

Abstract
The structure of Aq_328, an uncharacterized protein from hyperthermophilic bacteria Aquifex aeolicus, has been determined to 1.9 Å by using multi-wavelength anomalous diffraction (MAD) phasing. Although the amino acid sequence analysis shows that Aq_328 has no significant similarity to proteins with a known structure and function, the structure comparison by using the Dali server reveals that it: (1) assumes a histone-like fold, and (2) is similar to an ancestral nuclear histone protein (PDB code 1F1E) with z-score 8.1 and RMSD 3.6 Å over 124 residues. A sedimentation equilibrium experiment indicates that Aq_328 is a monomer in solution, with an average sedimentation coefficient of 2.4 and an apparent molecular weight of about 20 kDa. The overall architecture of Aq_328 consists of two noncanonical histone domains in tandem repeat within a single chain, and is similar to eukaryotic heterodimer (H2A/H2B and H3/H4) and an archaeal histone heterodimer (HMfA/HMfB). The sequence comparisons between the two histone domains of Aq_328 and six eukaryotic/archaeal histones demonstrate that most of the conserved residues that underlie the Aq_328 architecture are used to build and stabilize the two cross-shaped antiparallel histone domains. The high percentage of salt bridges in the structure could be a factor in the protein’s thermostability. The structural similarities to other histone-like proteins, molecular properties, and potential function of Aq_328 are discussed in this paper.
Keywords: structural genomics, MAD phasing, synchrotron radiation, histone fold, thermostability
INTRODUCTION
Like numerous other targets of Protein Structure Initiative Pilot Projects, the hypothetical protein Aq_328 shows no significant sequence similarity to proteins of a known structure and a function. The protein is encoded by an open reading frame (ORF) Aq_328 in A. aeolicus, and its homologs are found in both bacteria and archaea. The protein was selected for structure determination because it meets the goals of structural genomics studies of mapping protein-folding space and offering structure-based insight into their potential biochemical and biophysical functions.1,2 When structural information is available, it may be possible to deduce the functional information, even if the sequence similarity is distant.3,4
A. aeolicus is one of the most thermophilic bacteria known–it grows near hot springs in the deep ocean at temperatures between 85°C and 95°C.5 Aq_328 consists of 171 amino acids. A PSI-BLAST search indicates only four proteins with similar sequences that cluster with Aq_328 (E values are all below 1E-37). These proteins are from bacteria and archaea and are annotated as putative/hypothetical proteins. Among them, Aq_616 (gi|15606051) ORF is from the same species and shares the same domain with Aq_328 (Pfam-B_63624).
Here we report the 1.9-Å resolution crystal structure of Aq_328 from A. aeolicus. The protein shows high structural similarity to the histone fold protein, HMk (PDB code 1F1E, 147 residues), from a hyperthermophilic archaea Methanopyrus kandleri.6 Aq_328 is the second protein structure reported that contains a tandem repeat of two histone domains. It is noteworthy that both Aq_328 and HMk are from hyperthermophilic species. HMk is a structural homolog of methanogen and eukaryotic histones.6 The structural similarities to HMk suggest a possible physiological role for HMk in DNA packaging.7 Therefore, it may be inferred that Aq_328 is possibly a DNA-binding protein that is involved in DNA packaging. This function needs experimental verification.
MATERIALS AND METHODS
Cloning of AQ_328
The ORF of the A. aeolicus Aq_328 protein was amplified from genomic DNA with KOD DNA polymerase by using conditions and reagents provided by the vendor (Novagen, Madison, WI). The gene was cloned into a pMCSG7 vector8 by using a modified ligation-independent cloning protocol.9 This process generated an expression clone producing a fusion protein with an N-terminal His-6-tag and a TEV protease recognition site (ENLYFQ↓S). The fusion protein was over-produced in a E. coli BL21-derivative that harbored a plasmid pMAGIC encoding three rare E. coli tRNAs (Arg [AGG/AGA] and Ile [ATA]) as described earlier.10
Protein Expression and Purification
A selenomethionine (Se-Met) derivative of the expressed protein was prepared as described previously11 and purified according to standard protocol.12 The transformed BL21 cells were grown in M9 medium at 37°C. M9 medium is supplied with 0.4% sucrose, 8.5 mM NaCl, 0.1mM CaCl2, 2 mM MgSO4, and 1% thiamine. After OD600 reached 0.5, 0.01% (w/v) each of leucine, isoleucine, lysine, phenylalanine, threonine, and valine was added to inhibit the metabolic pathway of methionine and encourage Se-Met incorporation. Se-Met was then added at 6% (w/v), and 15 min later protein expression was induced by 1 mM isopropyl-β-D-thiogalactoside (IPTG). The cells were then incubated at 20°C overnight.
