

Introduction
The crystal structure of an uncharacterized conserved protein (residues 1–243) expressed by yfiH gene of Shigella flexneri 2a str.2457T (gi 30042248),1 has been determined and refined to 2.01 Å by single wavelength anomalous dispersion (SAD) method. The YfiH protein belongs to a vast protein family of at least 201 uncharacterized proteins which contain conservative Pfam motif PF02578 (DUF152) (residues 29 –243) and TIGR motif (TIGR00726)2,3 (residues 25–243). The YfiH protein belongs to the COG1496 which consists of 39 proteins widely distributed in 37 species from bacteria to human. The yfiH gene (DNA bases 101,672-102,403)1 is located between the two genes, clpB and sfhB. The clpB gene is thought to encode an adenosine triphosphatase subunit of an intracellular adenosine 5′-triphosphate-dependent protease which belongs to the ClpA/ClpB family and is induced by heat shock.4 The sfhB gene expresses a protein which is known as suppressor of ftsH mutation and is responsible for synthesis of pseudouridine from uracil at three positions in 23S ribosomal RNA.5 The function of YfiH protein is unknown, but preliminary data obtained for Brevibacterium lactofermentum ATCC 13869 show that the gene is not essential for the cell’s growth and viability.6
Results
The seleno-methionine derivative of YfiH protein crystallized in the P1 space group with unit cell dimensions of a = 43.87, b = 50.58, c = 55.35 Å, α = 90.32°, β = 96.32°, and γ = 90.40°. There are two protein molecules in the asymmetric unit related by an approximate 21 symmetry along the crystallographic c axis. Both molecules adopt nearly identical backbone conformation (σ = 0.31 Å using all Cα atoms) with only a few differences in side-chain conformations. A PQS7 search predicted a monomeric form for this protein consistent with weak interface interaction between the two molecules in the asymmetric unit. The monomeric character was confirmed by size-exclusion chromatography (see Materials and Methods), which indicates that YfiH is a monomer in solution and this form may represent the biologically relevant species in vivo.
The protein structure is an α/β/α fold: two layers of β-sheets, the smaller one with three antiparallel strands (S1, S10, and S4), and the bigger one with six antiparallel/parallel strands (S7, S5, S6, S3, and S2) are sandwiched between two α-helices (H2, H5) on one side and one α-helix (H1) on the other [Fig. 1(A, B)]. The N-terminal end of the H1 helix is connected to S1 in the smaller β-sheet via one of the long loops and the C-terminal end of this helix is linked to S2 in the larger β-sheet. The α-helix H2, located between S6 and S7 in the larger β-sheet, contains a kink at Gly-133 and connects to S7 by an α-helical turn. In addition, there is a small domain inserted between S7 in the larger β-sheet and the helix H5. This small domain contains a small helix-turn-helix motif (H3 and H4), followed by a β-hairpin (S8 and S9). The main α/β/α fold resumes with H5, a long loop containing a little more than one 3/10-helix H5, followed by the S10 located in the middle of the smaller β-sheet.
Fig. 1.
