

Abstract
AIMS
Although in vitro studies indicate that oxazepam is an isoform-selective substrate probe for UDP-glucuronosyltransferase 2B15, the utility of this drug as an in vivo probe is uncertain. The main aim of this study was to determine whether common missense polymorphisms in the UGT2B15 gene (D85Y and K523T) are associated with altered oxazepam pharmacokinetics and pharmacodynamics. We also determined the possible influence of a common deletion polymorphism in the gene encoding UGT2B17, which shows substantial substrate specificity overlap with UGT2B15.
METHODS
Thirty healthy male subjects were administered 15 mg of oxazepam by mouth followed by plasma oxazepam concentration monitoring for 36 h, and pharmacodynamic testing for 8 h. Genotypes were determined by genomic polymerase chain reaction and commercial 5′-nuclease assays.
RESULTS
Allele frequencies for D85Y, K523T, UGT2B17del were 47%, 23% and 19%, respectively. Median oxazepam apparent oral clearance was significantly lower in 85YY subjects (1.62 ml min−1 kg−1) compared with 85DD subjects (3.35 ml min−1 kg−1; P= 0.003, Student–Newman–Keuls test), whereas 85DY subjects were intermediate (2.34 ml min−1 kg−1; P= 0.018 vs. 85DD, P= 0.034 vs. 85YY). Regression analysis indicated that UGT2B15 D85Y genotype accounted for 34% of interindividual variability. However, neither UGT2B15 K523T nor UGT2B17del was associated with altered oxazepam disposition. Furthermore, no differences in pharmacodynamic measures, including quantitative electroencephalography, digit-symbol substitution test, self- or observer-rated visual analogue scales, could be demonstrated for any of the polymorphisms evaluated.
CONCLUSIONS
These results identify UGT2B15 D85Y as a major determinant of oxazepam clearance, and indicate that oxazepam may be useful as an in vivo probe for glucuronidation by UGT2B15.
Keywords: glucuronidation, oxazepam, pharmacogenetics, pharmacogenomics, pharmacokinetics, UGT2B15
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
UDP-glucuronosyltransferase (UGT) 2B15 is a major drug glucuronidation enzyme expressed in human liver.
Oxazepam is an isoform-selective probe drug that is being used for in vitro studies of UGT2B15.
The most common UGT2B15 missense polymorphisms (D85Y and K523T) are correlated with variable oxazepam glucuronidation in human liver bank samples.
UGT2B17 is also expressed in liver and has high sequence homology and substrate specificity overlap with UGT2B15.
WHAT THIS STUDY ADDS
UGT2B15 D85Y polymorphism is identified as a major determinant of oxazepam disposition, accounting for as much as 34% of interindividual variability in oxazepam apparent oral clearance.
An effect of the UGT2B15 K523T or the UGT2B17 deletion polymorphisms on oxazepam disposition could not be detected.
Provides evidence supporting the use of oxazepam as an isoform-selective in vivo probe for studies of variability in UGT2B15 activity.
Introduction
Glucuronidation mediated by the UDP-glucuronosyltransferase (UGT) family of conjugation enzymes is an important metabolic pathway for the elimination of xenobiotic and endobiotic compounds in humans and other mammalian species [1]. Consequently, interindividual variability in the rate of glucuronidation of drugs and endogenous compounds in people can contribute to altered risk for drug toxicity and disease susceptibility. Various factors may contribute to glucuronidation variability, including genetic polymorphisms and drug–drug interactions resulting from enzyme induction or inhibition [2]. Essential tools for the study of these factors are UGT isoform-selective substrate probes (also known as UGT phenotyping probes) [3]. Ideally, these probes are metabolized primarily through glucuronidation by a single UGT isoform and are either drugs or other nontoxic or endogenous compounds that can be used for both in vitro and in vivo studies. Examples of UGT phenotyping probes include bilirubin (endogenous metabolite of haeme and probe of UGT1A1) and zidovudine (an antiviral drug and probe of UGT2B7) [3]. However, as yet not all of the major UGT isoforms expressed in human liver, including UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, 2B15 and 2B17, have well-validated phenotyping probes.
UGT2B15 was originally identified as being important in the glucuronidation of androgenic steroids [4]. However, subsequent studies also indicated a role in the metabolism of drugs, drug metabolites and other xenobiotics, including E-4-hydroxytamoxifen, 5-hydroxyrofecoxib, eugenol, 8-hydroxyquinoline, phenolphthalein, 4′-hydroxyphenytoin, nandrolone, and bisphenol A [5–9]. In previous work we have identified oxazepam as a potential phenotyping probe for UGT2B15 primarily based on in vitro studies using human liver microsomes (HLM) and recombinant UGT isoforms [10].
Oxazepam is a 3-hydroxy-1,4-benzodiazepine derivative used in clinical practice as a sedative and anxiolytic agent [11–13]. Following administration, clearance of this drug is primarily via glucuronidation in liver and followed by excretion of a significant proportion of the glucuronide into urine [11–13]. The only available form is a mixture of R- and S-enantiomers that rapidly racemizes in physiological solutions, but is stabilized via glucuronidation in the liver to S-oxazepam-glucuronide (the major urinary metabolite) and R-oxazepam-glucuronide (a minor urinary metabolite) [14]. Through screening of 12 different recombinant UGTs, we identified UGT2B15 was the most active isoform mediating formation of S-oxazepam-glucuronide [10]. Importantly, we could detect no S-oxazepam glucuronidation activity with UGT2B17, a major hepatic isoform that is 94.5% identical in amino acid sequence to UGT2B15 and also shares substrates such as androgens. We also observed high concordance between UGT2B15 and HLM enzyme Km values for S-oxazepam glucuronidation. On the other hand, R-oxazepam-glucuronide was formed primarily by UGT1A9 and UGT2B7.
Using a bank of HLM, we observed 67% higher median rates of S-oxazepam glucuronidation (and unchanged R-oxazepam glucuronidation) in liver microsomes from male compared with female donors suggesting sex-dependent regulation of UGT2B15 expression [15]. This observation is supported by previous work that showed higher oral clearance of oxazepam in male compared with female subjects [16]. We also identified several common nonsynonymous polymorphisms in the UGT2B15 gene including D85Y (c.253g→t) and K523T (c.1568c→a) that appear to influence UGT2B15 activity. Using HLM, we determined that the D85Y polymorphism was associated with 67% lower median S-oxazepam glucuronidation activities. In support of this observation, recombinant UGT2B15 85Y allozyme was found to glucuronidate oxazepam and several other substrates much slower than the 85D allozyme. Although a clear effect of the K523T substitution was not found in the whole liver bank, about 30% higher S-oxazepam glucuronidation was observed in heterozygous compared with wild-type male livers. The effect of the K523T substitution on recombinant UGT2B15 has not yet been reported.