The harvested cells were resuspended in lysis buffer (500 mM NaCl, 5% glycerol, 50 mM HEPES, pH 8.0, 10 mM imidazole, 10 mM 2-mercaptoethanol). Lysozyme (1 mg/mL) and 100 μL of protease inhibitor cocktail (Sigma, P8849) were added per 2 g of wet cells, and the cells were kept on ice for 20 min before sonication. The lysate was clarified by centrifugation at 27,000 g for 1 h and then applied to a 5-mL HiTrap Ni-NTA column (Amersham Biosciences) on the AKTA EXPLORER 3D (Amersham Biosciences). His-tagged protein was eluted by using elution buffer (500 mM NaCl, 5% glycerol, 50 mM HEPES, pH 8.0, 250 mM imidazole, 10 mM 2-mercaptoethanol), and the tag was cleaved from the protein by treatment with recombinant His-tagged TEV protease (a gift from Dr. D. Waugh, NCI). A second Ni-NTA affinity chromatography was performed manually to remove the His-tag and His-tagged TEV protease. Protein was concentrated by using a Centricon 5k MW cutoff (Amicon) and stored at the temperature of liquid nitrogen.
Protein Crystallization and Data Collection
The protein was crystallized by vapor diffusion in hanging drops containing 1 μL of protein solution (3 mg/mL) and 1 μL of reservoir solution [0.1 M sodium cacodylate, pH 6.0; 5–15% PEG 3350; and 0.05M Zn(OAc)2]. The droplets were equilibrated at 20°C against the reservoir. Crystals appeared after two weeks. A single crystal of approximately 0.2 × 0.1 × 0.05 mm was cryoprotected by using 30% of PEG 3350 in the reservoir solution and flash-frozen in liquid nitrogen. The crystal belongs to space group P6522 with cell dimensions a = b = 55.92 Å, c = 244.37 Å, α = β = 90°, γ = 120° and contains one molecule in the asymmetric unit with solvent content 56%. The absorption edge of Se was determined by using an X-ray fluorescence scan of the crystal, followed by examination of the fluorescence data by using CHOOCH.13 A three-wavelength MAD dataset was collected at 100 K with 3 s/1°/frame and a 200-mm crystal-to-detector distance at the Structural Biology Center 19ID beamline of the Advanced Photon Source (APS), Argonne National Laboratory. Data were processed and scaled by using an HKL2000 suite14 and are summarized in Table I.
TABLE I.
Summary of Crystal MAD Data Collection
| Unit cell | a = b = 55.92 Å, c = 244.37 Å, α = β = 90°, γ = 120° | ||
| Space group | P6522 | ||
| MW Da (171 amino acids) | 19,795 D | ||
| Number of Se-Met | 7 | ||
| MAD data |
|||
| Edge | Peak | High | |
| Wavelength (Å) | 0.97945 | 0.97929 | 0.95372 |
| Resolution range (Å) | 50.0–1.9 | 50.0–1.9 | 50.0–1.9 |
| Number of unique reflections | 32,486 (2389) | 33,125 (2880) | 31,349 (1704) |
| Completeness (%) | 96.1 (71.1) | 98.0 (85.7) | 92.9 (50.9) |
| R merge (%) | 8.6 (43.8) | 8.5 (38.2) | 9.8 (57.0) |
Structure Determination and Refinement
The phases were determined by using SOLVE15 with MAD data and two out of seven selenium sites. The initial model was build automatically by RESOLVE,16 with 69% of total residues built and then refined by ARP/wARP,17 resulting in a continuous protein main chain. The final model was built manually by using the program TURBO-FRODO.18 Electron density calculated at 1 σ was well connected, except for the N-terminal residues 1–19, which are disordered in the crystal structure. The structure was initially refined with CNS (annealing, water molecule identification, minimization, and individual isotropic B factor refinement) and then improved by using REF-MAC519 to the final R factor 18.1% and R free 21.3% (Table II). Atomic coordinates and structure factors have been deposited into the PDB with ID 1R4V.
TABLE II.