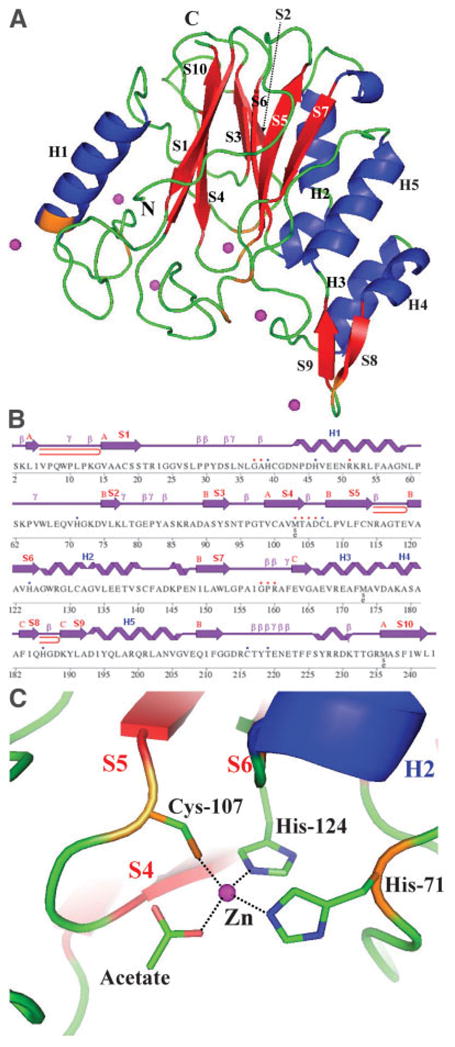
Crystal structure of YfiH protein. A: Ribbon diagram of YfiH protein. Helices are shown in blue, β-sheets in red, and Zn2 + ions are indicated in magenta; orange indicates protein residues interacting with Zn2+ ions, among eight Zn2+ ions in the asymmetric unit, six Zn2+ ions are shown here, four of these are shared between the two protein molecules. The two Zn2 + ions not shown interact with the other protein molecule in the asymmetric unit. Secondary structure elements are also indicated in black. B: Diagram showing the secondary structure elements in YfiH protein superimposed on its primary sequence. Residues interacting with Zn2+ ions are marked with blue dots and residues interacting with acetate molecules are marked with red. β-Hairpins are depicted as red loops. C: The potential active site of YfiH protein. The Zn2 + ions are coordinated to an acetate molecule, a cysteine and two histidine residues forming a tetrahedral configuration.
There are eight Zn+2 ions and five acetate molecules (both were included in crystallization conditions), found in two protein molecules in the asymmetric unit (molecules A and B). Each protein molecule holds two Zn+2 ions and the remainder of the Zn+2 ions are shared between two protein molecules. In molecule A, one of the Zn+2 ions is interacting with Nε2s of His-124 and His-71, Sγ of Cys-107, and an oxygen atom of one of five acetate molecules [Fig. 1(C)]. The second Zn+2 ion coordinates Oγ1 of Thr-219, Sγ of Cys-216, and an oxygen atom from one of the acetate molecules. In molecule B, these Zn+2 ions are involved in similar interactions. The remaining four Zn+2 ions contribute to crystal packing. Two are shared between the two protein molecules in the same asymmetric unit and are coordinated by N ε2s of His-186 and His-39 from the two different protein molecules, and two water molecules. The other two Zn+2 ions are coordinated by His-46, Glu-208, and Glu-147 (only for molecule A) from the two symmetry-related molecules.
In an effort to annotate the function of this protein, we searched a number of databases including BLAST,2 Pro-Func,3 PQS,7 DALI,8 and ISREC-TMpred9 servers. Residue conservation analysis showed 201 matching sequences found by PSI-BLAST with nearly all of them being conserved hypothetical proteins from different species except for a putative inner membrane protein from Salmonella enterica Typhimurium LT2.10 The ISREC-TMpred server9 identified two possible transmembrane helices: 1) inside to outside helix from residues 90 (94) to 114 (112) with a score 804, and 2) outside to inside helix from residues 95 to 113 with a score of 691 (scores above 500 considered significant). However, these overlapping regions are not in any of α-helix in the YfiH structure but, nonetheless, consist of S3, S4, S5, and the connecting loops located in the two layers of β-sheets surrounded by α-helices forming a part of a hydrophobic core.