The main objective of the current study was to determine the effect of UGT2B15 polymorphisms on the pharmacokinetics and pharmacodynamics of oxazepam in human volunteers. We limited our investigation to male subjects to minimize possible confounding effects of sex. In addition to UGT2B15 D85Y and K523T, subjects were also genotyped for the UGT2B17 deletion polymorphism that has been associated with altered androgen glucuronidation in vivo[17]. This work provides further justification for the use of oxazepam as an isoform-selective in vivo as well as an in vitro probe of UGT2B15 activity.
Materials and methods
Subjects
Thirty healthy male subjects aged 18–45 years were recruited from the local Boston area. Health status was based on medical history, physical examination, haematological profile, blood chemistries, urinalysis, and screens for hepatitis and human immunodeficiency virus infection. Subjects were excluded from the study if there was evidence of recent use of a medication that might alter drug glucuronidation including smoking, drinking alcohol, eating Brussels sprouts, cabbage or grapefruit, or taking phenytoin, carbamazepine, rifampin, valproic acid, diclofenac or probenecid. Also excluded were patients receiving sedative drugs, including benzodiazepines. All subjects provided written informed consent and the protocol and consent forms were approved by the Tufts Human Investigation Review Committee, which is the Institutional Review Board at Tufts University School of Medicine.
Study procedures
The study was conducted at the Clinical Psychopharmacology Research Unit at Tufts University School of Medicine. Prior to oxazepam dosing, pharmacodynamic tests (described below) were given to subjects and predose blood samples were obtained. A single 15-mg oral dose of oxazepam was administered with a glass of water and the subjects spent 12 h in the clinic, with blood samples drawn at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12 h post dose. Subjects returned to the study unit the next morning and night for 24- and 32-h blood samples.
Pharmacodynamic tests included subject sedative effect scores, digit symbol substitution test (DSST) and quantitative EEG, as previously described [18, 19]. All tests were administered twice prior to dosing and at each time point that blood samples were collected (up to 8 h). Briefly, sedative effect scores were recorded on 100-mm visual analogue scales by the subject (self-rated) and by a trained observer. For the DSST, subjects were asked to write down as many correct digit for symbol substitutions as possible within a 2-min period, and the number of correct substitutions was recorded. Quantitative eight-lead EEG recordings were made using a Stellate Harmonie-Routine EEG system (Stellate Systems Inc., Montreal, Canada). The EEG was recorded in 4-s epochs for as long as necessary to ensure (by visual inspection) collection of at least 2 min of artefact-free information within a time window of 5 min around each predetermined time point. The relative percent β band amplitude was calculated by dividing the β band amplitude by the total amplitude minus the delta wave amplitude. For data analysis, the two predose baseline ratings were averaged and all scores after administration were expressed as the increment or decrement relative to the mean predose value. The areas under the effect curve to 8 h (AUEC0–8 h) were then calculated from plots of effect values vs. time.
Plasma oxazepam assay
Plasma oxazepam concentrations were determined by high-pressure liquid chromatography with mass spectrometry (HPLC-MS) detection after liquid–liquid extraction. Briefly, 1-ml plasma samples were added to 15-ml polypropylene tubes containing 20 ng of D5-oxazepam (Cerilliant, Round Rock, TX, USA), vortexed for 2 min with 2 ml of toluene/isoamyl alcohol mix (98.5:1.5), and centrifuged at 4000 g for 15 min. The organic supernatant was transferred to HPLC vials and dried in a heated vacuum oven set at <40°C. Extracts were then resuspended in HPLC-MS mobile phase (a 1:1 mixture of 0.5% formic acid in water and acetonitrile). HPLC-MS apparatus consisted of a Surveyor HPLC connected to a Deca XP ion trap detector with electrospray source (Thermo Fisher Scientific, Waltham, MA, USA). Chromatographic separation was achieved using a 150 mm × 2 mm Synergi Fusion column (Phenomenex, Torrance, CA, USA) with a mobile phase run at 0.3 ml min−1. For oxazepam, M/Z+ ion transitions were 287 → 269 → 241, while ion transitions for D5-oxazepam were 292 → 274 → 246. Calibration samples were constructed using oxazepam (Cerilliant) added to blank bovine serum, which had identical oxazepam recovery compared with human plasma in preliminary studies. Oxazepam calibration curves were linear (R2 > 0.98) over the range 1–1000 ng ml−1. At the lowest quantifiable concentration (1 ng ml−1) the deviation from nominal concentration was consistently between −20% and +20%, while the coefficient of variation of duplicates was <30%. Quality control samples (100 and 1000 ng ml−1) were run in duplicate with each assay and showed deviations of −15% to 10% from nominal concentration and <15% coefficient of variation between duplicates.
UGT2B15 and UGT2B17 genotyping
Genomic DNA was isolated using a spin column kit (QiaAmp DNA Blood Mini Kit; Qiagen, Germantown, MD, USA) from whole blood buffy coat preparations collected from each subject. UGT2B15 D85Y (c.253g→t; rs1902023) and K523T (c.1568c→a; rs4148269) genotypes were determined for each sample using commercial Taqman assay kits (C_27028164-10 and C_9440184-20; Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions on a real-time polymerase chain reaction (PCR) instrument (Model 7300; Applied Biosystems). The accuracy of genotyping by this method was confirmed by assay of DNA samples with known UGT2B15 D85Y and K523T genotype from a previous study [15]. The presence of the UGT2B17del allele was ascertained by genomic PCR using the following primers that spanned the known deleted region: UGT2B17-del-forward: 5′-TGC ACA GAG TTA AGA AAT GGA GAG ATG TG-3′ and UGT2B17-del-reverse: 5′-GAT CAT CCT ATA TCC TGA CAG AAT TCT TTT G-3′[20]. The presence of the reference (nondeleted) allele was determined with the following set of primers designed to amplify within the known deleted region: UGT2B17-forward: 5′-TGA AAA TGT TCG ATA GAT GGA CAT ATA GTA-3′ and UGT2B17-reverse: 5′-GAC ATC AAA TTT TGA CTC TTG TAG TTT TC-3′[21]. PCR reactions (25 µl volume) included 5 ng genomic DNA and 800 nM of each primer with Platinum Taq Hifi Supermix (Invitrogen, Carlsbad, CA, USA). The PCR method for amplification of the UGT2B17del allele used initial denaturation at 95°C for 5 min, then 45 cycles of 95°C for 30 s, 55°C for 30 s and 72°C for 30 s, followed 72°C for 10 min and 4°C for 1 min. The method for the (nondeleted) UGT2B17 allele used the same conditions, except the 72°C extension time was 1 min instead of 30 s. The presence or absence of PCR products on an ethidium bromide-stained agarose gel with expected sizes of 172 bp (for the UGT2B17 nondeleted allele) and 884 bp (for the UGT2B17del allele) was used to infer the UGT2B17-deletion genotype for each subject. Derived genotype frequencies were evaluated for consistency with Hardy–Weinberg equilibrium using an exact test (https://www.pharmgat.org/pharmgat.org/Tools/exacthweform). The significance of linkage disequilibrium between polymorphisms was assessed by using a pair-wise likelihood-ratio test as implemented in the ARLEQUIN genetic analysis program [22].
