

Abstract
AIMS
To investigate in vivo the influence of the potent CYP2C19 and CYP3A4 inhibitor voriconazole on the pharmacokinetics and analgesic effects of tilidine.
METHODS
Sixteen healthy volunteers received voriconazole (400 mg) or placebo together with a single oral dose of tilidine (100 mg). Blood samples and urine were collected for 24 h and experimental pain was determined by using the cold pressor test. Noncompartimental analysis was performed to determine pharmacokinetic parameters of tilidine, nortilidine and voriconazole, whereas pharmacodynamic parameters were analysed by nonparametric repeated measures anova (Friedman).
RESULTS
Voriconazole caused a 20-fold increase in exposition of tilidine in serum [AUC 1250.8 h*ng ml−1, 95% confidence interval (CI) 1076.8, 1424.9 vs. 61 h*ng ml−1, 95% CI 42.6, 80.9; P < 0.0001], whereas the AUC of nortilidine also increased 2.5-fold. After voriconazole much lower serum concentrations of bisnortilidine were observed. The onset of analgesic activity occurred later with voriconazole, which is in agreement with the prolonged tmax of nortilidine (0.78 h, 95% CI 0.63, 0.93 vs. 2.5 h, 95% CI 1.85, 3.18; P < 0.0001) due to the additional inhibition of nortilidine metabolism to bisnortilidine. After voriconazole the AUC under the pain withdrawal–time curve was reduced compared with placebo (149 s h−1, 95% CI 112, 185 vs. 175 s h−1, 95% CI 138, 213; P < 0.016), mainly due to the shorter withdrawal time 0.75 h after tilidine administration.
CONCLUSIONS
Voriconazole significantly inhibited the sequential metabolism of tilidine with increased exposure of the active nortilidine. Furthermore, the incidence of adverse events was almost doubled after voriconazole and tilidine.
Keywords: CYP2C19, CYP3A4, inhibition, metabolism, nortilidine, tilidine
WHAT IS ALREADY KNOWN ABOUT THE SUBJECT
Tilidine, a World Health Organization level II analgesic, is a high extraction drug subject to pronounced first-pass metabolism, resulting in a low absolute bioavailability.
The analgesic activity of tilidine is almost exclusively exerted through its metabolite nortilidine, which easily penetrates the blood–brain barrier and binds to the µ-opioid receptor as a potent agonist.
In vitro, tilidine has been shown to be metabolized to nortilidine by N-demethylation via CYP3A4 and CYP2C19; furthermore, strong CYP3A4 and CYP2C19 inhibitors inhibited the formation of nortilidine, suggesting that these inhibitors will lead to a reduction of tilidine efficiency in vivo.
WHAT THIS PAPER ADDS
Co-administration of tilidine and the potent CYP3A4 and CYP2C19 inhibitor voriconazole resulted in a major pharmacokinetic interaction that was partly associated with changes in the analgesic effect.
Voriconazole inhibits both metabolic steps in the sequential metabolism of tilidine resulting in an increased exposure of the active nortilidine.
The incidence of adverse reactions was also significantly increased.
Introduction
The synthetic opioid tilidine is one of the most frequently prescribed analgesic drugs in Germany and Belgium, with a yearly production of 36 tons year−1[1]. Being classified as a World Health Organization class II analgesic it is used for treatment of moderate or strong pain or for long-term treatment of patients with chronic pain [2–4]. It is marketed as a fixed combination of tilidine and the opioid antagonist naloxone to prevent abuse. The therapeutic activity of tilidine is mainly related to its metabolite nortilidine [5]. Tilidine is presystemically metabolized via N-demethylation to nortilidine, which easily penetrates the blood–brain barrier and binds to the µ-opioid receptor as an agonist with a 100-fold higher µ-receptor affinity than tilidine itself [6]. Furthermore, nortilidine is metabolized to bisnortilidine and several polar metabolites (see Figure 1) [7, 8]. Recently, it was demonstrated that tilidine is a substrate of cytochrome P450 isozymes (CYP), especially of CYP3A4 and CYP2C19, and that strong CYP3A4 and CYP2C19 inhibitors almost completely inhibit the formation of nortilidine in vitro[9]. Consequently, drugs affecting the activity of these enzymes are expected to interact with tilidine in vivo. Given that tilidine is a prodrug and the formation of the active metabolite nortilidine can be decreased by potent CYP2C19 and CYP3A4 inhibitors, the central analgesic effect in humans might be reduced or even abolished.
Figure 1.
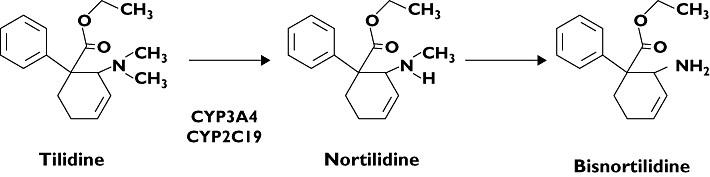
Metabolic pathway of tilidine in man
Voriconazole is a novel triazole antifungal agent used both intravenously and orally in order to treat a broad spectrum of fungal infections. It is rapidly and almost completely absorbed from the gastrointestinal tract [10] and undergoes metabolism involving mainly CYP2C19 and CYP3A4 [11]. Voriconazole also strongly inhibits CYP2C19 and CYP3A4, resulting in significant interactions with CYP3A4 substrates like midazolam and fentanyl [12, 13] and CYP2C19 substrates like omeprazole [14]. Hence, voriconazole might be ideal to inhibit tilidine N-demethylation to active nortilidine. We therefore investigated in a randomized, placebo-controlled clinical trial the potential pharmacokinetic and pharmacodynamic interaction of tilidine and voriconazole in healthy humans.