Crystallographic Statistics
| Parameter | Value |
|---|---|
| Resolution (Å) | 20~1.9 |
| Number of reflections (working set) | 16,877 |
| Number of reflections (test set) | 908 |
| Completeness for range (%) | 94.2 |
| σ cutoff | None |
| R-value (%) | 18.1 |
| Free R-value (%) | 21.3 |
| RMS deviations from ideal geometry | |
| Bond length | 0.014 Å |
| Angle | 1.722° |
| Number of atoms | 1456 in total |
| Protein | 1228 |
| Zn | 3 |
| Cacodylate | 1 |
| Water | 224 |
| Mean B value (Å2) | 23.191 |
| Ramanchandran plot statistics | |
| Residues in most favored regions | 96.3% |
| Residues in allowed regions | 3.7% |
| Residues in disallowed regions | 0 |
Sedimentation Velocity Analysis
Sedimentation velocity experiments were performed on a Beckman Optima Model XL-A analytical ultracentrifuge equipped with a four-place An-60Ti rotor and a two-channel aluminum cell. Protein sample (400 μL of 1 mg/mL; in 20 mM HEPES, 200 mM NaCl, 0.5 mM DTT, pH 8.0) with absorbance about 0.75 (O.D.) at 280 nm was loaded into the sample channel with the corresponding reference buffer in the reference channel. After equilibration at 3,000 rpm and 20°C, at which reference wavelength was determined, the rotor was accelerated to the selected experimental speed of 60,000 rpm. The scans of protein concentration profiles were collected at 15-min intervals for 20 h. The program UltraScan6.0 was used to calculate the distribution of sedimentation coefficient and apparent molecular weight.
RESULTS AND DISCUSSION
Description of Aq_328 structure
The Aq_328 protein consists of two domains [Fig. 1(a)], with each domain assuming a histone fold (a long α-helix is flanked by two short α-helices located on the same side). The N-terminal domain consists of four α-helices and one 310 helix, in which helix 1 (H1) is a short α-helix (F29–T38), H2 and H3 could be viewed as one long α-helix distorted by a short loop (L63–G66), H4 is a 310 helix (L81–D83), and H5 is another short α-helix (K88–Q98). H1 and H5 are located at the same side of H2 and H3. The C-terminal domain of Aq_328 consists of four α-helices, in which H6 is a short α-helix (V106–I113); H7 and H8 form an imperfect long α-helix (E125–A149) distorted by a one-residue turn (E130); and H9 is another short α-helix (R157–D168) located at the same side with H6. The N-terminal and C-terminal domains form an antiparallel cross-shape and are linked by a seven-residue loop (K99–G105). Since these two domains are arranged in the tandem repeat form, they are designated here as domain 1 and domain 2, respectively.
Fig. 1.

a: Ribbon diagram of Aq_328 structure. The N-terminal histone domain is colored in gold, C-terminal histone domain in green, and Zn2+ ions in blue. The residues coordinating Zn2+ are shown in ball-and-stick. b: Stereo-view of Aq_328. The orientation is the same as in (a). The α-helices of domain 1 are colored red, the α-helices of domain 2 blue. The loop bridges domain 1 and 2 is in green. Other loops are colored grey.
Three zinc ions and one cacodylate ion were found to bind to Aq_328 [Fig. 1(a)]. Two of the zinc ions are at the intermolecular surface of two symmetry-related molecules, with the cacodylate ion bridging them. One zinc ion is coordinated by D33 of one molecule, and the other is coordinated by E158 and E161 of a symmetry-related molecule [Figs. 1 and 2]. Thus, these two Zn2+ ions and one cacodylate probably help crystallization of Aq_328 by making very specific interactions between symmetry-related molecules. The functional role, if any, of the third Zn2+ ion, which is coordinated by E21 and D46, is difficult to deduce because the residues coordinating it are not well conserved.
Fig. 2.

Two Zn2+ ions from two symmetry-related molecules are located at the interface and are coordinated by three acidic residues. The cacodylate ion bridges the two Zn2+.
The structural alignment analysis using Dali shows that the Aq_328 structure is very similar to an ancestral two-domain histone protein HMk (Protein Data Bank ID 1F1E),6 which is from the hyperthermophilic archaea, Methanopyrus kandleri. HMk aligns to Aq_328 with Z (normalized statistical similarity weight) of 8.1 and RMSD of 3.6 Å for 124 Cα atoms (Fig. 3). Aq_328 and HMk contain 171 and 154 residues, respectively, which is about twice the length of a histone fold. Although a BLAST search showed little sequence similarity between HMk and Aq_328 (7.6% sequence identity), they share strong structural similarities and several conserved residues [Fig. 4(a)]. Moreover, Aq_328 has charged properties similar to those of Hmk, with pIs of 5.46 and 4.91, respectively. The architectures of both Aq_328 and HMk are very similar to the eukaryal histone two-chain heterodimers H2A/H2B and H3/H420–22 and the archaeal histone heterodimer HMfA/HMfB.23,24 However, they differ from the archaeal and eukaryotic histone proteins because they contain two histone-fold domains within a single chain.6 It is noteworthy that unlike HMk, the two histone-like domains of Aq_328 are noncanonical, with multiple helical segments and kinks. From the viewpoint of molecular evolution, the 7.6% of identical residues and a few conserved residues shared by Aq_328 and HMk may act as key sites that maintain the basic histone fold and possibly the same function. Because of the large differences in their amino acid composition, it is difficult to trace the phylogenomics of Aq_328 based on the sequences comparison; thus, Aq-328 can be designated as a histone variant, but not a histone protein. Also unlike HMk, Aq_328 contains an N-terminal extension (not included in the atomic structure), which is essential in the eukaryotic histones to down-regulate assembly and play a role in higher nucleosome assembly.25 This histone-tail region exists in eukaryotic histones, but not in archaeal histones. Perhaps Aq_328 is an intermediate in the transition from archeal to eukaryotic histone.