Pharmacokinetic parameter estimates
Pharmacokinetic parameters were determined by noncompartmental analysis of plasma oxazepam concentration data for each subject using WinNonlin Version 5 (Mountain View, CA, USA). Parameter estimates included observed maximum plasma concentration (Cmax), time of Cmax (Tmax), elimination half-life (t1/2), as well as the area under the plasma concentration–time curve extrapolated to infinity (AUCtotal). The amount of AUC that was extrapolated beyond the last measured time point (36 h after dose) averaged <3%. Weight-normalized apparent oral clearance (CL/F) was calculated by dividing dose by AUCtotal, and by subject weight with results expressed as ml min−1 kg−1 bodyweight. For descriptive purposes, data were summarized as median and interquartile range since some parameters were non-normally distributed.
Statistical analyses
Statistical analysis of differences in pharmacokinetic and pharmacodynamic parameters between genotype groups were performed using Sigmaplot 11 software (Systat, San Jose, CA, USA). Data were evaluated before analysis for normality of distribution and equal variance, and in cases where these prerequisites failed the data were either log transformed (Tmax, t1/2 and AUCtotal) or rank transformed (all AUEC values) for analysis. Initially data grouped by genotype were evaluated with analysis of variance (anova) and, if indicated, the Student–Newman–Keuls (SNK) method of multiple pair-wise comparisons was used to identify significantly different groups. Because of small homozygous variant genotype group size, the UGT2B15 K523T and UGT2B17del were each evaluated for differences between subjects that were homozygous reference genotype and those that carried at least one variant allele using the unpaired Student's t-test. Two-way anova was also used to detect possible effects of UGT2B15 K523T or UGT2B17del, controlling for UGT2B15 D85Y genotype. A P-value < 0.05 was considered statistically significant.
Finally, multiple linear regression analysis was used to explore the relative contributions of the three UGT2B polymorphisms and patient demographic factors to interindividual variability in oxazepam CL/F values. Dichotomous variables including UGT2B15 K523T and UGT2B17del polymorphism carrier genotypes and racial group were coded as 0 or 1, whereas age and body mass index were included as continuous variables. Two variables were used to describe UGT2B15 D85Y genotype, coded as 85DD/DY = 0 and 85YY = 1 for the recessive genetic model and also as 85DD = 0 and 85DY/YY = 1 for the dominant genetic model. This also allowed for the additive genetic model if both UGT2B15 D85Y variables were found to be significant. The percentage of variance explained by the model was based on the coefficient of determination (R2).
Results
Study population
Thirty male subjects were enrolled and successfully completed the study. Subjects included 24 Whites, four African-Americans, and two Asians with a mean (± SD) subject age of 31 ± 8 years, mean body weight of 78 ± 12 kg, and mean body mass index of 24 ± 3.
UGT genotype and allele frequencies
UGT2B15 D85Y and K523T genotypes could be determined in all 30 subjects, while UGT2B17del genotypes were obtained for all but one subject. DNA from subject 9503 failed to amplify with either the UGT2B17del or the exon 1 primer sets, despite successful amplification with the UGT2B15 D85Y and K523T primer sets. UGT2B15 85DD, 85DY and 85YY genotypes were identified in six (20%), 20 (67%) and four (13%) subjects, respectively. UGT2B15 523KK, 523KT and 523TT genotypes were found in 15 (50%), 14 (47%) and one (3%) subjects, respectively. UGT2B17del +/+, ± and −/− genotypes were established for 20 (69%), six (21%) and three (10%) subjects, respectively. These UGT2B15 D85Y and K523T and UGT2B17del genotype frequencies were consistent with the predicted Hardy–Weinberg distribution with exact test P-values of 0.14, 0.64 and 0.07, respectively. UGT2B15 D85Y, UGT2B15 K523T and UGT2B17del allele frequencies were 47%, 23% and 19% (respectively), which are consistent with frequencies previously reported for a primarily White population [15, 20]. None of the polymorphisms examined showed significant linkage disequilibrium with each other based on pair-wise likelihood-ratio tests (all exact P-values > 0.05).
Oxazepam pharmacokinetics and pharmacodynamics
Figure 1a shows a plot of mean (± SE) plasma oxazepam concentrations vs. time after oxazepam dose for all 30 subjects. Median (interquartile range) Cmax, Tmax, t1/2, AUCtotal and CL/F values for the 30 subjects were 202 ng ml−1 (164–259), 2 h (1.5–3), 5.9 h (4.9–7.8), 1.37 µg ml−1 h−1 (0.94–1.72) and 2.34 ml min−1 kg−1 (2.00–3.30), respectively. Figure 1b shows a plot of mean (± SE) change from baseline values for the EEG β amplitude effect in all 30 subjects. Median (interquartile range) AUEC0–8h values for EEG β amplitude, DSST, observer-rated sedation (OBS) and subject-rated sedation (SED) for the 30 subjects were 17.9 change h−1 (3.6–38.5), 61.3 change h−1 (27.8–101), 26.7 change h−1 (8.0–50.0) and 15.2 change h−1 (−45.8–71.0), respectively.
Figure 1.