Methods
The study was approved by the Competent Authority in Germany (EudraCT no. 2007-004666-41) and the Ethics Committee of the Medical Faculty of the University of Heidelberg (Ethics Committee registration no. AFmo-363/2007). It was conducted at the Department of Internal Medicine VI, Clinical Pharmacology and Pharmacoepidemiology, in accordance with the standards of Good Clinical Practice (as defined in the International Conference on Harmonization E6 Guideline for Good Clinical Practice), in agreement with the Declaration of Helsinki and the specific legal requirements in Germany.
Study population
Sixteen healthy male (n= 10) and female (n= 6) nonsmoking volunteers were included in this study (age 20–48 years; body mass index 20.3–26.3 kg m−2). A physical examination, appropriate laboratory tests and electrocardiography were carried out before the beginning of the study to assess the health status. Women were required to undergo pregnancy testing and were enrolled only if the result was negative and they were using a reliable contraception method. None of the volunteers received any continuous medication, except contraceptives.
Study design
In order to determine the influence of voriconazole on pharmacokinetics and analgesic effects of tilidine/nortilidine, a randomized, double-blind, placebo-controlled, cross-over study was conducted. On study day 1, after an overnight fast, each subject orally received in random order a single oral dose of 400 mg voriconazole (two capsules, Vfend®; Pfizer Pharma GmbH, Karlsruhe, Germany) or placebo 1 h before the intake of a single oral dose of 100 mg tilidine/naloxone solution (1.44 ml, Valoron N®; Pfizer Pharma). One hour time difference was chosen to avoid direct pharmaceutical interaction. After a wash-out period of 14 days the procedure was repeated with voriconazole or placebo interchanged. On study days 1 and 15, participants stayed at the Clinical Research Unit of the Department of Internal Medicine VI for 12 h after drug administration. During the study day they received standardized lunch (4 h after tilidine administration) and dinner meals. Alcohol, grapefruit juice and caffeinated beverages were not allowed throughout the study (from inclusion to the end of the study).
Blood sampling and urine collection
On days 1 and 15 blood samples (7.5 ml each) were collected before the intake of tilidine/naloxone and 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2.0, 2.25, 2.75, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12 and 24 h after administration of tilidine/naloxone. Blood samples were immediately centrifuged at 2500 g at 4°C, and separated plasma was stored at −20°C until analysis. Urine was collected completely during the 24-h period after drug administration; an aliquot of 10 ml was kept frozen at −20°C until analysis.
Determination of analgesic effects by the cold pressor test
The analgesic effect of tilidine was studied by using the cold pressor test as previously described [15–17]. The cold pressor test registers the evolution of pain sensation under unchanged pain stimulation, resulting from the unchanged water temperature by reference to pain threshold and withdrawal threshold [18].
The experiments were performed at pre-study visit (training session) and on study days 1 and 15 at 0.25 h before the intake of tilidine/naloxone and at 0.75, 1.75 and 2.75 h after the intake of tilidine/naloxone. The cold pressor test apparatus consisted of temperature-controlled water baths of 37 ± 0.5°C (warm-water bath) and 0 ± 0.5°C (ice-water bath). The nondominant forearm was placed into the warm bath for exactly 2 min; eyes of the participants were covered with eye patches. Fifteen seconds before transferring the forearm into the cold water bath, a blood pressure cuff was inflated to a pressure 20 mmHg below the diastolic blood pressure, which has been shown to increase the stability of the cold pressor test due to better control of temperature changes in the forearm by reduction of venous return [16]. The participants placed their forearm in a constant position with the fingers wide apart into the cold water for a maximum of 2 min and were instructed to indicate the time point of the first pain sensation (pain threshold) as well as the time point of intolerable pain, at which the forearm was removed from the cold water bath (withdrawal threshold). In addition, the subject had to quantify their subjective pain verbally on a scale from 0 to 100 (visual analogue scale).