Fig. 3.

Aq_238 (in green) is superimposed on HMk protein (in lime). The RMSD for 124 Cα atoms is 3.6 Å.
Fig. 4.
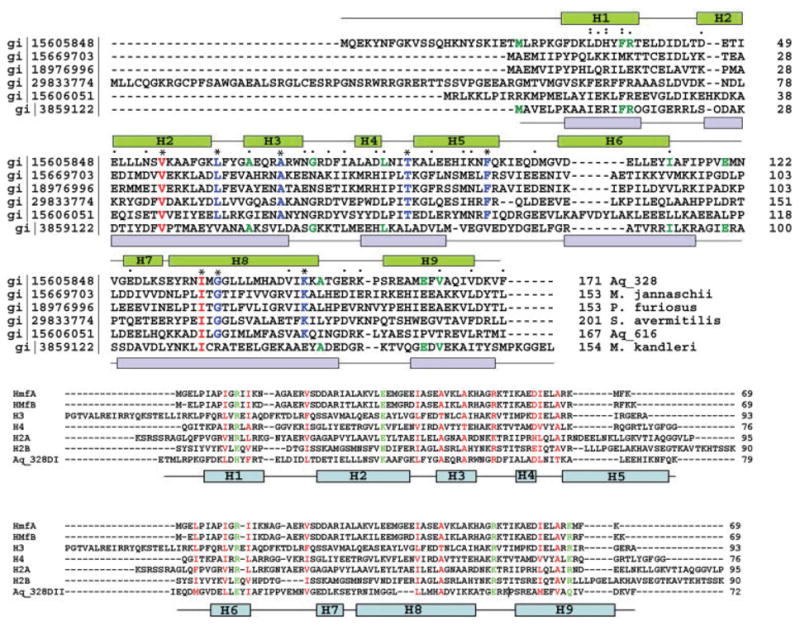

a: Multiple sequence alignment of AQ_328 sequence homologs and HMk (structural homolog) is created using ClustalW. Residues identical for all sequences are labeled as * and colored in red; residues identical for AQ_328 sequence homologs are labeled as * and colored in blue; conserved residues are labeled as “.” and “:”; residues identical in both Aq_238 and HMK are shown in green. The α-helices of Aq_328 are shown in green on the top, the α-helices of HMk are shown in blue on the bottom (there is an x in the HMk sequence, which is Met). b: ClustalW multiple sequence alignment of Aq_328 N-terminal domain (upper panel), C-terminal (lower panel) with histone proteins from eukaryotes (H2A, H2B, H3, and H4), and from archaea (HMfA and HMfB). Conservative residues are colored in red, residues with similar properties in green. Note: Aq_328 N-terminal sequence starts from residue 21 (residues 1~20 are missing in the crystal structure). The α helices of Aq_328 are shown in the diagram and colored in light blue. c: Ribbon diagram of N-terminal and C-terminal domains of Aq_328 with conserved residues shown in ball-and-stick (left, N-terminal domain; right, C-terminal domain). d: Surface electrostatic potential distributions of H3/H4 core structure (left) and Aq-328 (right). Aq-328 has the same orientation as H3/H4 heterodimer. The electrostatic potential is mapped on the molecular surfaces by GRASP (the coordinates of H3/H4 are taken from PDB 1kx5). Blue shows positive potential and red shows negative potential. The DNA-binding sites of H3/H4 and corresponding positive residues of Aq-328 are labeled.
