital, as clinicians often declined to allow their patients to participate in the trial and preferred to use UFH due to the perceived risk of bleeding with enoxaparin in this population. The publication by Green et al.[14] provides a better model with which to explain the CL of enoxaparin in subjects with renal impairment.
PK–PD
As little data are available to describe the dose–exposure–event relationship of enoxaparin, it is challenging to identify the impact that differing drug doses might have on bleeding events. We therefore linked the population PK model to a proportional-odds model to describe event occurrence and severity. The only other PK–PD study of enoxaparin that has quantified an adverse event as a function of drug exposure (excluding dose) used logistic regression to describe the presence of a bruise and incorporated subject age and maximum concentration (Cmax) as predictive variables of the event [15]. However, the severity of bruise was not investigated, as an event was defined as ‘multiple bruises of any magnitude’.
Bleeding has been previously modelled using a proportional-odds approach by Mould et al., who described the time course of bleeding events to the antiplatelet drug lotrafiban [34]. In this study the severity of an event was described as a function of increasing lotrafiban AUC, which enabled the identification of a toxic lotrafiban exposure and the subsequent design of a dosing strategy with a reduced risk of bleeding.
Our study has demonstrated that increasing cAUC and subject age best describe the occurrence and severity of bleeding and bruising events. It is not unexpected that age is a strong determinant of bleeding, as the risk of bleeding increases with age in both the presence and absence of an anticoagulant such as enoxaparin [35, 36]. Age has also been found to be predictive of bruising [15]. These findings are supported by our analysis when, after adjusting for exposure, age remains an independent predictor for bleeding.
It is expected that some metric of drug exposure such as Cmax increases the risk of bleeding and it has been previously linked to bleeding [13] and bruising [15]. The identification that cAUC was predictive of bleeding events over Cmax has not been previously reported for enoxaparin. cAUC incorporates time as an explanatory variable, and although this analysis cannot delineate between length of treatment and exposure, we believe it is likely to be a function of both. We recognize that treatment doses of enoxaparin are more likely to result in a bleeding event than prophylactic doses; however, other exposure variables such as Cmax may not fully describe the underlying physiological processes that result in an adverse event. It is of interest that the incidence of bleeding events following prophylactic dosing of enoxaparin (40 mg once daily) has been reported at 11.5% (28–35 days of therapy) in surgical subjects [37] and 12.6% in medical subjects (6–14 days of therapy) [38]. However, it should be noted that our study data were obtained from subjects administered treatment doses for a mean ± SD duration of therapy of 3.5 ± 2.3 days, therefore the current model can be used only in circumstances that are similar to the study.
There are limitations to this study. First, traditional binary data analyses require extensive data and a total of 63 events may be considered small. However, we are limited to a data from a RCT that cannot be enriched. To assess confidence in the results we have bootstrapped the PD data and added 90% CIs to the model predictions. The corresponding plots (Figure 5) show that both cAUC and age are good predictors of bleeding, albeit age being stronger that cAUC. Second, independent data would be necessary to evaluate this model, but unfortunately these are not available at this time. Third, therapeutic failure such as re-infarction was not modelled and therefore has not been included in our discussion; however, there was no significant increase in therapeutic failure in the RCT.
There is now compelling evidence to dose-individualize enoxaparin. Unfortunately, the current drug label [12] fails to consider body composition in its dose recommendations, and dichotomizes renal function that should be considered as a continuous variable. There is no sound physiological rationale to make this assumption and few data support this approach [39]. Other PK studies have recommended more flexible dosing strategies based on renal function, but the strategies have not been tested in confirmatory studies [30, 40]. It is of interest that the drug label for another hydrophilic anticoagulant, lepirudin [41], provides a detailed dose-adjustment strategy for subjects with varying renal function.
The occurrence and severity of bleeding and bruising events following administration of enoxaparin is best described as a function of cAUC and subject age. A description of the exposure–event relationship of enoxaparin has enabled the comparison of the probability of an event between an individualized and a conventional (product label) dosing strategy. This study has demonstrated that individualized dosing of enoxaparin based upon LBW and renal function reduces the risk of a major bruising or bleeding events.
Competing interests
J.A. has received sponsorship to attend conferences and consulting fees from Sanofi-Aventis.