Determination of tilidine and its metabolites in serum and urine
Study plasma (100 µl), calibration (range 1–250 ng ml−1) and quality control (QC) samples were transferred into 400 µl of acetonitrile, which contained the internal standard tramadol at a concentration of 100 ng ml−1. The samples were vortexed for 30 s and subsequently centrifuged (10 min, 16 000 g, 10°C). From the clear supernatant 450 µl was transferred into glass tubes. The organic phase was blown down to dryness with a gently stream of nitrogen in a water bath at 40°C and the residues were reconstituted with 200 µl (5 mM ammonium acetate/acetonitrile 1 : 1, v/v). Ten microlitres of this solution was injected into the liquid chromatography/tandem mass spectrometry (MS/MS). Details of the high-performance liquid chromatography/MS/MS instrumentations are described in [9]. The mass spectrometer was programmed to admit the protonated molecules [M+H] at m/z 274.1 (tilidine), 260.1 (nortilidine) and 264.0 (tramadol) via the first quadrupole filter (Q1), with collision-induced fragmentation at Q2 and monitoring the product ions via Q3 at m/z at 155.0 (tilidine and nortilidine) and m/z at 58.0 for tramadol. Peak area ratios obtained from the monitored ions were utilized for construction of calibration curves, using weighted linear least squares regression. Each analytical run included seven calibration samples at concentrations of 1–250 ng ml−1 for tilidine and nortilidine, respectively, and three QC samples at concentrations of 3.8 ng ml−1 (3.9 ng ml−1), 83.5 ng ml−1 (86.1 ng ml−1) and 161.1 ng ml−1 (166.1 ng ml−1), respectively. The QC samples were measured in duplicate. Data collection, peak integration, and calculations were performed using Analyst V 1.4.2 software (Applied Biosystems, Darmstadt, Germany). Prior to the quantification of the study samples the method was validated in accordance with Food and Drug Administration guidelines [19]. Calibration for both drugs (tilidine and nortilidine) was linear in the range from 1 to 250 ng ml−1. The coefficient of correlation (r2) was always >0.99. The lower limit of quantification was 1 ng ml−1 for both drugs. Data for nortilidine are given in parentheses. The day-to-day (n= 22) accuracy was between 102.3% (89.7%) and 103.8% (107.1%) and the coefficients of variation were between 5.9% (4.7%) and 11.5% (6.8%) (batch to batch). Within-batch accuracy (n= 6) was between 97.9% (103.9%) and 106.7% (107.3%) with coefficients of variation between 3.5% (0.4%) and 11.7% (5.8%). The extraction recovery was 109.0% (84.6%) at a concentration of 83.5 ng ml−1 (86.1 ng ml−1) and 98.3% (69.5%) at a concentration of 161.1 ng ml−1 (166.1 ng ml−1). The extraction recovery for the internal standard was 89.6% at a concentration of 100 ng ml−1.
Chromatographic peaks consistent with the metabolite bisnortilidine appeared also in plasma samples. A pure reference standard for bisnortilidine was not available. Therefore metabolite concentrations were determined under the assumption that calibration slopes for nortilidine were applicable to bisnortilidine. As such, bisnortilidine concentrations actually are expressed in relative units.
Calculation and statistical analysis
Data are presented as mean ± standard deviation. In the tables the 95% confidence intervals are given additionally. Noncompartmental analysis was performed by use of WinNonlin software (version 5.2; Pharsight, Mountain View, CA, USA) to determine the following pharmacokinetic parameters of tilidine and nortilidine: Cmax, tmax, terminal elimination half-life t1/2, AUC time from time 0 to the last measurable concentration (AUClast0–24) and AUC extrapolated to infinity (AUC0–∞), calculated by use of the linear trapezoidal rule. The renal clearance (CLR) of tilidine and nortilidine was determined as the amount of tilidine and nortilidine excreted in urine from 0 to 24 h (Ae0–24) divided by the corresponding AUC0–24 values. For each study day the area under the pain threshold–time curve and the area under the withdrawal threshold–time curve were calculated using pain threshold and withdrawal threshold data from 0 to 2.75 h corrected for the respective baseline value at t= 0 h. Differences in these pharmacokinetic and pharmacodynamic parameters between placebo and tilidine treatment were assessed by use of the nonparametric Wilcoxon matched pairs signed rank test (GraphPad Instat 3; San Diego, CA, USA). Pain threshold and withdrawal threshold at 0.75, 1.75 and 2.75 h after tilidine administration were compared against baseline by nonparametric repeated measures anova (Friedman) using the Instat 3 (GraphPad) statistics program.
Results
Tilidine
During treatment with voriconazole all 16 participants showed a higher exposure to tilidine, with AUC0–∞ increasing 20-fold (1250.9 ± 326.6 vs. 61.7 ± 36.0 h*ng ml−1, P < 0.0001, Figure 2a). The other pharmacokinetic parameters Cmax, tmax, t1/2 and Ae0–24 increased significantly as well (Table 1). Moreover, after intake of voriconazole a significant decrease of CLoral (92%, P < 0.0001), CLnonrenal (93%, P < 0.0001) and CLR (53%, P < 0.0001) of tilidine was observed (Table 1).
Figure 2.