The Dali search also revealed that the structure of Aq_328 has some similarity to domains of larger proteins from bacteria (DNA primerase from Desulfovibrio desulfuricans (z-score 5.1, RMSD 2.4 Å) and cell division protein FtsK (z-score 3.3, RMSD 3.2 Å). These structural similarities suggest the Aq_328 may be involved in DNA binding and may function like histone proteins. A. aeolicus genome codes for an Aq_328 sequence homolog Aq_616 [gi15606051, Fig. 4(a)] that shares 25% sequence identity with Aq_328. We speculate that these two proteins (Aq_328 and Aq_616) may form a paired association that favors the formation of the higher oligomers needed for packing DNA, and they are similar to other histone heterotetramers, such as the H2A/H2B association with H3/H4 found in eukaryotes.20–22
Conservation Patterns Underlying Aq_328 Architecture
The histone fold is the core protein structural unit of the nucleosome. The most well documented histone proteins are H2A, H2B, H3, and H4 from eukaryotes,20,21,22 and HMfA, HMfB from archaea.23,24,26 These histone proteins are usually produced as monomers that form dimers in solution27 and tetramers or higher oligomers in complexes with DNA.21,25,26,28 Their sequences vary from 69–93 residues in length, which is about half of the size of the Aq_328 sequence.
To trace the conservation patterns between the well documented histone proteins and Aq_ 328, we divided Aq_328 into its two parts—domain 1 containing α-helices H1–H5 (residues 21~99) and domain 2 containing α-helices H6–H9 (residues 100~171)—and then compared their sequences with six histone sequences (H2A, H2B, H3, H4, HMfA, and HMfB) using ClustalW [Fig. 4(b)].
The multiple sequence alignment shows that the domain 1 of Aq_328 has about 15% and domain 2 has about 11% of sequence similarity with all analyzed histone proteins. Figure 4(c) shows a ribbon diagram of both domain 1 and domain 2 of Aq_328, with homologous residues indicated by ball-and-stick representation. Several homologous residues appear to play important roles in forming and stabilizing the histone fold.
Residues L32, F36, and L44 may stabilize the orientation of the first and second helices in domain 1 by forming van der Waals contacts. In domain 2, the similar contacts are made by M160 and V163. In domain 1, R76 is located at the end of the second helix, and it interacts with D83 via a salt bridge and presumably defines the distance between the second and the third helices of the histone fold. However, there is no corresponding conserved salt bridge in domain 2. Most of the chemically similar residues of Aq_328 are used to build the cross-shaped architecture. M104, L109, and L139 in domain 2 are clustered to interact with the hydrophobic core formed by L32, F36, and L44 in domain 1. It is possible that this core may be one of the driving forces for forming and stabilizing the cross-shaped histone fold structure. Similarly, in another part of the structure, F115, V119, V123, and V127 in domain 2 interact with a second hydrophobic core formed by F60, F64, A67, I79, and L84 from domain 1. F64 and F115 form π–π stacking and interact with F60 nearby.
In the large hydrophobic cores formed between domain 1 and 2, a number of residues are homologous. In addition to hydrophobic interactions, there is one backbone–side chain hydrogen bond contributed by I113 in domain 2 and K57 in domain 1 and two backbone–backbone hydrogen bonds (I79–N122 and I79–G124) between the two domains. Three homologous residues are involved in these hydrogen bonds. Domain 1 has several more homologous residues than Domain 2. These residues (K57, A71, I79, and D83) play roles in stabilizing either the domain or overall structure. Additionally, there are 13 nonconserved hydrogen bonds providing cohesion forces for domain 1 and domain 2.
Since the structure of Aq_328 is not only similar to histone heterodimers, but also to some other DNA-binding proteins, it was important to analyze its surface charge distributions to ascertain whether the pattern is consistent with DNA-binding. Interestingly, Aq_328 and HMk are both acidic proteins, which is in contrast to most of the proteins that bind DNA nonspecifically, which typically are basic. It is thus of interest to further investigate and compare the charged properties between Aq_328 and histone proteins. Aq_328 has 24 positive residues (eight Arg and 16 Lys; four of the Lys are located in the N-terminal extension region) and 29 negative residues (12 Asp and 17 Glu, only one Glu in the N-terminal extension region). In the structure of Aq_328, the charged residues, which make up 28% of the sequence, are distributed unevenly on the protein surface and are solvent-accessible. Many are involved in formation of salt bridges. The HMk structure contains 23 basic amino acids and 30 acidic amino acids, which make up more than 34% of the total residues. Histone proteins generally have a large portion of positively charged amino acids (up to 30%). The core structure of H2A/H2B heterodimer has 29 positively charged residues distributed on the surface, while H3/H4 heterodimer has 33. The core structure of HMfA homodimer has 25 positively charged amino acids distributed on its surface, which is similar to Aq_328, but is fewer than those of eukaryotic histones.