We thank the Cardiology Research Unit at the Royal Brisbane and Women's Hospital, Queensland, Australia, in particular Linda Hindom, Tony Buxton, Leeanne Palethorpe and Lydia Liliwell for their assistance in subject identification and data collection. This study was funded by the Queensland Hospitals Drug Advisory Committee, Society of Hospital Pharmacists of Australia and the School of Pharmacy, University of Queensland. These bodies had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
REFERENCES
- 1.Cohen M, Demers C, Gurfinkel E, Turpie A, Fromell G, Goodman S, Langer A, Califf R, Fox K, Premmereur J, Bigonzi F, Stephens J, Weatherley B. A comparison of low molecular weight heparin with unfractionated heparin for unstable coronary artery disease. N Engl J Med. 1997;337:447–52. doi: 10.1056/NEJM199708143370702. [DOI] [PubMed] [Google Scholar]
- 2.Antman EM, McCabe C, Gurfinkel E, Turpie A, Alexander G, Bernink P, Salein D, de Luna A, Fox K, LaBlanche J-M, Radley D, Premmereur J, Braunwald E. Enoxaparin prevents death and cardiac ischaemic events in unstable angina/non-Q-wave myocardial infarction. Results of the thrombolysis in myocardial infarction (TIMI) 11B trial. Circulation. 1999;100:1593–601. doi: 10.1161/01.cir.100.15.1593. [DOI] [PubMed] [Google Scholar]
- 3.Antman EM, Morrow DA, McCabe CH, Murphy SA, Ruda M, Sadowski Z, Budaj A, Lopez-Candales J, Guneri S, Jiang F, White H, Fox K, Braunwald E. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med. 2006;354:1477–88. doi: 10.1056/NEJMoa060898. [DOI] [PubMed] [Google Scholar]
- 4.The SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy. JAMA. 2004;292:45–54. doi: 10.1001/jama.292.1.45. [DOI] [PubMed] [Google Scholar]
- 5.Merli G, Spiro T, Olsson C, Abildgaard U, Davidson B, Eldor A, Elias D, Grigg A, Musset D, Rodgers G, Trowbridge A, Yusen R, Zawilska K. Subcutaneous enoxaparin once or twice daily compared with intravenous heparin for the treatment of venous thromboembolic disease. Ann Intern Med. 2001;134:191–202. doi: 10.7326/0003-4819-134-3-200102060-00009. [DOI] [PubMed] [Google Scholar]
- 6.Quinlan DJ, McQuillan A, Eikelboom JW. Low-molecular-weight-heparin compared with intravenous heparin for treatment of pulmonary embolism. Ann Intern Med. 2004;140:175–83. doi: 10.7326/0003-4819-140-3-200402030-00008. [DOI] [PubMed] [Google Scholar]
- 7.Aroney CN, Aylward P, Kelly A-M, Chew DP, Clune E. Guidelines for the management of acute coronary syndromes 2006. Med J Aust. 2006;184(Suppl.):S1–S32. doi: 10.5694/j.1326-5377.2006.tb00304.x. [DOI] [PubMed] [Google Scholar]
- 8.Buller HR, Agnelli MD, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease. Chest. 2004;126(Suppl.):401S–28S. doi: 10.1378/chest.126.3_suppl.401S. [DOI] [PubMed] [Google Scholar]
- 9.Snow V, Qaseem A, Barry P, Hornbake ER, Rodnick JE, Tobolic T, Ireland B, Segal JB, Bass EB. Management of venous thromboembolism: a clinical practice guideline from the American College of Physicians and the Academy of Family Physicians. Ann Intern Med. 2007;146:204–10. doi: 10.7326/0003-4819-146-3-200702060-00149. [DOI] [PubMed] [Google Scholar]
- 10.Lovenox® prescribing information. Available at http://www.sanofi-aventis.us/ (last accessed 6 April 2008.
- 11.Product information Clexane®. Available at http://www.sanofi-aventis.co.uk/prodcuts/Clexane_spc.pdf (last accessed 6 April 2008.
- 12.Clexane® product information. Available at http://www.sanofi-aventis.com.au/products/aus_pi_clexane.pdf (last accessed 6 April 2008.