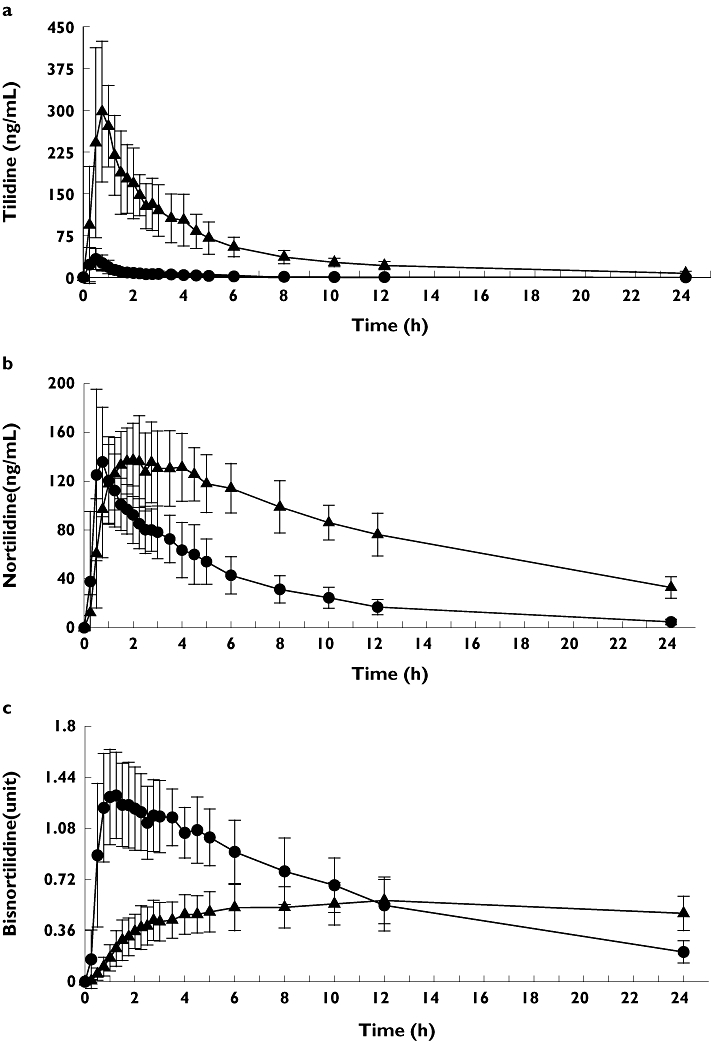
Mean (±SD) tilidine (a), nortilidine (b) and bisnortilidine (c) plasma concentration–time profile after single oral administration of 100 mg tilidine/naloxone solution in combination with single oral administration of placebo (circles) or 400 mg voriconazole (triangle) to 16 healthy participants
Table 1.
Pharmacokinetic parameters (mean ± SD) after noncompartmental analysis of tilidine, nortilidine and bisnortilidine during placebo and voriconazole administration in 16 healthy individuals
| Tilidine | Placebo | 95% CI | Voriconazole | 95%CI | P-value |
|---|---|---|---|---|---|
| Cmax (ng ml−1) | 37.4 ± 28 | 22.5, 52.4 | 359.8 ± 136.2 | 287.2, 432.4 | 0.0001 |
| tmax (h) | 0.6 ± 0.2 | 0.45, 0.68 | 0.8 ± 0.3 | 0.65, 0.94 | 0.0105 |
| AUC0–24 (h ng ml−1) | 55.8 ± 33.3 | 38.1, 73.6 | 1147.5 ± 286.5 | 994.8, 1300.2 | 0.0001 |
| AUC0–∞ (h ng ml−1) | 61.7 ± 36 | 42.60, 80.85 | 1250.9 ± 326.6 | 1076.8, 1425.0 | 0.0001 |
| AUC24–∞ (%) | 10.51 ± 5.2 | 7.73, 13.29 | 7.89 ± 4.9 | 5.3, 10.49 | |
| t1/2 (h) | 2.8 ± 1.7 | 1.9, 3.7 | 7.5 ± 1.7 | 6.7, 8.5 | 0.0001 |
| MRT (h) | 2.1 ± 0.6 | 1.7, 2.4 | 5.3 ± 0.5 | 5.0, 5.5 | 0.0001 |
| CL/F (ml min−1) | 39 775 ± 28 264 | 24 200, 60 863 | 1410 ± 322 | 1248, 1589 | 0.0001 |
| Ae0–24 (%) | 0.18 ± 0.11 | 0.12, 0.24 | 1.88 ± 1.49 | 1.08, 2.67 | 0.0001 |
| CLR (ml min−1) | 54.53 ± 30.87 | 38.09, 70.98 | 25.45 ± 22.14 | 13.65, 37.25 | 0.0021 |
| CLnonrenal (ml min−1) | 2 057.9 ± 1 545.0 | 1 234.8, 2 880.9 | 147.6 ± 87.1 | 101.1, 194.0 | 0.0001 |
| Nortilidine | Placebo | 95% CI | Voriconazole | 95%CI | P-value |
| Cmax (ng ml−1) | 153.7 ± 59.6 | 122.0, 185.5 | 160.0 ± 28.6 | 144.8, 175.3 | 0.3484 |
| tmax (h) | 0.8 ± 0.3 | 0.6, 0.9 | 2.5 ± 1.2 | 1.9, 3.2 | 0.0001 |
| AUC0–24 (h ng ml−1) | 758.7 ± 213.8 | 644.8, 872.7 | 1910 ± 297 | 1752, 2068 | 0.0001 |
| AUC0–∞ (h ng ml−1) | 801.7 ± 233.5 | 676.5, 923.4 | 2417 ± 476 | 2163, 2671 | 0.0001 |
| AUC24–∞ (%) | 5.09 ± 2.0 | 4.00, 6.18 | 20.05 ± 7.6 | 16.00, 24.09 | |
| t1/2 (h) | 5.9 ± 0.9 | 5.5, 6.4 | 10.0 ± 2.8 | 8.6, 11.6 | 0.0001 |
| MRT (h) | 5.9 ± 0.6 | 5.6, 6.2 | 9.1 ± 0.6 | 8.8, 9.4 | 0.0001 |
| Ae0–24 (%) | 5.06 ± 2.29 | 3.84, 6.29 | 8.67 ± 4.83 | 6.10, 11.24 | 0.0076 |
| CLR (ml min−1) | 94.1 ± 38.3 | 73.7, 114.5 | 63.8 ± 35.6 | 44.8, 82.7 | 0.0290 |
| Bisnortilidine | Placebo | 95% CI | Voriconazole | 95%CI | P-value |
| tmax (h) | 1.6 ± 1.2 | 1.0, 2.2 | 10.6 ± 6.3 | 7.3, 13.9 | 0.0001 |
| AUC0–24 | 15.2 ± 3.6 | 13.3, 17.1 | 11.7 ± 2.9 | 10.2, 13.3 | 0.0013 |
Differences between placebo and voriconazole treatment were assessed by use of the Wilcoxon matched pairs signed rank test. Cmax, maximum observed plasma concentration; tmax, time to reach maximum observed plasma concentration; AUC0–24, AUC from time 0 to last measurable concentration; AUC0–∞, AUC from zero to infinity; AUC24–∞, extrapolated AUC; t1/2, terminal elimination half-life; MRT, mean residence time; Ae0–24, amount excreted in urine from 0 to 24 h; CL/F, total body clearance for extravascular administration where F is the fraction of dose absorbed; CLR, renal clearance; CLnonrenal, nonrenal clearance.