The ProFunc3 and DALI8 searches found several structural homologs including uncharacterized protein from Bacillus stearothermophilus11 [Protein Data Bank (PDB) ID: 1t8h, Z = 11.0; sequence identity 31.0%], cytidine deaminase from Bacillus subtilis12 (PDB ID: 1ux1, Z = 4.4; 20.8%), and cytosine deaminase from Saccharomyces cerevisiae13,14 (PDB IDs: 1p6o, Z = 3.7; 21.2%, and 1uaq, Z = 3.6; 21.2%). The structure of conserved hypothetical protein from Salmonella enterica subsp enterica15 (PDB ID: 1rw0, sequence identity of 87%), which was not found in the DALI search because of late update of its library, is virtually identical (root-mean-square differences in Cα atoms are 0.45– 0.57 Å between chains) as expected from the sequence identity. The next closest structural homolog (1t8h) is also a conserved hypothetical protein of unknown function. Other structural homologs include cytidine and cytosine deaminases with Z scores between 4.4 –3.6. Both enzymes are the members of the pyrimidine salvage pathway, and both utilize a catalytic zinc ion and display similar mechanism. Reaction proceeds through the stereo-specific addition of a metal-bound hydroxyl group to the substrate, forming a tetrahedral transition state intermediate. Cytosine deaminase (or cytosine aminohydrolase) mediates reactions of cytosine to uracil. Cytidine deaminase correspondingly converts cytidine to uridine. These deaminases also adopt α/β/α sandwich folds with one α-helix flanked one side and two α-helices the other. However, they have only one β-sheet of four parallel strands and all α-helices are straight with no break. The structure of cytosine deaminase from S. cerevisiae (1p6o) barely superposes with the half of the large β-sheet (S7, S5, and S6) and two α-helices (H2 and H4) in the same side of the α/β/α sandwich of the YfiH structure (data not shown). In the active site of the cytidine deaminase, the Zn+2 ion was tetrahedrally coordinated by the Nγs of Cys-291 and Cys-294, the Nδ1 of the His-262, and by the hydroxyl oxygen O4 of the reaction transition state inhibitor 4(R)-hydroxyl-3,4-dihydropyrimidine (HPY).13 In the YfiH protein structure for both molecules in the asymmetric unit, one of Zn+2 ions, located in the similar area as in the structure 1p6o, is also tetrahedrally coordinated with the Nε2s of His-71 and His-124, the Sγ of Cys-107, and the OXT of an acetate molecule with the acetate molecule assuming the position occupied by HPY in the cytidine deaminase active site.13 Interestingly, the YfiH structure in the other crystal form, which is not included in this report, contains a similar Zn coordination, although the crystals were grown without zinc acetate. His-71, Cys-107, and His-124 are highly conserved in most of YfiH family proteins including proteins not only from archaea and bacteria but also from Homo sapiens (Q8IV20, 27% homology), filefish (CAG1329, 39%), frog (AAH78069, 31%), and mouse (Q8BZT9, 27%) (Fig. 2). However, the remainder of the residues in this potential active site are not similar to those in the structures 1p6o and of other reported cytosine/cytidine deaminases.
Fig. 2.

Multiple sequence alignment of YfiH protein homologs from various species. AAP17973.1 (Shigella flexneri), NP_754996.1 (Escherichia coli), NP_461592.1 (Salmonella typhimurium), AB0832 (Salmonella enterica subsp enterica), ZP_00156016.2 (Haemophilis influenzae), CAG13296.1 (Tetraodon nigroviridis), AAH78069.1 (Xenopus laevis), XP_233749.1 (Ratus norwegicus), AAH35749.1 (Homo sapiens), XP_417036.1 (Gallus gallus), NP_766076.1 (Mus musculus). Idt, percentage of identity. The conservations of His-71, Cys-107, and His-124, and identical amino acids are shown in red and blue, respectively.
Because there are limited functional data (other than structural studies) of cytosine and cytidine deaminases, it is difficult to determine if the two families are functionally related. Because the sfhB, the pseudouridine synthase gene, and yfiH gene with possible cytosine/cytidine deaminase function are neighbors, it is conceivable to say that YfiH protein could participate in the same pyrimidine salvage pathway as sfhB gene product.