- 13.Thrombolysis in Myocardial Infarction (TIMI) 11A Investigators. Dose-ranging trial of enoxaparin for unstable angina: results of TIMI 11A. J Am Coll Cardiol. 1997;29:1474–82. [PubMed] [Google Scholar]
- 14.Green B, Greenwood M, Saltissi D, Westhuyzen J, Kluver L, Rowell J, Atherton J. Dosing strategy for enoxaparin in patients with renal impairment presenting with acute coronary syndromes. Br J Clin Pharmacol. 2004;59:281–90. doi: 10.1111/j.1365-2125.2004.02253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green B, Duffull SB. Development of a dosing strategy for enoxaparin in obese patients. Br J Clin Pharmacol. 2003;54:96–103. doi: 10.1046/j.1365-2125.2003.01849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barras MA, Duffull SB, Atherton JJ, Green B. Individualized compared to conventional dosing of enoxaparin. Clin Pharmacol Ther. 2008;83:882–88. doi: 10.1038/sj.clpt.6100399. [DOI] [PubMed] [Google Scholar]
- 17.Montalescot G, Collet J, Tanguy ML, Ankri A, Payot L, Dumaine R, Choussat R, Beygui F, Gallois V, Thomas D. Anti-Xa activity relates to survival and efficacy in unselected acute coronary syndrome patients treated with enoxaparin. Circulation. 2004;110:392–98. doi: 10.1161/01.CIR.0000136830.65073.C7. [DOI] [PubMed] [Google Scholar]
- 18.Cockroft DW, Gault H. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 19.Green B, Duffull SB. Prospective evaluation of a D-Optimal designed population pharmacokinetic study. J Pharmacokinet Pharmacodyn. 2003;30:145–61. doi: 10.1023/a:1024467714170. [DOI] [PubMed] [Google Scholar]
- 20.Berkowitz S, Stinnett S, Cohen M, Fromell G, Bigonzi F. Prospective comparison of hemorrhagic complication after treatment with enoxaparin versus unfractionated heparin for unstable angina pectoris or non-ST-segment elevation acute myocardial infarction. Am J Cardiol. 2001;88:1230–34. doi: 10.1016/s0002-9149(01)02082-3. [DOI] [PubMed] [Google Scholar]
- 21.Beal SL, Boeckmann A, Sheiner L. NONMEM User's Guide. Ellicot City, MD: Icon Development Solutions; 1999. –2006. [Google Scholar]
- 22.Green B, Duffull S. What is the best size descriptor to use for pharmacokinetic studies in the obese? Br J Clin Pharmacol. 2004;58:119–33. doi: 10.1111/j.1365-2125.2004.02157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han P, Duffull SB, Kirkpatrick CMJ, Green B. Dosing in obesity: a simple solution to a big problem. Clin Phamacol Ther. 2007;82:505–8. doi: 10.1038/sj.clpt.6100381. [DOI] [PubMed] [Google Scholar]
- 24.Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44:1051–65. doi: 10.2165/00003088-200544100-00004. [DOI] [PubMed] [Google Scholar]
- 25.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481–504. doi: 10.1023/a:1012299115260. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn. 2003;30:387–404. doi: 10.1023/b:jopa.0000012998.04442.1f. [DOI] [PubMed] [Google Scholar]
- 27.Ettte EI, Williams P, Kim Y, Lane J, Liu M-J, Capparelli E. Model appropriateness and population pharmacokinetic modeling. J Clin Pharmacol. 2003;43:610–23. [PubMed] [Google Scholar]
- 28.Parke J, Holford NH, Charles BG. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects populations models. Comput Methods Programs Biomed. 1999;59:19–29. doi: 10.1016/s0169-2607(98)00098-4. [DOI] [PubMed] [Google Scholar]
- 29.Feng Y, Green B, Duffull SB, Kane-Gill SL, Bobek MB, Bies RR. Development of a dosage strategy in patients receiving enoxaparin by continuous intravenous infusion using modelling and simulation. Br J Clin Pharmacol. 2006;62:165–76. doi: 10.1111/j.1365-2125.2006.02650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hulot J-S, Vantelon C, Urien S, Bouzamondo A. Effect of renal function on the pharmacokinetics of enoxaparin and consequences on dose adjustment. Ther Drug Monit. 2004;26:305–10. doi: 10.1097/00007691-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 31.Bruno R, Baille P, Retout S, Vivier N. Population pharmacokinetics and pharmacodynamics of enoxaparin in unstable angina and non-ST-segment elevation myocardial infarction. Br J Clin Pharmacol. 2002;56:407–14. doi: 10.1046/j.1365-2125.2003.01904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janmahasatian S, Duffull SB, Chagnac A, Kirkpatrick CMJ, Green B. Lean body mass normalizes the effect of obesity on renal function. Br J Clin Pharmacol. 2008;65:964–65. doi: 10.1111/j.1365-2125.2008.03112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HK, Duffull SB, Green B. The effect of study design on pharmacokinetics in patients with impaired renal function. In: PAGE (2008) Abstr 1354. Available at http://wwwpage-meetingorg/?abstract=1354 (last accessed 8 November 2008.