Nortilidine
In every participant AUC0–24 and AUC0–∞ of nortilidine increased after treatment with voriconazole in comparison with treatment with placebo (average 2.5- and threefold), but the formation of nortilidine was significantly delayed (tmax 2.5 ± 1.2 h vs. 0.8 ± 0.3 h, P < 0.0001, Figure 2b). Cmax did not differ significantly, but a significant increase in tmax, t1/2 and Ae0–24 was observed (Table 1). After intake of voriconazole, CLR of nortilidine was significantly reduced to 68% of renal clearance after intake of placebo (P < 0.0001).
Bisnortilidine
Compared with placebo, after treatment with voriconazole AUC0–24 of bisnortilidine decreased significantly (11.7 ± 2.9 vs. 15.2 ± 3.6 h*ng ml−1, P < 0.0013, Figure 2c), and maximum concentrations observed were delayed by 9 h (Table 1).
Cold pressor test
There was a significant reduction of pain and withdrawal threshold compared with placebo 0.75 h after administration of tilidine (pain threshold: 2.75 ± 3.59 s vs. 6.69 ± 5.72 s, P < 0.0131; withdrawal threshold: 18.0 ± 24.1 s vs. 36.6 ± 28.7 s, P < 0.001. At this point in time plasma concentration of nortilidine was significantly reduced after administration of voriconazole in comparison with placebo (97.0 ± 39.6 vs. 135.8 ± 44.9 ng ml−1, P= 0.0017). At later points in time (1.75 h and 2.75 h), plasma concentration of nortilidine after intake of voriconazole was even higher compared with placebo. No significant difference in time until pain threshold rather withdrawal threshold between placebo and voriconazole could be observed 1.75 h and 2.75 h after administration of tilidine (Figure 3). The area under the withdrawal threshold curve corrected for the respective baseline was significantly smaller after administration of voriconazole in comparison with placebo (35.9 ± 60.9 h*s vs. 66.0 ± 51.2 h*s, P < 0.0215), whereas the area under the pain threshold–time curve was not significantly different (11.4 ± 14.0 h*s vs. 13.8 ± 19.9 h*s, P= 0.95).
Figure 3.
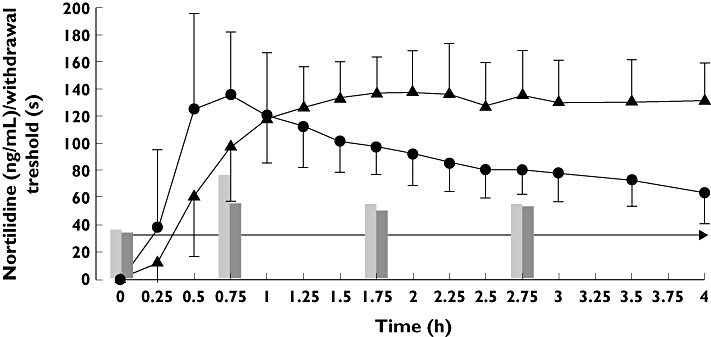
Mean (±SD) plasma concentrations of nortilidine over the first 4 h in relation to withdrawal threshold (cold pressure test) after single oral administration of 100 mg tilidine/naloxone solution in combination with single oral administration of placebo (circles/grey bars) or 400 mg voriconazole (triangle/black bars) to 16 healthy participants
Safety and tolerability
The study drugs were well tolerated and no serious adverse drug events occurred. All adverse drug events (ADE) were mild and transient. On average the volunteers reported at least one ADE per study day. After administration of voriconazole the incidence of ADEs doubled from 40 after placebo to 79 (P < 0.0001). The most frequent ADEs after administration of tilidine and voriconazole were mild dizziness (93.8%), nausea (75%), headache (56.3%), visual disturbances/photophobia (50%), vomiting (37.5%) and itching (31.3%). Intermittent nausea occurred about 5.4 h (individual differences 0.25–10.5 h) after administration of tilidine, and two of the volunteers had to be treated with dimenhydrinate because of recurrent vomiting. Visual disturbances were reported on average 80 min after administration of voriconazole (range 10–195 min) until at least 125 min. No visual disturbances were observed during tilidine alone. After administration of tilidine with placebo the most common ADEs were mild and transient dizziness (75%), headache (43.8%), itching (31.3%), sensation of heat (31.3%) and intermittent nausea with recurrent vomiting (25%).
