

Abstract
L-Proline is an amino acid that plays an important role in proteins uniquely contributing to protein folding, structure, and stability, and this amino acid serves as a sequence-recognition motif. Proline biosynthesis can occur via two pathways, one from glutamate and the other from arginine. In both pathways, the last step of biosynthesis, the conversion of Δ1-pyrroline-5-carboxylate (P5C) to L-proline, is catalyzed by Δ1-pyrroline-5-carboxylate reductase (P5CR) using NAD(P)H as a cofactor. We have determined the first crystal structure of P5CR from two human pathogens, Neisseria meningitides and Streptococcus pyogenes, at 2.0Å and 2.15Å resolution, respectively. The catalytic unit of P5CR is a dimer composed of two domains, but the biological unit seems to be species-specific. The N-terminal domain of P5CR is an α/β/α sandwich, a Rossmann fold. The C-terminal dimerization domain is rich in α-helices and shows domain swapping. Comparison of the native structure of P5CR to structures complexed with L-proline and NADP+ in two quite different primary sequence backgrounds provides unique information about key functional features: the active site and the catalytic mechanism. The inhibitory L-proline has been observed in the crystal structure.
Keywords: structural genomics, MAD phasing, SAD phasing, proline biosynthesis, P5C reductase
Introduction
L-Proline is an amino acid that plays an important role in protein structure and cell signaling, uniquely contributing to protein folding, structure, and stability and serving as a sequence recognition motif, as well as a component of signal transduction pathways.1,2 Free proline has been shown to accumulate in response to osmotic stress in a wide variety of organisms, implying a role in stress tolerance and osmoregulation.3 Whereas proline accumulation is a primitive response to osmotic stress, the source of this accumulation seems to be different. In eubacteria, proline accumulation occurs via enhanced uptake from activated transcription of the operons encoding proline permease (proP and proU).1,4 Nonetheless, some bacteria can synthesize proline de novo under osmotic stress when proline is not present in the environment.5 In eukaryota, the mechanism for proline accumulation is mainly through de novo synthesis.6
Proline biosynthesis can occur via two pathways: one from glutamate and the other from arginine (Figure 1).7,8 The glutamate pathway is the main mechanism of proline biosynthesis in bacteria, while eukaryotes use this route predominantly under stress and limited nitrogen conditions. Under high nitrogen input, the pathway from arginine appears to be prominent and is mainly found in higher plants.9,10 Three enzymes and four reaction steps are required to convert glutamate to proline (Figure 1).11 In the first step, glutamate is phosphorylated by γ-glutamyl kinase (EC 2.7.2.11), forming γ-glutamyl phosphate. Then, γ-glutamyl phosphate is converted by γ-glutamyl phosphate reductase (EC 1.2.1.41) to glutamate γ-semialdehyde. This step is followed by the immediate cyclization of glutamate γ-semialdehyde to Δ1-pyrroline-5-carboxylate. In the final step, Δ1-pyrroline-5-carboxylate is reduced by Δ1-pyrroline-5-carboxylate reductase (P5CR; EC 1.5.1.2. L-proline: NAD(P)-5-oxidoreductase) to yield L-proline. The enzymes arginase (EC 3.5.3.1), ornithine amino transferase (EC 2.6.1.13), and P5CR are needed for biosynthesis of proline from arginine.12 Thus, the final step of biosynthesis for both pathways, the conversion of Δ1-pyrroline-5-carboxylate (P5C) to L-proline, is catalyzed by P5CR using NAD(P)H as cofactor.1,13
Figure 1.

Pathways of proline biosynthesis.