- 34.Mould D, Chapelsky M, Aluri J, Swagzdis J, Samuels R, Granett J. A population pharmacokinetic–pharmacodynamic and logistic regression analysis of lotrafiban in patients. Clin Phamacol Ther. 2001;69:210–22. doi: 10.1067/mcp.2001.114925. [DOI] [PubMed] [Google Scholar]
- 35.Moscucci M, Fox KA, Cannon CO, Klein W. Predictors of major bleeding in acute coronary syndromes: the global registry of acute coronary events (GRACE) Eur Heart J. 2003;24:1815–23. doi: 10.1016/s0195-668x(03)00485-8. [DOI] [PubMed] [Google Scholar]
- 36.Granger C, Goldberg RJ, Dabbous O, Pieper KS. Predictors of hospital mortality in the global registry of acute coronary events. Arch Intern Med. 2003;163:2345–53. doi: 10.1001/archinte.163.19.2345. [DOI] [PubMed] [Google Scholar]
- 37.Eriksson B, Dahl O, Rosencher N, Kurth A, van Dijk C, Frostick S, Prins MH, Hettiarachchi R, Hantel S, Schnee J, Buller HR. Dabigatran etexilate versus enoxaparin for the prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet. 2007;370:949–56. doi: 10.1016/S0140-6736(07)61445-7. [DOI] [PubMed] [Google Scholar]
- 38.Samama MM, Cohen AT, Darmon J-Y, Desjardins L, Eldor A, Janbon C, Leizorovicz A, Nguyen H, Olsson C-G, Turpie AG, Weisslinger N. A comparison of enoxaparin with placebo for the prevention of venous thromboembolism in acutely ill medical patients. N Engl J Med. 1999;341:793–800. doi: 10.1056/NEJM199909093411103. [DOI] [PubMed] [Google Scholar]
- 39.Brophy D, Sica D. Use of enoxaparin in patients with chronic kidney disease. Drug Safety. 2007;30:991–94. doi: 10.2165/00002018-200730110-00001. [DOI] [PubMed] [Google Scholar]
- 40.Hulot J-S, Montalescot G, Lechat P, Collet J-P, Ankri A, Urien S. Dosing strategy in patients with renal failure receiving enoxaparin for the treatment of non-ST-segment elevation acute coronary syndrome. Clin Pharmacol Ther. 2005;77:542–52. doi: 10.1016/j.clpt.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 41.Refludan® product information. Available at http://www.drugs.com/pro/refludan.html (last accessed 26 August 2009.
- 42.Sanchez-Pena P, Hulot J-S, Urien S, Ankri A, Collet J-P, Choussat R, Lechat P, Montalescot G. Anti-factor Xa kinetics after intravenous enoxaparin in patients undergoing percutaneous coronary intervention: a population model analysis. Br J Clin Pharmacol. 2005;60:364–73. doi: 10.1111/j.1365-2125.2005.02452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kane-Gill SL, Feng Y, Bobek MB, Pruchnicki MC, Dasta JF. Administration of enoxaparin by continuous infusion in a naturalistic setting: analysis of renal function and safety. J Clin Pharm Ther. 2005;30:207–13. doi: 10.1111/j.1365-2710.2005.00634.x. [DOI] [PubMed] [Google Scholar]
- 44.Berges A, Laporte S, Epinat M, Zufferey P, Tranchand B, Decousus H, Mismetti P. Anti-factor Xa activity of enoxaparin administered at prophylactic dosage to patients over 75 years old. Br J Clin Pharmacol. 2007;64:428–38. doi: 10.1111/j.1365-2125.2007.02920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]