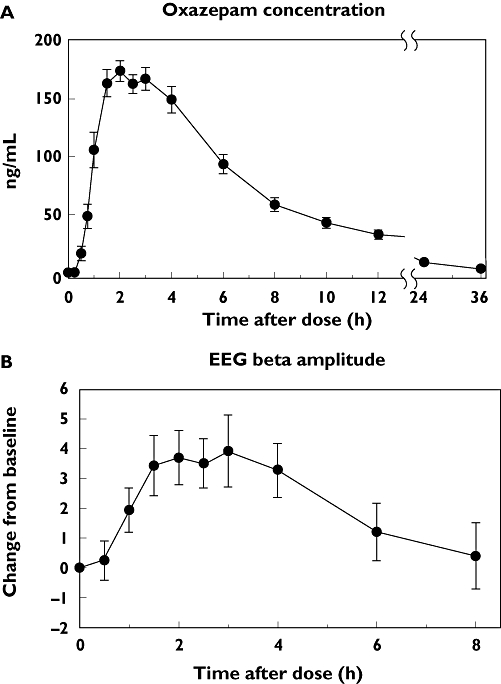
Plasma oxazepam concentrations and associated effect on electroencephalogram (EEG) activity in 30 subjects administered 15 mg oxazepam by mouth. (A) The mean (± SE) concentrations for all 30 subjects at each time point after drug administration. (B) The mean (± SE) changes in EEG β wave amplitudes from predose values at each time point after drug administration for the same 30 subjects
Effect of UGT genotype on oxazepam pharmacokinetics
As shown in Figure 2, plasma oxazepam concentrations differed between subjects grouped by UGT2B15 D85Y genotype, with higher mean concentrations observed in 85YY subjects compared with 85DD subjects, with intermediate values observed in heterozygous DY subjects. These differences were also reflected by pharmacokinetic parameters (given in Table 1), with 85YY subjects showing >140% higher median AUCtotal values compared with 85DD subjects (P < 0.05, SNK test), and 60% higher values compared with DY subjects (P < 0.05). As shown in Figure 3, the most distinct differences between D85Y genotype groups were observed for CL/F values, which showed 52% lower median CL/F values in 85YY subjects compared with 85DD subjects [P= 0.003, SNK test; 95% confidence interval (CI) for the difference in means 0.93, 3.00]. 85DY subjects showed a distinct intermediate phenotype with lower CL/F values compared with 85DD subjects (P= 0.018, SNK test; 95% CI for the difference in means 0.11, 1.81) and higher CL/F values compared with 85YY subjects (P= 0.034, SNK test; 95% CI for the difference in means 0.41, 1.60). Smaller differences between D85Y genotype groups were observed for Cmax and t1/2 values, with higher median Cmax and t1/2 values in 85YY vs. DY subjects (P < 0.05, SNK test), but there were no statistically significant differences for other group comparisons. Tmax did not differ between subjects (P > 0.05, anova).
Figure 2.
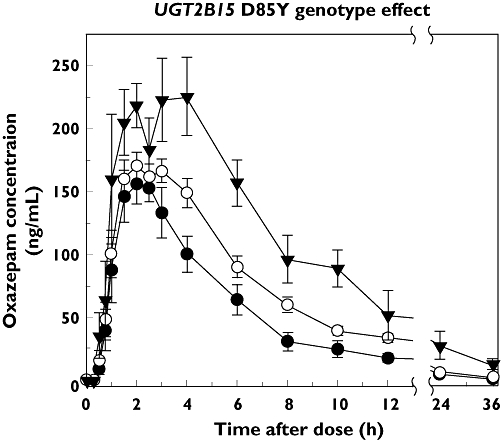
Effect of UGT2B15 D85Y genotype on plasma oxazepam concentrations in 30 subjects administered a 15-mg oral dose of oxazepam. Shown at each time point after administration are the mean (± SE) concentrations for subjects with either 85DD (n= 6), DY (n= 20) or 85YY (n= 4) genotypes. Derived pharmacokinetic parameters are given in Table 1. D/D (–•–); D/Y (–○–); Y/Y (–▾–)
Table 1.
Influence of UGT2B15 D85Y genotype on oxazepam pharmacokinetic and pharmacodynamic parameters measured in 30 male subjects
| UGT2B15 D85Y genotype | ||||
|---|---|---|---|---|
| Parameter (units) | DD (n= 6) | DY (n= 20) | YY (n= 4) | anova (P-value) |
| Cmax (ng ml−1) | 177 (152–200) | 208a (169–257) | 269a (228–288) | 0.011 |
| Tmax (h) | 2.0 (1.4–2.6) | 2.0 (1.5–2.9) | 2.5 (1.3–3.0) | 0.65 |
| t1/2 (h) | 5.5 (4.7–9.7) | 5.5 (4.6–7.4)b | 9.5 (6.8–11.2)b | 0.040 |
| AUCtotal (µg ml−1 h−1) | 0.93cd (0.77–1.20) | 1.37ce (0.98–1.69) | 2.26de (1.76–3.48) | <0.001 |
| CL/F (ml min−1 kg−1) | 3.35fg (2.84–4.17) | 2.34fh (2.00–3.30) | 1.62gh (0.95–2.10) | 0.003 |
| EEG AUEC0–8h (change h−1) | 25.7 (8.3–38.2) | 17.9 (4.1–50.0) | −6.26 (−36.3–49.0) | 0.67 |
| DSST AUEC0–8h (change h−1) | 37.3 (−8.4–87) | 62.8 (30.2–105) | 56.1 (−28.4–103) | 0.53 |
| OBS AUEC0–8h (change h−1) | 18.3 (6.0–40.3) | 32.9 (7.9–62.6) | 33.5 (5.3–257) | 0.68 |
| SED AUEC0–8h (change h−1) | 24.4 (−62.4–82.6) | 14.6 (−65.6–86.1) | 5.2 (−107–51.1) | 0.83 |
AUEC, area under the effect curve; EEG, electroencephalogram relative β wave amplitude; DSST, digit-symbol substitution test; OBS, observer-rated sedation score; SED, subject-rated sedation score.
Identifies median values that are significantly different from each other (P < 0.05) by Student–Newman–Keuls pair-wise multiple comparisons testing. Tmax, t1/2 and AUCtotal values were log transformed and AUEC values were rank transformed since data were not normally distributed. Shown for each parameter are the genotype group median (interquartile range) values and the P-value of group comparisons by anova.
Figure 3.