P5CR and proline metabolism have been linked to a number of cellular processes. In some organisms, the activity of P5CR is osmoregulated concomitantly with proline accumulation.14-16 For higher plants like soybean, the mRNA level of P5CR increased after osmotic stress, and transgenic plants expressing P5CR were more stress-resistant to heat and drought.17 In insects like Drosophila melanogaster, the transcription of P5RC is increased in response to cold stress.18 In cultured cells, P5C and the proline cycle activate the pentose phosphate shunt and increase nucleotide synthesis by using the salvage and de novo pathways.2,19,20 P5CR has been shown to play a role in the regulation of de novo purine biosynthesis through the generation of NADP+, which is required for the synthesis of the purine precursor ribose 5-phosphate.21 In human erythrocytes, the level of P5CR activity is comparable with the activity levels of major erythrocyte enzymes. This is consistent with the interpretation that the function of the enzyme in human erythrocytes may be to generate an oxidizing potential in the form of NADP+.21 In some mammalian cells, the inter-conversion of proline and P5C provides a metabolic shuttle of redox equivalents between the cytosol and mitochondria. P5C can be transported into cells as a source of oxidizing potential where its reduction to proline generates NADP+.22,23 The process is mediated by significant amounts of P5CR found in the cytosol and some membrane-containing fractions. The proline can then be transported into the mitochondria, in which proline oxidase mediates its conversion back to P5C with the concomitant production of NADPH and ATP, thereby completing a proline cycle. In this process, proline mediates the generation of reactive oxygen species that have been linked to apoptosis.24,25 To date, a large number of P5CR enzymes from different sources have been studied, showing considerable diversity with respect to reaction rate with the nicotinamide coenzymes (NADPH or NADH), molecular mass and affinity to substrate, as well as a product inhibition. It was proposed that maybe this diversity reflects the different role the enzyme plays in regulation and metabolism.10
P5CR was selected as a part of the Midwest Center for Structural Genomics Protein Structure Initiative project. P5CR is a member of a very large family, with over 400 family members across all kingdoms of life, including humans.26 To increase the chance of obtaining a crystal structure from a member of this large and important family of enzymes, orthologues from 14 organisms have been cloned. Here, we report the first crystal structures of P5CR from Neisseria meningitides (Nm-P5CR) and Streptococcus pyogenes (Sp-P5CR) at 2.0Å and 2.15Å resolution, respectively. The proteins share 22% sequence identity and 47% sequence similarity. The crystal structure of free enzyme is compared to enzyme complexes with L-proline and NADP+ cofactor.
Result and Discussion
Quality of models of P5CRs
The crystal structure of Nm-P5CR has been determined by selenomethionine (SeMet) MAD phasing, and has been refined using CNS-SOLVE version 1.126 and REFMAC527 to 2.0Å resolution, yielding a model with an R-factor of 18.8% (Rfree of 22.1%). The data collection and refinement statistics and the indicators of model quality are listed in Tables 1 and 2. The final model contains all 263 amino acid residues, 249 water molecules, and one sulfate ion. The electron density map is generally of excellent quality, except for the C terminus of a molecule. The last residue (Gln263) is traceable for only the main-chain and is modeled as alanine. All non-glycine and non-proline residues of the model lie either in the most favorable region (95.6% of residues) or in the additionally allowed region (4.4% of residues) of the Ramachandran plot. The model of the P5CR/NADP+ complex was refined by using REFMAC5 at 2.1Å resolution with a final R-factor 17.7% and Rfree 25.2%. Maps were of good quality, except for the density for the NADP+ molecule, which is not well defined, and parts of the density are missing and show a high B-factor.
Table 1.
Data collection statistics
| Crystals | Nm-P5CR | Nm-P5CR/ NADP+ |
Sp-P5CR/L-pro- line |
Sp-P5CR/ NADP+ |
|
|---|---|---|---|---|---|
| Wavelength (Å) | 0.97984 | 0.97970 | 0.97917 | 0.97933 | 0.97933 |
| Space group | P21212 | P21212 | P21212 | C2 | C2 |
| Cell parameters (Å, deg.) |
a=89.52 b=49.93 c=65.60 |
a=89.52 b=49.84 c=65.53 |
a=89.70 b=49.83 c=65.45 |
a=171.82 b=109.96 c=84.16 β=95.38 |
a=171.63 b=109.65 c=84.03 β=96.08 |
| Resolution (Å) | 50–2.0 | 50–2.0 | 50–2.1 | 50–2.20 | 50–2.15 |
| Total reflections | 227,384 | 227,177 | 37,608 | 152,468 | 162,858 |
| Unique reflections | 20,333 | 20,462 | 14,057 | 78,923 | 83,006 |
| Completeness (%) | 99.9 (99.7) | 99.6 (98.9) | 78.4 (35.2) | 98.5 (92.5) | 98.98 (92.0) |
| I/sigma | 37.2 (4.4) | 35.3 (3.2) | 14.1(1.7) | 14.2(2.0) | 23.10(2.17) |
| Rmergea(%) | 10.1 (46.7) | 11.5 (52.9) | 8.6 (39.1) | 6.1 (32.6) | 6.7(32.3) |
The data in parentheses refer to the highest-resolution shell.