Materials and Methods
Protein cloning, expression, and purification
The YfiH gene was cloned in pMCSG7 vector16 and overexpressed in Escherichia coli BL21 (DE3)—Gold (Stratagene) harboring an extra plasmid encoding three rare tRNAs (AGG and AGA for Arg, ATA for Ile). The pMCSG7 vector bearing a TEV protease cleavage site creates a construct with cleavable His6-tag fused into N-terminus of the target protein and adds three artificial residues (SerAsnAla) on that end. The cells were grown using SeMet-containing enriched M9 medium and conditions known to inhibit methionine biosynthesis.17,18 The cells were grown at 37°C to an OD600 of ~0.6 and protein expression induced with 1 mM isopropyl-β-D-thiogalactopyransoid. After induction, the cells were incubated overnight with shaking at 20°C. The harvested cells were resuspended in 5 volumes of lysis buffer (50 mM HEPES pH 8.0, 500 mM NaCl, 10 mM imidazole, 10 mM β-mercaptoethanol, and 5% v/v glycerol) and stored at −20°C.
The thawed cells were lysed by sonication after the addition of inhibitors of proteases (Sigma, P8849) and 1 mg/mL lysozyme. The lysate was clarified by centrifugation at 30,000g (RC5C-Plus centrifuge, Sorval) for 20 min, followed by filtration through a 0.45-μm filter and 0.22-μm in-line (Gelman).
The standard purification protocol was thoroughly described previously.19 Immobilized metal affinity chromatography (IMAC-I) using a 5-mL HiTrap Chelating HP column charged with Ni+2 ions and buffer-exchange chromatography on a HiPrep 26/10 desalting column (both Amersham Biosciences) were performed using AKTA EX-PLORE 3D (Amersham Biosciences). His6-tag was cleaved using the recombinant TEV protease expressed from the vector pRK508.20 The protease was added to the target protein in a 1:30 ratio and the mixture was incubated at 4°C for 48 h. The YfiH protein was then purified using a 1-mL HiTrap Chelating column (Amersham Biosciences) charged with Ni+2 ions. The protein was dialyzed in 20 mM Tris-HCl pH 7.1, 50 mM NaCl, 2 mM DTT, and concentrated using a Centricon Plus-20 Centrifugal Concentrator (Millipore).
Protein assays
The molecular weight of YfiH protein in solution was determined by size-exclusion chromatography on Superdex-75 10/30 column (Amersham Biosciences) calibrated by ribonuclease (13.7 kDa), chymotrypsinogen (25 kDa), ovalbumine (43 kDa), albumin (67 kDa), as standards. The calibration curve of Kav = Ve − Vo/Vt − Vo was used, where Ve is the elution volume for the protein, Vo is the column void volume, and Vt is the total bed volume.
Protein crystallization
The protein crystals initially were obtained by using crystallization screen Index (Hampton Research) and Wizard I and II crystallization screens (Emerald Biostructures) with Cartesian robot system (Cartesian Technologies) using sitting drop technique. The crystals appeared in 1 day in five conditions: Index #4 and #93 and Wizard II #4, #11, and #42. The protein was then crystallized by optimization of the Index condition #93 by vapor diffusion in hanging drops by mixing 2 μL of the protein solution (30 mg/mL) with 1 μL of 0.1 M zinc acetate and 14% (w/v) polyethylene glycol 3350, and equilibrated at 23°C over 1 mL of this solution. Crystals, which appeared the next day, were flash-frozen in liquid nitrogen with crystallization solution containing 15% (v/v) glycerol as cryoprotectant before data collection.
Data collection
Diffraction data were collected at 100° K at the 19ID beam line of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory. The SAD data at 0.9793 Å (peak energy: 12.6603 KeV) near the Se absorption edge up to 2.01 Å were collected from a single (0.1 × 0.02 × 0.05 mm) Se-Met labeled protein crystal in the cold stream maintained around 100° K. The crystal was exposed for 8 s per 1.5° rotation of ω with the crystal to detector distance of 200 mm. The data were recorded on an SBC-2 detector by the two scannings of 225° on ω, at ϕ = 0° and ϕ = 180°. The space group was P1 with cell dimension of a = 43.87, b = 50.58, c = 55.35 Å, α = 90.32°, β = 96.32°, and γ = 90.40°. All data were processed and scaled with HKL2000 suite21 (Table I) to an Rmerge of 7.1%.