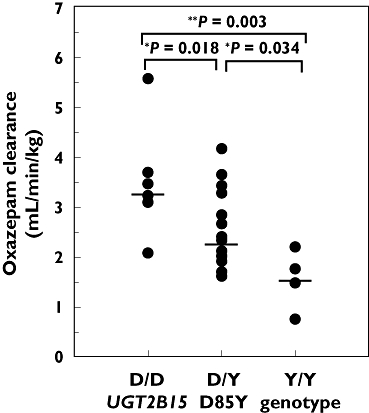
Effect of UGT2B15 D85Y genotype on oxazepam oral clearance in 30 subjects administered a 15-mg oral dose of oxazepam. Shown are the body weight normalized apparent oral clearance values determined for each subject and grouped by UGT2B15 85DD (n= 6), DY (n= 20) and 85YY (n= 4) genotypes. Also shown are the genotype group median values (horizontal line) and P-values for group comparisons by Student–Newman–Keuls test
In contrast to UGT2B15 D85Y, neither UGT2B15 K523T (Table 2) nor UGT2B17del (Table 3) was associated with altered oxazepam pharmacokinetic values (P > 0.05, anova). Since for both UGT2B15 K523T and UGT2B17del the numbers of homozygous variant subjects were somewhat limited (one and three subjects, respectively), analyses were also performed with an unpaired t-test by grouping subjects into either reference genotype or variant carrier groups for each UGT variant. However, neither UGT2B15 K523T nor UGT2B17del was found to influence any of the pharmacokinetic parameters (P > 0.05, unpaired t-test). Analyses by two-way anova to control for the UGT2B15 D85Y effect also failed to identify any effect of either UGT2B15 K523T or UGT2B17del. Finally, analyses were then limited to the 20 identified 85DY (heterozygous) subjects, also to control for the UGT2B15 D85Y effect, but again no influence of UGT2B15 K523T or the UGT2B17del could be detected.
Table 2.
Influence of UGT2B15 K523T genotype on oxazepam pharmacokinetic and pharmacodynamic parameters measured in 30 male subjects
| UGT2B15 K523T genotype | ||||
|---|---|---|---|---|
| Parameter (units) | KK (n= 15) | KT (n= 14) | TT (n= 1) | anova (P-value) |
| Cmax (ng ml−1) | 200 (188–218) | 227 (158–276) | 163 | 0.14 |
| Tmax (h) | 2.5 (2.0–3.0) | 2.0 (1.5–2.5) | 1.5 | 0.10 |
| t1/2 (h) | 6.5 (4.8–7.8) | 5.7 (4.7–8.8) | 5.3 | 0.73 |
| AUCtotal (µg ml−1 h−1) | 1.40 (0.94–1.63) | 1.37 (0.87–1.82) | 1.29 | 0.52 |
| CL/F (ml min−1 kg−1) | 2.35 (2.03–3.48) | 2.37 (1.89–3.32) | 2.26 | 0.45 |
| EEG AUEC0–8h (change h−1) | 17.9 (1.96–38.7) | 12.6 (2.06–36.7) | 56.1 | 0.34 |
| DSST AUEC0–8h (change h−1) | 45.3 (–7.7–84.9) | 65.1 (35.0–102) | 82.1 | 0.48 |
| OBS AUEC0–8h (change h−1) | 24.1 (8.00–44.0) | 23.5 (6.3–54.4) | 88.3 | 0.41 |
| SED AUEC0–8h (change h−1) | 14.9 (−45.8–77.9) | 13.1 (−75.5–57.5) | 119 | 0.33 |
AUEC, area under the effect curve; EEG, electroencephalogram relative β wave amplitude; DSST, digit-symbol substitution test; OBS, observer-rated sedation score; SED, subject-rated sedation score. Tmax, t1/2 and AUCtotal values were log transformed and AUEC values were rank transformed since data were not normally distributed. Shown for each parameter are the genotype group median (interquartile range) values except for the TT genotype, which had only one subject. Also shown are the P-values of group comparisons by anova on ranks.
Table 3.
Influence of UGT2B17del genotype on oxazepam pharmacokinetic and pharmacodynamic parameters measured in 29 male subjects
| UGT2B17del genotype | ||||
|---|---|---|---|---|
| Parameter (units) | +/+ (n= 20) | +/− (n= 6) | −/− (n= 3) | anova (P-value) |
| Cmax (ng ml−1) | 201 (170–258) | 181 (157–221) | 263 (201–276) | 0.18 |
| Tmax (h) | 2.0 (1.5–3.0) | 2.3 (1.9–2.5) | 1.5 (1.0–3.0) | 0.12 |
| t1/2 (h) | 5.7 (5.0–7.4) | 6.8 (5.0–9.6) | 7.9 (4.8–11) | 0.27 |
| AUCtotal (µg ml−1 h−1) | 1.37 (0.98–1.70) | 1.08 (0.78–1.44) | 2.17 (0.98–2.35) | 0.13 |
| CL/F (ml min−1 kg−1) | 2.34 (1.94–3.39) | 2.95 (2.02–3.52) | 2.21 (1.77–3.48) | 0.82 |
| EEG AUEC0–8h (change h−1) | 18.4 (2.0–38.4) | 8.9 (−24.7–50.8) | 20.6 (11.8–61.4) | 0.50 |
| DSST AUEC0–8h (change h−1) | 65.1 (31.8–101) | 43.9 (0.0–100) | 44.5 (−53–116) | 0.64 |
| OBS AUEC0–8h (change h−1) | 37.8 (10.2–50.3) | 12.9 (−5.8–48.5) | 17.1 (8.0–326) | 0.45 |
| SED AUEC0–8h (change h−1) | 14.3 (−57.2–61.7) | 42.2 (−39.7–174) | 61.1 (−140–96.9) | 0.30 |
AUEC, area under the effect curve; EEG, electroencephalogram relative β wave amplitude; DSST, digit-symbol substitution test; OBS, observer-rated sedation score; SED, subject-rated sedation score. Tmax, t1/2 and AUCtotal values were log transformed and AUEC values were rank transformed since data were not normally distributed. Shown for each parameter are the genotype group median (interquartile range) values and the P-value of group comparisons by anova on ranks.