Table 2.
Refinement statistics
| Nm-P5CR | Nm-P5CR/NADP+ | Sp-P5CR/L-proline | Sp-P5CR/NADP+ | |
|---|---|---|---|---|
| Resolution range (Å) | 50–2.00 | 50–2.1 | 50–2.20 | 50–2.15 |
| Rcryst (%) | 19.0 | 17.7 | 17.3 | 17.5 |
| Rfree (%) | 23.0 | 25.2 | 21.4 | 21.0 |
| A. r.m.s. deviations from ideality | ||||
| Bond lengths (Å) | 0.018 | 0.023 | 0.012 | 0.012 |
| Angles (deg.) | 1.52 | 1.94 | 1.33 | 1.35 |
| No. of protein atoms | 1980 | 1980 | 9592 | 9592 |
| No. of hetero atoms | 5 | 48 | 45 | 260 |
| No. of solvent atoms | 249 | 247 | 739 | 668 |
| B. Average B-factor | ||||
| Main-chain atoms (Å2) | 27.9 | 31.0 | 34.4 | 43.8 |
| Side-chain atoms (Å2) | 31.3 | 33.9 | 36.3 | 46.6 |
| Protein atoms (Å2) | 29.5 | 32.3 | 35.0 | 45.2 |
| Heteroatoms63(Å2) | – | 50.9 | 43.1 | – |
| Heteroatoms (Å2) | ||||
| NADP+ | 60.1 | |||
| The adenine ribose P/νmax64 | – | 66.7 | – | 136.5/33.9 |
| The nicotinamide ribose P/νmax | 147.9/27.08 | |||
| L-Pro 1 and 2 | – | – | 30.9a/56.4b | – |
| SO4 | 90.8 | – | – | – |
| Na | – | – | 34.9 | 42.8 |
| FMT | – | – | – | 47.8 |
| Solvent atoms (Å2) | 40.2 | 46.4 | 41.4 | 40.4 |
| C. Ramachandran statistics | ||||
| Favored/allowed/generous | 95.6/4.4/0.0/ | 95.1/4.9/0.0/ | 95.0/5.0/0.0 | 94.4/5.5/0.1/ |
| Disallowed (%) | 0.0 | 0.0 | 0.0 | 0.0 |
P, the phase angle of pseudo-rotation; νmax, maximum out-of-plane pucker; FMT, formic acid.
B-factor value for L-proline 1 (reaction product).
B-factor value for L-proline 2 (product inhibition).
The structures of Sp-P5CR in complex with L-proline and NADP+ have been determined independently by SeMet SAD phasing and refined against the peak data with REFMAC527 to 2.15Å (R-factor 17.3%, Rfree 21.4%) and 2.20Å (R-factor 19.4%, Rfree 22.8%) resolution, respectively. The quality of the data and refined structures are summarized in Tables 1 and 2. The refined models consist of: (1) 1285 residues of five molecules A, B, C, D and E; five molecules of NADP+; five Na ions; five molecules of formic acid; and 668 water molecules in the structure of the Sp-P5CR/NADP+ complex; and (2) 1285 residues belonging to five monomers, ten molecules of L-proline, five Na ions, and 739 water molecules in the structure in the Sp-P5CR/L-proline complex. The stereochemistry of the structures was checked with PROCHECK.28 Final refinement statistics are presented in Table 2.
Overall structure of P5CRs and structural homology
Although Nm-P5CR and Sp-P5CR structures show low sequence identity (22%), their structures are remarkably similar and superimpose with an rmsd value of 1.75Å over 244 Cα atoms for monomers and 2.19Å on 457 Cα atoms for dimers (Figures 2, 3(a) and (b)).29 Here, we use the higher resolution structure of Nm-P5CR to characterize the overall structure of P5CRs. The P5CR monomer consists of 11 helices and eight strands arranged in two well-defined domains. The N-terminal α/β/α domain includes residues 1 to 153 and assumes a canonical NADP-binding Rossmann fold. The hook-shaped C-terminal domain includes residues 154 to 263 (Figure 3(a) and (b)). It is involved in dimer formation and contributes in the active site formation. Interestingly, although both P5CR structures are very similar, there are some distinct differences in the secondary structure elements (Figure 2). For example Sp-P5CR α-helix H3 is not present in Nm-P5CR and α-helix H7 of Sp-P5CR is two turns longer than equivalent α-helix H6 in Nm-P5CR.