TABLE I.
Summary of the YfiH Crystal Data and SAD Data Collection
| Unit cell parameters | a = 43.874, b = 50.583, c = 55.347 Å |
| α = 90.32°, β = 96.32°, γ = 90.40° | |
| Space group | PI |
| Molecular weight [243 residues (SeMet)] | 26,380 Da |
| Molecules per asymmetric unit (a.u.) | 2 |
| SeMet residues per a.u. | 8 |
| SAD data collection | |
| Wavelength (Å)/energy (keV) | 0.9793/12.6603 |
| Resolution range (Å) | 30–2.01 (2.08–2.01)a |
| No. of unique reflections | 30,856 (2,849) |
| Completeness (%) | 97.4 (89.6) |
| Rmergeb | 0.071 (0.21) |
In parentheses are statistics for the highest resolution shell.
R merge = (|Ihkl − 〈I〉|)/Ihkl, where the average intensity 〈I〉 is taken over all symmetry equivalent measurements and Ihkl is the measured intensity for any given reflection.
Structure determination and refinement
The structure was determined by SAD phasing utilizing the anomalous signal from Se atoms using HKL2000_PH (W. Minor, University of Virginia, personal communication) and RESOLVE22 and refined to 2.0 Å using REFMAC 5.223 in CCP4.24 The initial model was completed by using ARP/wARP25 and manual tailoring using Coot.26 The structure 1rw015 was used as a guide and to confirm the models. The final R was 0.18 with the free R of 0.25 with all data (Table II). Electron density calculated at 1.2 σ is well connected for most of the main-chain except a few areas on the surface of the molecules.
TABLE II.
Refinement Statistics for YfiH Structure
| Resolution range (Å) | 55.05–2.01 |
|---|---|
| No. of unique reflections | 27,582 |
| Sigma cutoff | 0.0 |
| R-valuea | 0.179 |
| R-freeb | 0.252 (3071)c |
| RMS deviations from ideal geometry | |
| Bond length (1–2) (Å) | 0.015 |
| Angle (°) | 1.517 |
| No. of atoms | |
| Protein | 4,148 |
| Acetate | 20 |
| Glycerol | 12 |
| Zn | 8 |
| Water | 420 |
| Mean B-factor (Å2) | |
| all atoms | 22.5 |
| Protein atoms | |
| Protein main-chain | 20.5 |
| Protein side-chain | 21.8 |
| Acetate | 21.6 |
| Glycerol | 19.4 |
| Zn | 29.0 |
| Water | 33.2 |
| Ramachandran plot statistics (%) | |
| Residues in allowed regions | 99.0 (479) |
| Residues in disallowed region | 1.0 (4) |
R-value = ||Fo| − |Fc||/||Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
R-free is equivalent to R value but is calculated for 10% of the reflections (number in the parentheses).
Chosen at random and omitted from the refinement process.
Validation and deposition
The stereochemistry of the structure was checked with PROCHECK27 and the Ramachandran plot. The main-chain torsion angles for all residues except four are in allowed regions. The main-chain conformations of these four residues (Asp-106 and Lys-231 per molecule), although, appear not in allowed regions, are well supported by the electron density (2fo-fc map contoured at 1.5 σ) suggesting that they are also in proper conformation. Atomic coordinates and experimental structure factors of YfiH have been deposited with the PDB and are accessible under the code 1xaf.
Acknowledgments
National Institutes of Health; Grant number: GM62414-01; Grant sponsor: U.S. Department of Energy, Office of Biological and Environmental Research; Grant number: W-31-109-Eng-38.
