

Azathioprine (AZA), a prodrug of 6-mercaptopurine, is prescribed in evolving forms of inflammatory bowel disease (IBD), affecting predominantly White adolescents and young adults. Thiopurine S-methyltransferase (TPMT) catabolizes AZA and its genetic polymorphism affects the balance between active 6-thioguanine nucleotides (6-TGN) intracellular concentrations [recommended target 235–400 pmol per 8 × 108 red blood cells (RBC)] and hepatotoxic 6-methylmercaptopurine ribonucleotides (6-MMP) [1]. Three polymorphisms (TPMT*2, *3B, *3C; rs1800462, rs1800460, rs1142345, respectively) account for >95% of variant alleles, but additional rare variants are described (TPMT*1S to TPMT*25) [2]. Approximately 90% of Whites are extensive metabolizers with two wild-type TPMT alleles, 6–11% are intermediate metabolizers and 0.2–0.6% are TPMT-deficient patients, exposed to severe myelotoxicity as they accumulate high 6-TGN concentrations under AZA conventional doses [3].
This case reports a pancytopenia under AZA treatment for IBD. As 6-TGN and 6-MMP concentrations and TPMT genotype based on the three predominant polymorphisms were discordant, we conducted additional genetic analysis and identified a new, never described TPMT mutation affecting TPMT activity.
Case report
In December 2005, an Iranian girl aged 12 years, presenting with abdominal pain, fever, vomiting and proctorrhagia, was diagnosed with IBD. She improved under 5-aminosalicylic acid (5ASA, 2 g day−1) remaining symptom-free for 15 months. Relapse occurred with permanent abdominal pain and nausea. Initial blood and liver tests were normal, C-reactive protein was negative. Cortisone and 5ASA (2 g day−1) were started, but clinical symptoms persisted. Genotyping using allelic discrimination showed that the patient was heterozygous for TPMT*2, wild-type for TPMT*3B and TPMT*3C, and AZA was started (2 mg kg−1 day−1). Two months latter, blood test showed pancytopenia (haemoglobin 8.5 g per 100 ml, leucocytes 2900 mm−3, platelets 82 000 mm−3) and positive polymerase chain reaction for Parvovirus B19 (negative for Epstein–Barr virus and cytomegalovirus). Myelogram showed a rich, regenerative bone marrow. 6-TGN and 6-MMP concentrations were, respectively, 3414 pmol per 8 × 108 RBC and below the quantification limit (20 pmol per 8 × 108 RBC). TPMT activity was 3.45 nmol h−1 ml−1 RBC. AZA was switched to 5ASA (2 g day−1). After 2 weeks, blood tests were normalized and 6-TGN was 906 pmol per 8 × 108 RBC. Three months later, AZA was restarted at 25 mg day−1 (0.5 mg kg−1 day−1) as the patient was considered TMPT heterozygous (TPMT phenotype was not taken into account). After 3 weeks, 6-TGN levels were high (1918 pmol per 8 × 108 RBC), AZA treatment was definitely stopped and the patient received infliximab.
Additional genetic analyses were performed to investigate the discordance between metabolite concentrations and TPMT genotype. We sequenced exons 3 to 10 of TPMT gene and their consensus flanking sequences. The patient was heterozygous for TPMT*2 (c.238G→C) in exon 5, an inactivating false-sense variant, homozygous TT for TPMT*1S (c.474C→T) in exon 7, polymorphism without functional consequences, heterozygous for a new mutation TPMT IVS8-1G→A in the splice acceptor site (AG) of intron 8, not archived in the database dbSNP [2]. We also sequenced TPMT gene for both parents: the father was heterozygous for TPMT*2 and TPMT*1S and the mother was homozygous mutant for TPMT*1S and heterozygous for TPMT IVS8-1G→A. Therefore, the child was a carrier of a heterozygous composite TPMT genotype (Figure 1), carrying two TPMT variants (TPMT*2, TPMT IVS8-1G→A) responsible of complete TPMT deficiency, with severe myelosuppression and very high 6-TGN concentrations at standard AZA doses, remaining high after dose reduction.
Figure 1.