Figure 2.

Multiple sequence alignment of a selected representative of the pyrroline-5-carboxylate reductase family. Sequence identities are highlighted in red and similarities are shown as red letters. The corresponding secondary structures of P5CR from S. pyogenes and N. meningitides are shown on the top (black) and the bottom (blue), respectively. Helices (H, α-helix; h, 310 helix) appear as small squiggles, beta strands (S, α-strand) as arrows. The following abbreviations were used, with Unit-Prot accession numbers indicated in parentheses: SP, Streptococcus pyogenes M1 GAS (Q9A1S9); LP, Lactobacillus plantarum (Q88Z19); EC, Escherichia coli O6 (Q8FKE0); HS, Homo sapiens (P32322); SOY, Glycine max (Soybean) (P17817); PEA, Pisum sativum (Q04708); MB, Methanococcoides burtonii; SC, Staphylococcus aureus strain COL (Q5HFR9); BA, Bacillus anthracis Ames (Q81M92); VC, Vibrio cholerae (Q9KUQ5); PL, Photorhabdus luminescens subsp. Laumondii (Q7N7H0); NM, Neisseria meningitides (Q9K1N1).
Figure 3.

Cylinder and ribbon drawing of the P5CR dimers. (a) An overall view of the monomer of Nm-P5CR. (b) An overall view of the dimer of Sp-P5CR complexed with NADP+ (green) with the proline residues (yellow) docked into the active center. The conserved residues are highlighted in red. (c) Diagram of the C-terminal dimerization domain in Nm-P5CR.
As determined by the Dali server,30 the N-terminal domain is a classic nucleotide-binding domain showing structural similarity to the nucleotide-binding fold observed in other oxidoreductases, such as L-3-hydroxyacyl CoA dehydrogenase (pdb code, 2hdh; Z-score 15; sequence identity (seqid), 15%),31 ketol-acid reductoisomerase (pdb code, 1np3; Z-score 13.7; seqid, 15%),32 glycerol-3-phosphate dehydrogenase (pdb code, 1yj8; Z-score, 13.1; seqid, 17%), hydroxyisobutyrate dehydrogenase (pdb code, 1j3v; Z-score, 12.8; seqid, 18%), 2-dehydropantoate 2-reductase (pdb code, 1ks9; Z-score, 12.1; seqid, 16%),33 and L-lactate dehydrogenase (pdb code, 1oc4; Z-score, 12.0; seqid, 17%).34 This domain consists of an eight-stranded, highly twisted mixed β-sheet (S3, 47–49; S2, 26–30; S1, 2–6; S4, 60–63; S5, 83–86; S6, 106–110, S7, 120–125; S8, 146–150) flanked by six α-helices (H1, 10–22; H2, 34–43; H5, 113–117; and H6, 132–143, on one side and H3, 67–74, and H4, 93–99, on the opposite side of the sheet) (Figure 2).
The C-terminal/dimerization domain is highly α-helical (99 out of 110 residues being in α-helices) (Figure 3(c)). This domain consists of six α-helices (H6, 156–163; H7, 167–184; H8, 189–210; H9, 214–220; H10, 227–237; H11, 240–260) and makes extensive interaction with the C-terminal domain from a symmetry-related monomer (Figure 3(c)). The DALI server search revealed structural homology only to ketol-acid reductoisomerase (pdb code, 1np3; Z-score, 6.3; seqid, 11%).32 Although the Z-score indicates some structural homology and both structures have comparable topologies, the superimposition of the dimerization domains indicated rather poor overlap between the structures, with some α-helices of the C-terminal domain of Sp-P5CR being superimposed over the N-terminal domain of ketol-acid reductoisomerase.