The authors thank all members of the Structural Biology Center at Argonne National Laboratory for their help in conducting experiments and L. Keller for her help with the preparation of the manuscript.
Footnotes
This article is a US government work and, as such, is in the public domain in the United States of America
This article was created by the University of Chicago as Operator of Argonne National Laboratory (“Argonne”) under Contract No. W-31-109-ENG-38 with the U.S. Department of Energy. The U.S. Government retains for itself, and others acting on its behalf, a paid-up, nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
References
- 1.Wei J, Goldberg MB, Burland V, et al. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect Immun. 2003;71:2775–2786. doi: 10.1128/IAI.71.5.2775-2786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laskowski RA, Watson JD, Thornton JM. From protein structure to biochemical function? J Struct Funct Genomics. 2003;4:167–177. doi: 10.1023/a:1026127927612. [DOI] [PubMed] [Google Scholar]
- 4.Lee S, Sowa ME, Watanabe YH, et al. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 5.Raychaudhuri S, Conrad J, Hall BG, Ofengand J. A pseudouridine synthase required for the formation of two universally conserved pseudouridines in ribosomal RNA is essential for normal growth of Escherichia coli. RNA. 1998;4:1407–1417. doi: 10.1017/s1355838298981146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honrubia MP, Ramos A, Gil JA. The cell division genes ftsQ and ftsZ, but not the three downstream open reading frames YFIH, ORF5 and ORF6, are essential for growth and viability in Brevibacterium lactofermentum ATCC 13869. Mol Genet Genomics. 2001;265:1022–1030. doi: 10.1007/s004380100497. [DOI] [PubMed] [Google Scholar]
- 7.Henrick K, Thornton SM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 8.Holm L, Sander C. Touring protein fold space with Dali/FSSP. Nucleic Acids Res. 1998;26:316–319. doi: 10.1093/nar/26.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann K, Stoffel W. TMbase—a database of membrane spanning proteins segments. Biol Chem Hoppe Seyler. 1993;374:166. [Google Scholar]
- 10.McClelland M, Sanderson KE, Spieth J, et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 11.Minasov G, Shuvalova L, Mondragon A, et al. 1.8 A Crystal structure of an uncharacterized B. stearothermophilus protein. Forthcoming. [Google Scholar]
- 12.Johansson E, Neuhard J, Willemoes M, Larsen S. Structural, kinetic, and mutational studies of the zinc ion environment in tetrameric cytidine deaminase. Biochemistry. 2004;43:6020–6029. doi: 10.1021/bi035893x. [DOI] [PubMed] [Google Scholar]
- 13.Ireton GC, Black ME, Stoddard BL. The 1.14 A crystal structure of yeast cytosine deaminase: evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure. 2003;11:961–972. doi: 10.1016/s0969-2126(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 14.Ko T-P, Lin J-J, Hu C-Y, Hsu Y-H, Wang AH-J, Liaw S-H. Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J Biol Chem. 2003;278:19111. doi: 10.1074/jbc.M300874200. [DOI] [PubMed] [Google Scholar]
- 15.Seetharaman J, Swaminathan S. Crystal structure of hypothetical protein Yfih. Forthcoming. [Google Scholar]
- 16.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 17.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 18.Walsh MA, Dementieva I, Evans G, Sanishvili R, Joachimiak A. Taking MAD to the extreme: ultrafast protein structure determination. Acta Crystallogr D Biol Crystallogr. 1999;D55:1168–1173. doi: 10.1107/s0907444999003698. [DOI] [PubMed] [Google Scholar]
- 19.Kim Y, Dementieva I, Zhou M, et al. Automation of protein purification for structural genomics. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kapust RB, Waugh DS. Controlled intracellular processing of fusion proteins by TEV protease. Protein Expr Purif. 2000;19:312–318. doi: 10.1006/prep.2000.1251. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 22.Terwilliger TC. Automated main-chain model-building by template-matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;D59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 24.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 25.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]