Contribution of UGT2B15 D85Y to variability in oxazepam clearance
Multiple linear regression analysis was used to evaluate the possible contributions of UGT2B15 D85Y, UGT2B15 K523T and UGT2B17del genotypes and subject demographic descriptors to variability in oxazepam clearance. Of these parameters, only the UGT2B15 D85Y genotypes 85DD (P= 0.018) and 85YY (P= 0.033) were identified as significant predictors of CL/F (P= 0.003, anova). Inclusion of both UGT2B15 D85Y genotype terms in the final model is consistent with a simple additive model of phenotypic expression. Based on the coefficient of determination (R2) for the regression, 34% of the total variance in CL/F was explained by UGT2B15 D85Y genotype.
Effect of UGT genotype on oxazepam pharmacodynamics
As shown in Figure 4, there was a trend for lower mean EEG β amplitude values in UGT2B15 85YY subjects compared with 85D and 85DD genotype subjects. However, median AUEC0–8h values for EEG as well as for DSST, OBS and SED (all given in Table 1) showed no significant differences between UGT2B15 D85Y genotype groups (P > 0.05, anova). Furthermore, neither UGT2B15 K523T (Table 2) nor the UGT2B17del (Table 3) showed effects on any of the pharmacodynamic parameters evaluated (P > 0.05, anova).
Figure 4.
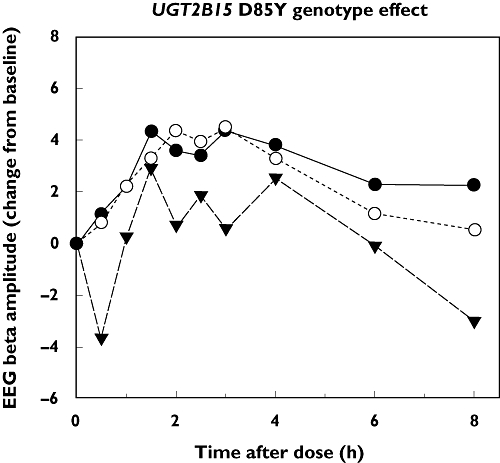
Effect of UGT2B15 D85Y genotype on electroencephalogram (EEG) activity in 30 subjects administered a 15-mg oral dose of oxazepam. Plotted values represent the mean change from predose values in EEG β wave amplitude for subjects grouped by UGT2B15 85DD (n= 6), DY (n= 20) and 85YY (n= 4) genotypes. D/D ( ); D/Y (
); D/Y ( ); Y/Y (
); Y/Y ( )
)
Discussion
The results of this study indicate that UGT2B15 D85Y polymorphism is a major determinant of oxazepam pharmacokinetics in healthy male volunteers. Specifically, lower oxazepam clearance was observed in direct proportion to the number of 85Y alleles that were carried by the subjects. This effect is probably explained by the presence of an amino acid substitution (tyrosine for aspartate) in the N-terminal region of the UGT protein that we previously showed to decrease recombinant enzyme protein levels by >60% and also decrease intrinsic enzyme activity by 80% [10]. However, our previous work also suggested that the effect of this polymorphism may be substrate-dependent in that the glucuronidation of dihydrotestosterone was reduced by only 33%, whereas glucuronidation of five other substrates of varied structures was reduced by 70–92% [10]. The D85Y substitution is located within the substrate binding domain of UGT2B15 (residues 61–194) previously identified by Lewis et al.[23]. However, since a complete crystal structure of UGT2B15 (or any other UGT) has not yet been published, the exact mechanism by which this polymorphism alters enzyme catalysis is unknown.
Regression analysis indicated that as much as 34% of the variability in oxazepam CL/F values was explained by the D85Y polymorphism, leaving 66% of the variability unexplained. Consequently, subjects were also genotyped for the second most common nonsynonymous polymorphism in the UGT2B15 gene that results in an amino acid substitution (K523T) in the C-terminal domain of the enzyme protein. Our previous work using a human liver bank had suggested that carriers of this substitution had 30% higher S-oxazepam glucuronidation activity [15]. The results of the present study did not indicate an effect of K523T on oxazepam pharmacokinetics even when D85Y effects were controlled for by either statistical methods (two-way anova and multiple linear regression analysis) or by subpopulation analysis (analysis of D85Y heterozygotes only). However, it should be pointed out that the study was primarily powered to discern effects of the UGT2B15 D85Y polymorphism, and any definitive conclusion regarding possible effects of the K523T polymorphism will require study of a larger number of subjects with the 523TT genotype. In vitro studies of recombinant UGT2B5 K523T allozyme would also help clarify the functional significance of this polymorphism.
We also genotyped our study subjects for UGT2B17del. Since this is a null allele it should result in complete loss of UGT2B17 activity in homozygous variant individuals. However, again our results showed no effect of this polymorphism on oxazepam pharmacokinetics, which is consistent with our previous in vitro data that demonstrated a lack of oxazepam glucuronidation by recombinant UGT2B17. This result is somewhat surprising given the high amino acid sequence identity (94.5%) between UGT2B15 and UGT2B17, and the ability of both isoforms to glucuronidate androgenic steroids, albeit with differing capacities [24]. However, a closer examination of amino acid sequence homology indicates that sequence identity is lower in the N-terminal half (residues 1–265: 91.3% identity) compared with the C-terminal half (residues 266–530: 97.7% identity), probably reflecting the role of these domains in binding the (structurally varying) aglycone substrates and the (nonvarying) UDP-glucuronic acid co-substrate, respectively. Once structure-based molecular models of UGT2B15 and UGT2B17 become available, substrate docking studies using oxazepam could be performed to better understand the catalytic mechanism of both enzymes.
UGT2B15 and UGT2B17 are encoded by separate genes in close proximity to each other on chromosome 4q13.2 [20]. It has been previously suggested that UGT2B17del is in significant genetic linkage with UGT2B15 D85Y [20], which may account for discrepancies between disease risk genetic association studies. For example, the UGT2B15 D85Y polymorphism was originally identified as a prostate cancer susceptibility marker [25, 26]. However, susceptibility was associated with the higher activity allele that should have conferred protection, whereas other studies have failed to find any significant gene–disease association [27]. We evaluated possible linkage disequilibrium between the UGT2B17del, UGT2B15 D85Y and UGT2B15 K523T and found no significant association. Although this analysis is limited by our small sample size, in a previous study we also found no linkage between UGT2B15 D85Y and UGT2B15 K523T, suggesting that these variants may exist on separate haplotype blocks [15].