Dimer interface
In the Nm-P5CR and Sp-P5CR crystal structures, monomers associate into a tight dimer through the domain swapping and interactions of H6-H11 (Nm-P5CR numbering used) of one subunit with their counterparts of the opposite subunit in an antiparallel orientation, thereby forming a three-layer dimer interface (Figure 3(c)). An interface of the center layer of the dimer is formed by H6, H7, and H9 and their equivalents from the symmetry-related subunit. It is sandwiched on the bottom by the layer formed by H8 and H8′ and on the top by the layer arranged by H10, H11 and H10′, and H11′. Moreover, layers are positioned so the helices cross over each other at an angle of ~50° between the bottom and the central layers and at an angle of ~40° between the central and the top layers. This arrangement makes the bottom layer approximately perpendicular to the top layer and appears to result in a highly stable structure. The stability of this assembly is achieved through a wide variety of interactions, including α-helical dipole–dipole, hydrophobic, and salt bridges interactions (see below).
The α-helices interact in two different ways, generating well-defined pairs of parallel and antiparallel α-helical dipoles both within a layer and between layers. For instance, helices H6 and H7 interact constructively to form one long dipole of 45Å in length. The longest parallel dipole–dipole interaction is observed for helices H10 and H11, which generate a dipole of 53Å. Helices H6, H7, H10, and H11 from each monomer arrange together into a multi-helix bundle (Figure 3(c)). The bundle is joined together by a hook-like assembly of helices H8 and H9 and its homologs from the opposite monomer. These helices swap to the adjacent monomers and line up perpendicularly to the bundle. This results in tighter packing of layers and contributes to stability of the dimer. Therefore, the C-terminal domain serves as a rigid bridge between N-terminal domains.
Most of interactions between α-helices are hydrophobic, with a large number of non-polar side-chains pointed toward the interior of the center layer, forming a very well defined hydrophobic core. More than 50% of the dimerization domain residues (112 residues out of 204 and 108 residues out of 210 in the structures of Sp-P5CR and Nm-P5CR, respectively) are non-polar, with ~70% of the residues located within the hydrophobic core.
Apart from the dipole–dipole and hydrophobic interaction, the dimer interface is stabilized by several salt bridges. There are six intra-subunit (Glu174/Lys187, Lys189/Glu192, and Glu232/Arg235 in each subunit) and two inter-subunit (Lys182/Asp247′ and Lys182′/Asp247) salt bridges formed in the structure of Sp-P5CR. The structure of Nm-P5CR is, in turn, stabilized by two intra-subunit (Glu233/Arg236 in each subunit) and four inter-subunit (Arg193/Glu212′Obackbone and Lys201/Glu208′, Arg193′/Glu212 and Lys201′/Glu208) salt bridges.
The solvent-accessible surface for a dimer is 21,990 Å2, where ~4300 Å2 of solvent-accessible surface is buried by the dimer interface. This corresponds to ~28.3% of the total accessible-solvent surface area of each monomer and 47.7% of the C-terminal domain buried surface. This analysis suggests that each C-terminal domain is unlikely to be able to exist independently, and the domain becomes stable only when a dimer is assembled. Therefore, the dimer can be considered to consist of three domains: two N-terminal domains and a dimerization domain. A similar arrangement has been observed for many other dimeric proteins.35,36
Nm-P5CR forms a dimer and Sp-P5CR assembles into decamer