Discussion
In this clinical study, major interaction between tilidine and voriconazole was observed resulting in a 20-fold increase of tilidine exposure. Consequently, the apparent oral clearance was reduced by 96.7%, indicating an almost abolished first-pass metabolism. Tilidine is characterized as a high extraction drug subject to pronounced first-pass metabolism resulting in a low absolute bioavailability of 7.6% [7]. Based on in vitro data previously published [9], it was proposed that the antifungal voriconazole might inhibit the CYP3A4- and CYP2C19-mediated N-demethylation of tilidine to nortilidine.
The therapeutic activity of tilidine is mainly related to its metabolite nortilidine. Maximum plasma concentrations of nortilidine were observed within an hour (0.8 h) after administration of the tilidine solution. These plasma concentrations were almost fivefold higher than the maximum plasma concentrations of tilidine itself. During voriconazole treatment tmax of nortilidine was prolonged to 2.5 h, suggesting inhibition of nortilidine formation. However, Cmax of nortilidine was unchanged compared with placebo, whereas Cmax of tilidine was almost 10-fold increased. Furthermore, nortilidine AUC was threefold increased during voriconazole treatment. This was unexpected since inhibition of metabolism will usually result in a reduced rate of formation of the metabolite and reduced exposure if the metabolite elimination is not altered. It must therefore be proposed that the elimination of nortilidine is also influenced by voriconazole.
Hajda et al.[7] have shown that only one-third of the tilidine dose was available in the systemic circulation as nortilidine, although two-thirds of the dose were metabolized to nortilidine. This is due to the fact that during the first-pass metabolism of tilidine to nortilidine in the liver half of the amount of the formed nortilidine is further metabolized to bisnortilidine before it is released to the systemic circulation (sequential metabolism). So far it is not known which enzymes are involved in the secondary N-demethylation step. This has not been studied yet due to lack of bisnortilidine reference substance, which is no longer available. Our data suggest that the sequential first-pass metabolism of tilidine is inhibited after administration of voriconazole. Although no quantification of bisnortilidine could be carried out, bisnortilidine concentrations in relative units could be compared between the two study parts (placebo and voriconazole). A marked delay of tmax (9 h) of bisnortilidine following voriconazole administration and a significant reduction of the AUC0–24 of bisnortilidine were observed. Inhibition of the nortilidine N-demethylation to bisnortilidine would support these findings. In addition, this also can explain the increased AUC of nortilidine after voriconazole treatment.
With tilidine there is a rather complex situation, as it seems to be the same enzymes involved in the sequential first-pass metabolism to bisnortilidine, with nortilidine as an intermediate that is the active principle of this drug. Inhibition of both N-demethylation steps results in accumulation of the active metabolite nortilidine, as its metabolism to bisnortilidine seems to be the crucial step for the rate of overall metabolism.
The interaction between voriconazole and the opioid fentanyl is much more simple [13]. The antifungal voriconazole inhibits the N-dealkylation of fentanyl to norfentanyl via CYP3A4, which is the primary step of fentanyl elimination. The AUC of fentanyl increased 1.5-fold, whereas the AUC of norfentanyl decreased 2.2-fold after intake of voriconazole in comparison with the control group. Therefore, caution should be exercised during long-lasting fentanyl treatment, because elevated fentanyl concentrations by metabolic inhibition with voriconazole may lead to respiratory depression.
Both renal clearances of tilidine and nortilidine significantly decreased twofold and 1.5-fold, respectively, after intake of voriconazole. We are confident that this is not a result of the voriconazole treatment, but rather an artefact due to the study conditions. Considering the short half-life of tilidine of 3–5 h, urine collection over 24 h is sufficient for almost complete renal excretion. During voriconazole, however, terminal elimination half-life was extended to 8–10 h. It can be assumed that excretion of tilidine will not be complete after 24 h. This is supported by the plasma concentration data after 24 h. Although the extrapolated AUC of tilidine was similar with 7.9% after voriconazole and 10.5% after placebo (Table 1), the absolute AUC24–∞ after voriconazole was higher than the total AUC after placebo (103.4 vs. 61.7).
In addition, voriconazole reduced the nonrenal clearance of tilidine by 93% and markedly inhibited the tilidine metabolism, which is mediated by hepatic CYP3A4 and CYP2C19 [9]. Unfortunately, the study design does not allow any conclusions on the contribution of CYP2C19 and CYP3A4 to the overall tilidine metabolism. CYP2C19 genotype is responsible for considerable interindividual differences in pharmacokinetics of other drugs, which might lead either to impaired drug metabolism [20] or subtherapeutic drug exposure [21, 22]. Although the involvement of polymorphic CYP2C19 was known from in vitro data [9], we did not stratify the participants according to their genotype. However, their CYP2C19 genotype was assessed using standard methods described elsewhere [23]; 14 participants were identified as homozygous CYP2C19 wild-type, two were heterozygous for *1/*2, and none was a poor metabolizer.