Using our liver bank samples, we have also reported that UGT2B15 D85Y is associated with reduced glucuronidation of lorazepam, a structural analogue of oxazepam [3]. In support of this finding, a study of the pharmacokinetics of intravenously administered lorazepam in healthy Koreans demonstrated 42% lower clearance of lorazepam in 85YY subjects compared with 85DD subjects [28]. This is similar to, although somewhat smaller than, the difference in oxazepam oral clearance between these same genotype groups that we observed (52% lower). Since that study recruited only subjects with either 85YY or 85DD genotypes, it was not possible to determine effects of other polymorphisms, including UGT2B15 K523T and UGT2B17del. In a later study, the same group investigated whether a common nonsynonymous polymorphism in the UGT2B7 gene (H268Y) was associated with altered lorazepam clearance in subjects with the 85YY genotype, but found no effect of UGT2B7 H268Y genotype.
Despite showing a relatively large effect of UGT2B15 D85Y on oxazepam pharmacokinetics, we were unable to show any significant association between genotype and oxazepam effect using a variety of methods (quantitative EEG, DSST and visual analogue scales) that have been validated previously in our laboratory for the quantification of the neurological effects of other benzodiazepines [18]. This is probably a consequence of the relatively small size of the homozygous reference and variant genotype groups we studied (six and four subjects, respectively) coupled with limitations in the precision of the available measures of benzodiazepine pharmacodynamic effects. Although we observed positive median AUEC0–8h values (for the entire study group) consistent with the sedative effects of oxazepam, there was high intersubject variability as reflected by interquartile ranges that were as much as eightfold that of the median values. In the previous study by Chung et al.[28], they were also unable to demonstrate differences in pharmacodynamic effects of lorazepam between 85DD and 85YY subjects even with a larger number of subjects (nine and 15 subjects, respectively). However, they did observe a small but statistically significant difference in lorazepam sedative effects when the same subjects were administered rifampicin (an enzyme inducer), despite having a smaller difference in plasma lorazepam concentrations between genotype groups. It is also likely there are factors other than drug concentration that influence benzodiazepine pharmacodynamics, such as polymorphisms in genes encoding the γ-aminobutyric acid receptor subunit proteins that mediate benzodiazepine effects [29, 30].
In conclusion, the UGT2B15 D85Y polymorphism is identified as a major determinant of oxazepam pharmacokinetics, accounting for about one-third of interindividual variability in drug clearance. This work provides evidence supporting the use of oxazepam as an isoform-selective probe of UGT2B15 activity for in vivo as well as for in vitro studies.
Competing interests
None to declare.
M.H.C. was supported by grant GM-061834 from the National Institute of General Medical Sciences (NIGMS), National Institutes of Health (Bethesda, MD, USA). D.J.G. was supported by grants AG-17880 from the National Institute on Aging and AI-58784 from the National Institute on Allergy and Infectious Disease, National Institutes of Health (Bethesda, MD, USA). L.M.H. was supported by an individual National Research Service Postdoctoral Fellowship Award F32-AA015647 from the National Institute of Alcohol Abuse and Alcoholism, National Institutes of Health (Bethesda, MD, USA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Mackenzie PI, Bock KW, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, Miners JO, Owens IS, Nebert DW. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics. 2005;15:677–85. doi: 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- 2.Miners JO, Mackenzie PI. Drug glucuronidation in humans. Pharmacol Ther. 1991;51:347–69. doi: 10.1016/0163-7258(91)90065-t. [DOI] [PubMed] [Google Scholar]
- 3.Court MH. Isoform-selective probe substrates for in vitro studies of human UDP-glucuronosyltransferases. Methods Enzymol. 2005;400:104–16. doi: 10.1016/S0076-6879(05)00007-8. [DOI] [PubMed] [Google Scholar]
- 4.Chen F, Ritter JK, Wang MG, McBride OW, Lubet RA, Owens IS. Characterization of a cloned human dihydrotestosterone/androstanediol UDP-glucuronosyltransferase and its comparison to other steroid isoforms. Biochemistry. 1993;32:10648–57. doi: 10.1021/bi00091a015. [DOI] [PubMed] [Google Scholar]
- 5.Green M, Oturo E, Tephly T. Stable expression of a human liver UDP-glucuronosyltransferase (UGT2B15) with activity toward steroid and xenobiotic substrates. Drug Metab Dispos. 1994;22:799–805. [PubMed] [Google Scholar]
- 6.Nishiyama T, Ogura K, Nakano H, Ohnuma T, Kaku T, Hiratsuka A, Muro K, Watabe T. Reverse geometrical selectivity in glucuronidation and sulfation of cis- and trans-4-hydroxytamoxifens by human liver UDP-glucuronosyltransferases and sulfotransferases. Biochem Pharmacol. 2002;63:1817–30. doi: 10.1016/s0006-2952(02)00994-2. [DOI] [PubMed] [Google Scholar]
- 7.Kuuranne T, Kurkela M, Thevis M, Schanzer W, Finel M, Kostiainen R. Glucuronidation of anabolic androgenic steroids by recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos. 2003;31:1117–24. doi: 10.1124/dmd.31.9.1117. [DOI] [PubMed] [Google Scholar]
- 8.Zhang JY, Zhan J, Cook CS, Ings RM, Breau AP. Involvement of human UGT2B7 and 2B15 in rofecoxib metabolism. Drug Metab Dispos. 2003;31:652–8. doi: 10.1124/dmd.31.5.652. [DOI] [PubMed] [Google Scholar]
- 9.Hanioka N, Naito T, Narimatsu S Human UD. P-glucuronosyltransferase isoforms involved in bisphenol A glucuronidation. Chemosphere. 2008;74:33–6. doi: 10.1016/j.chemosphere.2008.09.053. [DOI] [PubMed] [Google Scholar]