The crystal structure of a DNA-histone octamer complex reveals that there are 14 contact sites (3.5 sites for a histone heterodimer), but only a few positive charged residues are making direct contact with the DNA.22 In eukaryotic histones, the DNA-protein interaction sites are located in the loop regions at each end of the heterodimer (L1L2 sites) and the two first α-helices (α1α1 sites), where Arg and Thr are making contact with DNA.22 This provides a sequence-independent mode of interaction between the DNA phosphate groups and the protein side chains and establishes the significance of the histone’s cross-shaped architecture in packaging DNA.
The Aq_328 monomer aligns to the core structures of H2A/H2B and H3/H4 heterodimers with RMSD 1.01 Å and 1.15 Å over 100 residues, respectively. It presents eight positively charged residues (K31, R37, R76, K88, K95, K99, R153, and K154) and four Thr (T38, T45, T87, and T150) in regions similar to those of the H2A/H2B and H3/H4 histones. Figure 4(d) shows the electrostatic potential mapped on the molecular surfaces of H3/H4 core structure (left) and Aq_328 (right). The DNA-binding sites of H3/H4 heterodimer are labeled. In the corresponding positions, several positive charged residues are arranged on the molecular surface of Aq_328. It appears that the charge distribution of Aq_328 is more similar to H3/H4 than to H2A/H2B heterodimer.
Although Aq_328 is structurally related to histone proteins, its real function is currently unknown. The similarities of Aq_328 with the noncanonical histone HMk, which has been identified in a hyperthermophilic prokaryote Methanopyrus kandleri,29 suggest that the function of Aq_328 maybe related to DNA packaging in A. aeolicus. The acidity of both Aq_328 and HMk would help prevent the nonspecific self-aggregation that has been reported for other basic histones under physiological conditions. Therefore, in eukaryotes, an acidic chaperone called a nucleoplasmin is needed to prevent histone self-aggregation.30,31
Since Aq_328 originates from a bacterium, A. aeolicus, which lives in an environment that is rich in inorganic components (such as mineral salts),5 there is a possibility that the metal cations may play a role in affecting the function of A. aeolicus proteins. In the presence of salts at high-concentration, the electrostatic contribution to binding free energy of protein can be effected by the nonspecific binding of cations to the protein surface; therefore, the penalty of unfavorable charged residues will be compensated for and the forming of complexes favored.32,33
A sedimentation velocity experiment revealed that Aq_328 forms a monomer in solution with a sedimentation coefficient about 2.4 (data not shown). Aq_328 monomer is like a product of dimerization of a single histone-fold domain. Dimerization is not only the common feature of histone proteins, but it also the primary determinant in forming stable oligomeric aggregates and then packing DNA.21 Therefore, we presume that Aq_328 may have the tendency to aggregate into a homodimer or interact with other histone-like proteins to form a heterodimer. And the most likely candidate for the second kind of interaction may be Aq_616. However, this hypothesis should be tested experimentally.
Thermostability and Possible Function of Aq_328
A. aeolicus is one of the most thermophilic bacteria known. Determining the molecular basis for protein thermostability is an active area of investigation. Thus, it is of interest to analyze the structure of Aq_328 for features that may contribute to its thermostability. Salt bridges have been shown to play an important role in protein thermostability by improving electrostatic interactions.34,35 Hyperthermophilic enzymes, in general, possess a much higher number of ion pairs per residue. In enzymes from mesophilic organisms, about 4% of the ion pairs per residue are involved in salt bridges, but this value is doubled in some enzymes, like aldehyde ferredoxin oxidoreductase from hyperthermophilic Pyrococcus furious.34 The structure of Aq_328 contains seven pairs of salt bridges (D33-R37, D41-R153, K62-D168, R76-D83, K88-E91, D107-R132, and R157-E158), giving 4.6% of ion pairs per residue. Additionally, there are three ion pair networks (E21-R25-D46, K31-D103-E108, and R157-E158-E161), which form tertiary salt bridges to enhance thermostability significantly by stabilizing the α-helix dipole36 and are energetically more favorable than isolated ion pairs.37 In total, the number of ion pairs per residue for Aq_328 is 8.6%.
Another distinct structural feature that may help improve thermostability is the packing mode of the domain 1 and domain 2. In Aq_328, the cross-shaped architecture produces 34.4% of interface-accessible surface area between the two histone-like domains. But the same surface areas calculated for histone heterodimers in mesophilic eukaryotes are only 23.4% (H2A/H2B) and 26.6% (H3/H4), which are far less than that of Aq_328. The higher value of interface-accessible surface area indicates that the cross-shaped two histone-like domains of Aq_328 are more rigid and more stable. Furthermore, the cross-shaped architecture brings together some residues that form salt bridges (D41-R153, K62-D168, and K31-D103-E108). The hyper-thermophilic histone-like protein, HMk, also exploits a more rigid packing mode, with 32.1% of the interface-accessible surface area between the two histone-fold domains. It seems that gene duplication of histone-like domain maybe an adaptation for a high-temperature environment.