In the present study we also investigated the alteration of the pharmacodynamics of the opioid tilidine in combination with voriconazole by using the cold pressor test. To exclude significant differences in pain perception between study days 1 and 15, we compared baseline pain threshold and withdrawal threshold before administration of tilidine. No significant differences were found, indicating baseline pain sensation was unchanged throughout the study. Following administration of tilidine and placebo individuals showed a significantly longer tolerance of pain stimulus, which affected the pain threshold and withdrawal threshold at all times (0.75 h, 1.75 h, 2.75 h) compared with baseline. The reduction of the ΔAUC of the withdrawal threshold (reduced pain tolerance) after administration of voriconazole is mainly due to a significant difference in pain sensation at 0.75 h (pain threshold and withdrawal threshold). At this time the mean plasma concentrations of nortilidine after voriconazole were reduced, which also indicates a lower pain tolerance due to a lower concentration of the analgesic active metabolite nortilidine. At the later time points (1.75 h, 2.75 h) the plasma concentrations of nortilidine following voriconazole administration were even higher, but the pain and withdrawal thresholds were similar to placebo. One possible reason for the fact that the pain sensation did not correspond well to plasma concentrations of nortilidine could be a missing relationship between the nortilidine plasma concentrations and its concentrations at the µ-opioid receptors in the cerebral tissue. During the first 3 h voriconazole caused a delay of analgesic effect after tilidine that was obvious at 0.75 h and abated subsequently (1.75 h, 2.75 h). In general the combination of tilidine and naloxone in an oral formulation does not reduce or antagonize the analgesic activity. In this interaction study theoretically the antagonist naloxone could have influenced the pharmacokinetics and pharmacodynamics of tilidine and its metabolites. However, literature shows that naloxone is exclusively and extensively metabolized by glucuronyltransferases [24], resulting in very low bioavailability after oral administration of 2% [25]. Thus it is unlikely that naloxone could have influenced the results of the interaction between tilidine and voriconazole.
Another objective of this study was to assess the tolerance of the combination of the opioid tilidine and the triazole antimycotic voriconazole. The results of the reported ADEs argue for a major increase of ADEs in frequency and intensity after voriconazole. Following the administration of voriconazole, the incidence of ADEs doubled (79 vs. 40). ADEs which could not be judged as a direct consequence of the intake of the study drugs, i.e. fatigue, were excluded from analysis. Because plasma concentrations of tilidine and nortilidine were higher after administration of voriconazole, it is possible that a correlation exists between higher plasma concentrations of the drug and incidence of ADEs. Unfortunately, the sample size was too small to make a definitive statement in the present study. Vomiting occurred in six volunteers about 8 h after the intake of voriconazole or placebo (8.5 h after voriconazole and 12.5 h after placebo, individual differences 3–13 h). It can be anticipated that there was no influence on the pharmacokinetics of tilidine since absorption of the tilidine solution is very rapid with tmax always <1 h.
In summary, a major pharmacokinetic interaction between tilidine and voriconazole was observed, which was also partly associated with changes in the analgesic effect. Voriconazole inhibits both metabolic steps in the sequential metabolism of tilidine, resulting in an increased exposure of the active nortilidine. Furthermore, the number of ADEs doubled after administration of these two drugs in typically used clinical doses. Therefore we recommend avoiding the combination of tilidine/naloxone and voriconazole in clinical practice.
Competing interests
None to declare.
We thank Ms Magdalena Longo, Ms Brigitte Tayrouz, Ms Jutta Kocher and Ms Marlies Stützle-Schnetz for their skilful technical and consultative assistance.
REFERENCES
- 1.International Narcotics Control Board. Narcotic Drugs: Estimated World Requirements for 2007 and Statistics for 2005 (E/INCB/2006/2) New York: United Nations; 2007. [Google Scholar]
- 2.Nikolaus T, Zeyfang A. Pharmacological treatments for persistent non-malignant pain in older persons. Drugs Aging. 2004;21:19–41. doi: 10.2165/00002512-200421010-00003. [DOI] [PubMed] [Google Scholar]
- 3.Flöter T, Brunnmuller U. Tilidine/naloxon retard in long-term administration in chronic pain and multimorbidity. Multicenter study of long-term tolerance and effectiveness in 2 years observation. Fortschr Med Orig. 2002;120:29–35. [PubMed] [Google Scholar]
- 4.Wörz R, Wörz E. Long-term treatment of chronic pain with tilidine-naloxone. An analysis of 50 patients with chronic pain conditions of non-malignant origin. Fortschr Med. 1995;113:388–92. [PubMed] [Google Scholar]