- 10.Court MH, Duan SX, Guillemette C, Journault K, Krishnaswamy S, von Moltke LL, Greenblatt DJ. Stereoselective conjugation of oxazepam by human UDP-glucuronosyltransferases (UGTs): S-oxazepam is glucuronidated by UGT2B15, while R-oxazepam is glucuronidated by UGT2B7 and UGT1A9. Drug Metab Dispos. 2002;30:1257–65. doi: 10.1124/dmd.30.11.1257. [DOI] [PubMed] [Google Scholar]
- 11.Greenblatt DJ. Clinical pharmacokinetics of oxazepam and lorazepam. Clin Pharmacokinet. 1981;6:89–105. doi: 10.2165/00003088-198106020-00001. [DOI] [PubMed] [Google Scholar]
- 12.Abernethy DR, Greenblatt DJ, Divoll M, Shader RI. Enhanced glucuronide conjugation of drugs in obesity: studies of lorazepam, oxazepam, and acetaminophen. J Lab Clin Med. 1983;101:873–80. [PubMed] [Google Scholar]
- 13.Abernethy DR, Greenblatt DJ, Ochs HR, Weyers D, Divoll M, Harmatz JS, Shader RI. Lorazepam and oxazepam kinetics in women on low-dose oral contraceptives. Clin Pharmacol Ther. 1983;33:628–32. doi: 10.1038/clpt.1983.85. [DOI] [PubMed] [Google Scholar]
- 14.Patel M, Tang BK, Grant DM, Kalow W. Interindividual variability in the glucuronidation of (S) oxazepam contrasted with that of (R) oxazepam. Pharmacogenetics. 1995;5:287–97. doi: 10.1097/00008571-199510000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Court MH, Hao Q, Krishnaswamy S, Bekaii-Saab T, Al-Rohaimi A, Von Moltke LL, Greenblatt DJ. UDP-glucuronosyltransferase (UGT) 2B15 pharmacogenetics: UGT2B15 D85Y genotype and gender are major determinants of oxazepam glucuronidation by human liver. J Pharmacol Exp Ther. 2004;310:656–65. doi: 10.1124/jpet.104.067660. [DOI] [PubMed] [Google Scholar]
- 16.Greenblatt DJ, Divoll M, Harmatz JS, Shader RI. Oxazepam kinetics: effects of age and sex. J Pharmacol Exp Ther. 1980;215:86–91. [PubMed] [Google Scholar]
- 17.Jakobsson J, Ekstrom L, Inotsume N, Garle M, Lorentzon M, Ohlsson C, Roh HK, Carlstrom K, Rane A. Large differences in testosterone excretion in Korean and Swedish men are strongly associated with a UDP-glucuronosyl transferase 2B17 polymorphism. J Clin Endocrinol Metab. 2006;91:687–93. doi: 10.1210/jc.2005-1643. [DOI] [PubMed] [Google Scholar]
- 18.Greenblatt DJ, Gan L, Harmatz JS, Shader RI. Pharmacokinetics and pharmacodynamics of single-dose triazolam: electroencephalography compared with the Digit-Symbol Substitution Test. Br J Clin Pharmacol. 2005;60:244–8. doi: 10.1111/j.1365-2125.2005.02409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scavone JM, Greenblatt DJ, Harmatz JS, Engelhardt N, Shader RI. Pharmacokinetics and pharmacodynamics of diphenhydramine 25 mg in young and elderly volunteers. J Clin Pharmacol. 1998;38:603–9. doi: 10.1002/j.1552-4604.1998.tb04466.x. [DOI] [PubMed] [Google Scholar]
- 20.Wilson W, 3rd, Pardo-Manuel de Villena F, Lyn-Cook BD, Chatterjee PK, Bell TA, Detwiler DA, Gilmore RC, Valladeras IC, Wright CC, Threadgill DW, Grant DJ. Characterization of a common deletion polymorphism of the UGT2B17 gene linked to UGT2B15. Genomics. 2004;84:707–14. doi: 10.1016/j.ygeno.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Gallagher CJ, Kadlubar FF, Muscat JE, Ambrosone CB, Lang NP, Lazarus P. The UGT2B17 gene deletion polymorphism and risk of prostate cancer. A case–control study in Caucasians. Cancer Detect Prev. 2007;31:310–5. doi: 10.1016/j.cdp.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider S, Roessli D, Excofier L. Arlequin. A software for population genetics data analysis. Version 2.1. Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva.
- 23.Lewis BC, Mackenzie PI, Elliot DJ, Burchell B, Bhasker CR, Miners JO. Amino terminal domains of human UDP-glucuronosyltransferases (UGT) 2B7 and 2B15 associated with substrate selectivity and autoactivation. Biochem Pharmacol. 2007;73:1463–73. doi: 10.1016/j.bcp.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Belanger A, Pelletier G, Labrie F, Barbier O, Chouinard S. Inactivation of androgens by UDP-glucuronosyltransferase enzymes in humans. Trends Endocrinol Metab. 2003;14:473–9. doi: 10.1016/j.tem.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 25.MacLeod SL, Nowell S, Plaxco J, Lang NP. An allele-specific polymerase chain reaction method for the determination of the D85Y polymorphism in the human UDP-glucuronosyltransferase 2B15 gene in a case–control study of prostate cancer. Ann Surg Oncol. 2000;7:777–82. doi: 10.1007/s10434-000-0777-3. [DOI] [PubMed] [Google Scholar]
- 26.Okugi H, Nakazato H, Matsui H, Ohtake N, Nakata S, Suzuki K. Association of the polymorphisms of genes involved in androgen metabolism and signaling pathways with familial prostate cancer risk in a Japanese population. Cancer Detect Prev. 2006;30:262–8. doi: 10.1016/j.cdp.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Cunningham JM, Hebbring SJ, McDonnell SK, Cicek MS, Christensen GB, Wang L, Jacobsen SJ, Cerhan JR, Blute ML, Schaid DJ, Thibodeau SN. Evaluation of genetic variations in the androgen and estrogen metabolic pathways as risk factors for sporadic and familial prostate cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:969–78. doi: 10.1158/1055-9965.EPI-06-0767. [DOI] [PubMed] [Google Scholar]
- 28.Chung JY, Cho JY, Yu KS, Kim JR, Jung HR, Lim KS, Jang IJ, Shin SG. Effect of the UGT2B15 genotype on the pharmacokinetics, pharmacodynamics, and drug interactions of intravenous lorazepam in healthy volunteers. Clin Pharmacol Ther. 2005;77:486–94. doi: 10.1016/j.clpt.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Bowser DN, Wagner DA, Czajkowski C, Cromer BA, Parker MW, Wallace RH, Harkin LA, Mulley JC, Marini C, Berkovic SF, Williams DA, Jones MV, Petrou S. Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [gamma 2(R43Q)] found in human epilepsy. Proc Natl Acad Sci USA. 2002;99:15170–5. doi: 10.1073/pnas.212320199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iwata N, Cowley DS, Radel M, Roy-Byrne PP, Goldman D. Relationship between a GABAA alpha 6 Pro385Ser substitution and benzodiazepine sensitivity. Am J Psychiatry. 1999;156:1447–9. doi: 10.1176/ajp.156.9.1447. [DOI] [PubMed] [Google Scholar]