The histone fold is a common fold found in all three kingdoms of life. It is becoming apparent that the histones are a heterogeneous protein family as more examples of histone and histone fold protein sequences are continually added to public histone databases (http://research.nhgri.nih.gov/histones/).38 In this paper, we provide the structural evidence for a new small family of histone-like proteins in bacteria and archaea. These proteins can be classified as single-chain heterodimeric histone variants. It will help guide the further biochemical and biophysical experiments that will be needed to decipher the specific function of Aq_328.
Acknowledgments
Atomic coordinates have been deposited in the Protein Data Bank (PDB) with PDB-ID 1R4V and accession number RCSB020439. The authors wish to thank all members of the Structural Biology Center at Argonne National Laboratory for their help in conducting these experiments. This work was supported by National Institutes of Health Grant GM62414 and by the U.S. Department of Energy, Office of Biological and Environmental Research, under contract W-31-109-Eng-38.
Footnotes
The submitted manuscript has been created by the University of Chicago as Operator of Argonne National Laboratory (“Argonne”) under Contract No. W-31-109-ENG-38 with the U.S. Department of Energy. The U.S. Government retains for itself, and others acting on its behalf, a paid-up, nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
References
- 1.Vitkup D, Melamud E, Moult J, Sander C. Completeness in structural genomics. Nat Struct Biol. 2001;8:559–566. doi: 10.1038/88640. [DOI] [PubMed] [Google Scholar]
- 2.Zarembinski TI, Hung L-W, Mueller-Dieckmann H-J, Kim K-K, Yokota H, Kim R, Kim S-H. Structure-based assignment of the biochemical function of a hypothetical protein: a test case of structural genomics. Proc Natl Acad Sci USA. 1998;95:15189–1593. doi: 10.1073/pnas.95.26.15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murzin AG, Patthy L. Sequences and topology: from sequence to function. Curr Opin Struct Biol. 1999;9:359–362. [Google Scholar]
- 4.Zhang R, Grembecka J, Vinokour E, Collart F, Dementieva I, Minor W, Joachimiak A. Structure of Bacillus subtilis YXKO—a member of UPF0031 family—a putative kinase. J Struct Biol. 2002;139:161–170. doi: 10.1016/s1047-8477(02)00532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deckert G, Warren PV, Gaasterland T, Young WG, Lenox A, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, et al. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 6.Fahrner RL, Cascio D, Lake JA, Slesarev A. An ancestral nuclear protein assembly: crystal structure of the Methanopyrus kandleri histone. Protein Sci. 2001;10:2002–2007. doi: 10.1110/ps.10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musgrave D, Forterre P, Slesarev A. Negative constrained DNA supercoiling in archaeal nucleosomes. Mol Microbiol. 2000;35:341–349. doi: 10.1046/j.1365-2958.2000.01689.x. [DOI] [PubMed] [Google Scholar]
- 8.Dieckman L, Gu M, Stols L, Donnelley MI, Collart FR. High throughput methods for gene cloning and expression. Protein Expr Purif. 2002;25:1–7. doi: 10.1006/prep.2001.1602. [DOI] [PubMed] [Google Scholar]
- 9.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelley MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 10.Wu R, Zhang R, Dementieva I, Maltzev N, Laskowski R, Gornicki P, Joachimiak A. Crystal structure of Enterococcus faecalis SlyA-like transcriptional factor. J Biol Chem. 2003;278:20240–20244. doi: 10.1074/jbc.M300292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh MA, Dementieva I, Evans G, Sanishvili R, Joachimiak A. Taking MAD to the extreme: ultrafast protein structure determination. Acta Crystallogr D Biol Crystallogr. 1999;55:1168–1173. doi: 10.1107/s0907444999003698. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A. Automation of protein purification for structural genomics. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans G, Pettifer RF. CHOOCH: a program for deriving anomalous-scattering factors from X-ray fluorescence spectra. J Appl Cryst. 2001;34:82–86. [Google Scholar]
- 14.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in enzymology. part A. Vol. 276. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 15.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terwilliger TC. Automated main-chain model-building by template-matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2002;59:34–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamzin VS, Wilson KS. Automated refinement for protein crystallography. Methods Enzymol. 1997;277:269–305. doi: 10.1016/s0076-6879(97)77016-2. [DOI] [PubMed] [Google Scholar]