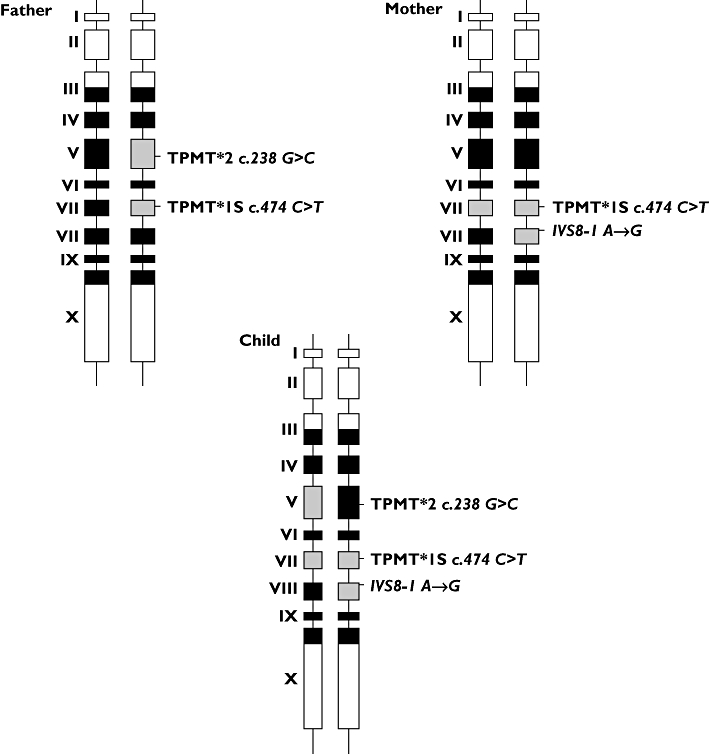
Polymorphism of thiopurine S-methyltransferase gene for parents and their child. Black boxes represent exons that encode open reading frame (ORF) sequences; white boxes represent exons out of ORF; grey boxes represent exons carrying a polymorphism
This report illustrates the major role of TPMT polymorphism in response to AZA including myelotoxicity. Parvovirus B19 infection was excluded in absence of typical symptoms (erythema infectiosum, arthralgia). Before treatment, determination of the three predominant TPMT polymorphisms remains crucial to identify high-risk patients requiring dosage adjustment (dose reduction of 30–50% for heterozygous patients and >90% for TPMT-deficient patients), avoiding life-threatening toxicity [4]. In addition, blood cell counts and measurement of 6-TGN and 6-MMP concentrations at steady-state (after 3 weeks at a stable AZA daily dose) are an important tool for thiopurine monitoring. It allows identifying a limited number of high-risk patients carrying rare TPMT mutations and making dosing decisions. In our observation, the patient was obviously TPMT homozygous deficient and AZA should have been restarted at a lower dose (0.2 mg kg−1 day−1). In case of phenotype/genotype discordance, sequence analysis of TPMT gene is interesting to identify rare inactivating variants. Additional gene polymorphisms in the AZA pathway such as xanthine oxidase and inosine triphosphate pyrophosphatase could also be tested.
Competing interests
None declared.
REFERENCES
- 1.Dubinsky MC, Lamothe S, Yang HY, Targan SR, Sinnett D, Théorêt Y, Seidman EG. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705–13. doi: 10.1016/s0016-5085(00)70140-5. [DOI] [PubMed] [Google Scholar]
- 2.Garat A, Cauffiez C, Renault N, Lo-Guidice JM, Allorge D, Chevalier D, Houdret N, Chavatte P, Loriot MA, Gala JL, Broly F. Characterisation of novel defective thiopurine S-methyltransferase allelic variants. Biochem Pharmacol. 2008;76:404–15. doi: 10.1016/j.bcp.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Colombel JF, Ferrari N, Debuysere H, Marteau P, Gendre JP, Bonaz B, Soulé JC, Modigliani R, Touze Y, Catala P, Libersa C, Broly F. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn's disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025–30. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]
- 4.Cheok MH, Evans WE. Acute lymphoblastic leukaemia: a model for the pharmacogenomics of cancer therapy. Nat Rev Cancer. 2006;6:117–29. doi: 10.1038/nrc1800. [DOI] [PubMed] [Google Scholar]