- 5.Vollmer KO, Thomann P, Hengy H. Pharmacokinetics of tilidine and metabolites in man. Arzneimittelforschung. 1989;39:1283–8. [PubMed] [Google Scholar]
- 6.Thierry C, Boeynaems JM, Paolo M. Actions of tilidine and nortilidine on cloned opioid receptors. Eur J Pharmacol. 2005;506:205–8. doi: 10.1016/j.ejphar.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 7.Hajda JP, Jähnchen E, Oie S, Trenk D. Sequential first-pass metabolism of nortilidine: the active metabolite of the synthetic opioid drug tilidine. J Clin Pharmacol. 2002;42:1257–61. doi: 10.1177/009127002762491352. [DOI] [PubMed] [Google Scholar]
- 8.Seiler KU, Jähnchen E, Trenk D, Brennscheidt U, Heintz B. Pharmacokinetics of tilidine in terminal renal failure. J Clin Pharmacol. 2001;41:79–84. doi: 10.1177/00912700122009863. [DOI] [PubMed] [Google Scholar]
- 9.Weiss J, Sawa E, Riedel K-D, Haefeli WE, Mikus G. In vitro metabolism of the opioid tilidine and interaction of tilidine and nortilidine with CYP3A4, CYP2C19, and CYP2D6. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:275–82. doi: 10.1007/s00210-008-0294-7. [DOI] [PubMed] [Google Scholar]
- 10.Purkins L, Wood N, Greenhalgh K, Allen MJ, Oliver SD. Voriconazole, a novel wide-spectrum triazole: oral pharmacokinetics and safety. Br J Clin Pharmacol. 2003;56:10–6. doi: 10.1046/j.1365-2125.2003.01993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyland R, Jones BC, Smith DA. Identification of the cytochrome P450 enzymes involved in the N-oxidation of voriconazole. Drug Metab Dispos. 2003;31:540–7. doi: 10.1124/dmd.31.5.540. [DOI] [PubMed] [Google Scholar]
- 12.Saari TI, Laine K, Leino K, Valtonen M, Neuvonen PJ, Olkkola KT. Effect of voriconazole on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Clin Pharmacol Ther. 2006;79:362–70. doi: 10.1016/j.clpt.2005.12.305. [DOI] [PubMed] [Google Scholar]
- 13.Saari TI, Laine K, Neuvonen M, Neuvonen PJ, Olkkola KT. Effect of voriconazole and fluconazole on the pharmacokinetics of intravenous fentanyl. Eur J Clin Pharmacol. 2008;64:25–30. doi: 10.1007/s00228-007-0398-x. [DOI] [PubMed] [Google Scholar]
- 14.FDA Antiviral Drugs Advisory Committtee. Briefing Document for Voriconazole (Oral and Intravenous Formulations) 4 October 2001. Available at http://www.fda.gov/ohrms/dockets/ac/01/briefing/3792b2_02_FDA-voriconazole.pdf (last accessed 15 June 2009.
- 15.Jones SF, McQuay HJ, Moore RA, Hand CW. Morphine and ibuprofen compared using the cold pressor test. Pain. 1988;34:117–22. doi: 10.1016/0304-3959(88)90156-X. [DOI] [PubMed] [Google Scholar]
- 16.Garcia de Jalon PD, Harrison FJ, Johnson KI, Kozma C, Schnelle K. A modified cold stimulation technique for the evaluation of analgesic activity in human volunteers. Pain. 1985;22:183–9. doi: 10.1016/0304-3959(85)90178-2. [DOI] [PubMed] [Google Scholar]
- 17.Eckhardt K, Li S, Ammon S, Schänzle G, Mikus G, Eichelbaum M. Same incidence of adverse drug events after codeine administration irrespective of the genetically determined differences in morphine formation. Pain. 1998;76:27–33. doi: 10.1016/s0304-3959(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 18.Blasco T, Bayes R. Unreliability of the Cold Pressor Test Method in pain studies. Methods Find Exp Clin Pharmacol. 1988;10:767–72. [PubMed] [Google Scholar]
- 19.Shah VP, Midha KK, Findlay JW, Hill HM, Hulse JD, McGilveray IJ, McKay G, Miller KJ, Patnaik RN, Powell ML, Tonelli A, Viswanathan CT, Yacobi A. Bioanalytical method validation – a revisit with a decade of progress. Pharm Res. 2000;17:1551–7. doi: 10.1023/a:1007669411738. [DOI] [PubMed] [Google Scholar]
- 20.Herrlin K, Massele AY, Jande M, Alm C, Tybring G, Abdi YA, Wennerholm A, Johansson I, Dahl ML, Bertilsson L, Gustafsson LL. Bantu Tanzanians have a decreased capacity to metabolize omeprazole and mephenytoin in relation to their CYP2C19 genotype. Clin Pharmacol Ther. 1998;64:391–401. doi: 10.1016/S0009-9236(98)90070-4. [DOI] [PubMed] [Google Scholar]
- 21.Sim SC, Risinger C, Dahl M-L, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther. 2006;79:103–13. doi: 10.1016/j.clpt.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Baldwin RM, Ohlsson S, Pedersen RS, Mwinyi J, Ingelman-Sundberg MA, Eliasson E, Bertilsson L. Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol. 2008;65:767–74. doi: 10.1111/j.1365-2125.2008.03104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss J, Ten Hoevel MM, Burhenne J, Walter-Sack I, Hoffmann MM, Rengelshausen J, Haefeli WE, Mikus G. CYP2C19 genotype is a major factor contributing to the highly variable pharmacokinetics of voriconazole. J Clin Pharmacol. 2009;49:196–204. doi: 10.1177/0091270008327537. [DOI] [PubMed] [Google Scholar]
- 24.Handal KA, Schauben JL, Salamone F. Naloxone. Ann Emerg Med. 1983;12:438–45. doi: 10.1016/s0196-0644(83)80343-6. [DOI] [PubMed] [Google Scholar]
- 25.Albeck H, Woodfield S, Kreek MJ. Quantitative and pharmacokinetic analysis of naloxone in plasma using high-performance liquid chromatography with electrochemical detection and solid-phase extraction. J Chromatogr. 1989;488:435–45. doi: 10.1016/s0378-4347(00)82967-9. [DOI] [PubMed] [Google Scholar]