The analysis of crystal packing and ligand and cofactor binding revealed that a dimer is a basic catalytic unit for both proteins (Figure 3(b)), although they assume different quaternary structures. The basic unit, a dimer, is formed by swapping the C-terminal domains. The overall shape of a dimer resembles a bean molecule, which measures 90Å in the longest dimension, 40Å in diameter in the widest point, and ~30Å in diameter in the narrowest point (Figure 3(a)). The two monomers interact very tightly using several α-helices of the C-terminal domain (Figure 3(b) and (c)). An analysis of crystal packing of Nm-P5CR and Sp-P5CR showed different quaternary structures, the former being a dimer and the latter forming a decamer. These oligomeric states observed in the crystal are consistent with the experimentally determined molecular mass of enzymes in solution as estimated from size-exclusion chromatography: 58.8 kDa (dimer) for Nm-P5CR and 284.6 kDa (decamer) for Sp-P5CR (data not shown). To date, a broad range of oligomeric states of P5CR has been reported, extending from 125 kDa (suggesting tetramer) for yeast P5CR37 to 200–340 kDa (octamer–dodecamer) observed for P5CRs purified from Clostridium sticklandii, Raltus norvegicus, Homo sapiens, Escherichia coli, and Spinacia oleracea.7,10,38-40
As shown by the crystal structures, these enzymes can exist as a dimer or as multimers of dimers. In Sp-P5CR, five dimers are arranged into a decamer resembling an hourglass-shaped barrel (Figure 4). The decamer, with overall dimensions of 87Å×98Å×99Å, is organized as a pentamer of dimers, with the dimers arranged tightly around the 5-fold symmetry axis and the active sites facing the outside of the barrel. The monomers in each of the five dimers are related by the 2-fold symmetry axis, which is perpendicular to the 5-fold symmetry axis. The decamer is formed entirely by side-by-side (between dimer 1, subunit A/D, and dimer 2, subunit B/C) and cross contacts (between (1) subunit A of dimer 1 with subunit C of dimer 2 and (2) subunit D of dimer 1 with subunit B of dimer 2) contributed by residues of the C-terminal part of H8; N terminus of H9; helices H10, H11; N-terminal part of H12; and the symmetry counterparts. Therefore, these helices contribute both to the dimer and decamer formation. Calculation of the accessible surface area indicated that dimers contribute to the dimer–dimer formation by burying the 2104Å2 surface area.
Figure 4.

Ribbon drawing of the dodecamer of Sp-P5CR in two views with NADP+. (a) A view down the non-crystallographic 5/2-fold relating the subunits. (b) The dodecamer viewed in a side orientation. NADP+ molecule is shown as a space-filling model in green, blue, and red.
There are several important interactions that help to stabilize the dimer–dimer interface and contribute to decamer formation. The interface is stabilized by a small hydrophobic patch that includes Leu171, Ala175, Val181, Ile227, Leu230 and Met231. Apart from the hydrophobic interactions, the rest of the interface is stabilized by the extensive solvent-mediated interactions and two salt bridges formed by Lys187/Asp217′ and Lys187′/Asp217. An interesting π–π interaction is observed between imidazol rings of His213 of helix H10 and H10′ of the neighboring dimer. These helices are part of active sites in two neighboring dimers. The imidazole rings of His213 of helices H10 and His213′ of H10′ stack over each other, contributing to the stability of dimer–dimer interface. This interaction may act as a sensor that allows communication between active sites. The majority of the residues involved in decamer formation are not strongly conserved among the family, which is consistent with different oligomeric states of P5CR in different species.
P5CR active site
NADP+ binding
The structures of Nm-P5CR and Sp-P5CR complexed with the NADP+ cofactor were obtained by co-crystallization. Although B-factors for NADP+ atoms are quite high (~60 Å2), the electron density of the coenzyme is well defined in Sp-P5CR and allowed for accurate placement of the entire molecule (Figure 5). In the structure of Nm-P5CR complexed with NADP+, the cofactor molecules appear to be bound in a similar conformation as that in the structure of Sp-P5CR. However, the electron density is of rather poor quality, allowing only unambiguous positioning of part of the coenzyme. The nicotinamide ring is disordered. Therefore, the structure of Sp-P5CR+NADP+ is used here to characterize binding of NADP+.
Figure 5.
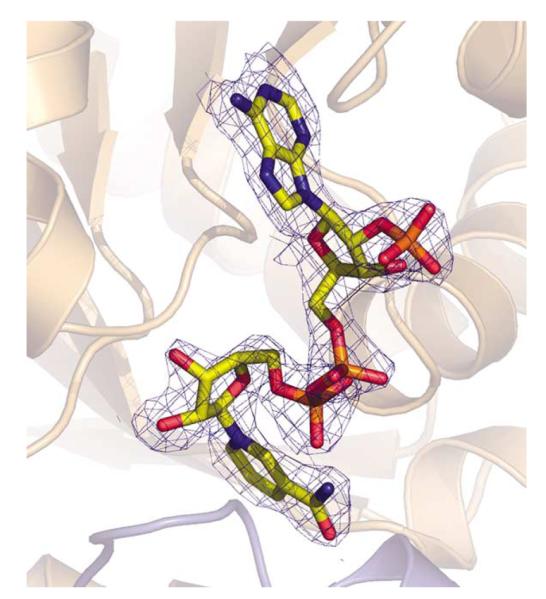
Electron density maps (2Fo–Fc) contoured at 1σ (blue) around NADP+ cofactor in the structure complex of Sp-P5CR/NADP+.