- 18.Jones TA. A graphics model building and refinement system for macromolecules. Appl Cryst. 1978;11:268–272. [Google Scholar]
- 19.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 20.Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN. The nucleosomal core histone octamer at 3.1 Å resolution: a tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci USA. 1991;88:10148–10152. doi: 10.1073/pnas.88.22.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arents G, Moudrianakis EN. The histone fold: a ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc Natl Acad Sci USA. 1995;92:11170–11174. doi: 10.1073/pnas.92.24.11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 23.Sandman K, Krzycki JA, Dobrinski B, Lurz R, Reeve JN. HMf, a DNA-binding protein isolated from the hyperthermophilic archaeon Methanothermus fervidus, is most closely related to histones. Proc Natl Acad Sci USA. 1990;87:5788–5791. doi: 10.1073/pnas.87.15.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Decanniere K, Babu AM, Sandman K, Reeve JN, Heinemann U. Crystal structures of recombinant histones HMfA and HMfB from the hyperthermophilic archaeon Methanothermus fervidus. J Mol Biol. 2000;303:35–47. doi: 10.1006/jmbi.2000.4104. [DOI] [PubMed] [Google Scholar]
- 25.de la Barre AE, Gerson V, Gout S, Creaven M, Allis CD, Dimitrov S. Core histone N-termini play an essential role in mitotic chromosome condensation. EMBO J. 2000;19:379–391. doi: 10.1093/emboj/19.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marc F, Sandman K, Lurz R, Reeve JN. Archaeal histone tetramerization determines DNA affinity and the direction of DNA supercoiling. J Biol Chem. 2002;277:30879–30886. doi: 10.1074/jbc.M203674200. [DOI] [PubMed] [Google Scholar]
- 27.Eickbush TH, Moudrianakis EN. The histone core complex: an octamer assembled by two sets of protein-protein interactions. Biochemistry. 1978;17:4955–4964. doi: 10.1021/bi00616a016. [DOI] [PubMed] [Google Scholar]
- 28.Sandman K, Reeve JN. Structure and functional relationships of archaeal and eukaryal histones and nucleosomes. Arch Microbiol. 2000;173:165–169. doi: 10.1007/s002039900122. [DOI] [PubMed] [Google Scholar]
- 29.Slesarev AI, Belova GI, Kozyavkin SA, Lake JA. Evidence for an early prokaryotic origin of histones H2A and H4 prior to the emergence of eukaryotes. Nucleic Acids Res. 1998;26:427–430. doi: 10.1093/nar/26.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dingwall C, Laskey RA. Nucleoplasmin: the archetypal molecular chaperone. Semin Cell Biol. 1990;1:11–17. [PubMed] [Google Scholar]
- 31.Dutta S, Akey IV, Dingwall C, Hartman KL, Laue T, Nolte RT, Head JF, Akey CW. The crystal structure of nucleoplasmin-core: implications for histone binding and nucleosome assembly. Mol Cell. 200;8:841–853. doi: 10.1016/s1097-2765(01)00354-9. [DOI] [PubMed] [Google Scholar]
- 32.Murray D, Honig B. Electrostatic control of the membrane targeting of C2 domains. Mol Cell. 2002;9:145–154. doi: 10.1016/s1097-2765(01)00426-9. [DOI] [PubMed] [Google Scholar]
- 33.Misra VK, Hecht JL, Sharp KA, Friedman RA, Honig B. Salt effects on protein-DNA interactions: the λcI repressor and EcoRI endonuclease. J Mol Biol. 1994;238:133–295. doi: 10.1006/jmbi.1994.1286. [DOI] [PubMed] [Google Scholar]
- 34.Chan MK, Mukund S, Kletzin A, Adams MW, Rees DC. Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science. 1995;267:1463–1469. doi: 10.1126/science.7878465. [DOI] [PubMed] [Google Scholar]
- 35.Shin DH, Yokota H, Kim R, Kim S-H. Crystal structure of conserved hypothetical protein Aq1575 from Aquifex aeolicus. Proc Natl Acad Sci USA. 2002;99:7980–7985. doi: 10.1073/pnas.132241399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das R, Gerstein M. The stability of thermophilic proteins: a study based on comprehensive genome comparison. Funct Integr Genomics. 2000;1:76–88. doi: 10.1007/s101420000003. [DOI] [PubMed] [Google Scholar]
- 37.Horovitz A, Serrano L, Avron B, Bycroft M, Fersht AR. Strength and co-operativity of contributions of surface salt bridges to protein stability. J Mol Biol. 1990;216:1031–1044. doi: 10.1016/S0022-2836(99)80018-7. [DOI] [PubMed] [Google Scholar]
- 38.Sullivan S, Sink DW, Trout KL, Makalowska I, Taylor PM, Baxevanis AD, Landsman D. The histone database. Nucleic Acids Res. 2002;30:341–342. doi: 10.1093/nar/30.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]