The NADP+ molecule binds in a groove between the N-terminal domain and the dimerization domain in an extended conformation with the 2′ phosphate group of the adenosine ribose of the NADP+ anchored at the N terminus of H2 and the nicotinamide moiety of the coenzyme buried inside the active center pocket. The nucleotide-binding domain contains the characteristic dinucleotide-binding motif (GXGXXA/G) (part of the Rossmann fold), which is well conserved throughout the P5CR enzyme family (Figure 2; residues 7–12 in Sp-P5CR). This glycine-rich region forms a loop between H1 and S1 that imposes the correct position of the central part of the coenzyme and provides the partial charge stabilization of the phosphate group by the dipole of H1. There are eight direct interactions between the coenzyme and seven amino acid residues of the protein, which are contributed almost primarily by the NADP+-binding domain. In addition to direct interactions, the coenzyme is bridged with the protein via five solvent-mediated interactions. The interactions are shown schematically in Figure 6. There is only a single hydrogen bond interaction between AN1 atom of the adenine moiety of the coenzyme and the NE2 of the imidazole ring of His51 of the protein. Similar to the adenine moiety, the ribose moiety is bridged only with the backbone amide group of Lys66 via a water molecule. The NH1and NE atoms of Arg35 form a hydrogen bond with the oxygen atoms of the monophosphate of the ribose moiety of NADP+. The position of the sulfate ion found in Nm-P5CR corresponds to a phosphate group (AP2) of NADP+ (see below). In contrast to the adenyl and ribose moieties, the pyrophosphate is involved in more extensive interactions with protein and solvent. It hydrogen bonds to the peptide bond amides of Lys10 and Ser221′ and three water molecules. One of these water molecules bridges the pyrophosphate with the glycine-rich loop between strand S1 and helix H1, thus forming hydrogen bonds with Gly9, Ala12, and Gly64 of strand S3. This water molecule has been shown to be conserved in a large number of Rossmann fold domains and was proposed to be a structural feature typical to the classic Rossmann dinucleotide-binding fold.41
Figure 6.

Schematic representation of NADP+ interactions with the surrounding protein residues (continuous blue lines) and solvent molecules (broken black line) or hydrophobic contacts (semi-circle) in molecule A of Sp-P5CR. Distances and hydrogen bond donors and acceptors are shown as subscripts and superscripts, respectively.
The nicotinamide is in a syn conformation, and its pro-S face is exposed for substrate positioning and pro-S hydrite transfer to P5C. The pro-R face of the nicotinamide ring packs against the hydrophobic part of the active center pocket and is flanked by methionine residues Met11, Met86, Met109, and Met112. It is stabilized by a single hydrogen bond interaction between a highly ordered water molecule and the NO7 of the carboxyamide group of the nicotinamide. The highly ordered water molecule, in turn, is fixed in position by hydrogen bond interaction with the backbone amide of Met112 and a peptide carbonyl of Gly163. Besides this contact, an intra-molecular hydrogen bond is formed between the carboxamide nitrogen atom of the nicotinamide ring and the nearby phosphoryl oxygen of the pyrophosphate moiety, thereby adding to the stability of NADP molecule and protein/NADP complex.
Sulfate binding
In the structure of native Nm-P5CR, an unassigned electron density was found on the surface of a protein that was too large for a water molecule. Sulfate was fitted into this density (the protein was crystallized in the presence of ammonium sulfate). Sulfate is bound exclusively to the N-terminal domain of one subunit in the shallow cavity on the surface and is positioned to interact with the positively charged N terminus of H2. The interactions with protein include solvent-mediated contact with Asn31, a hydrogen bond with an amide of Gly33, and a hydrogen bond with Lys36 (Figure 7). The position of sulfate is defined by the interaction, with the positively charged surface formed by the dipole of the H2, Lys36, Arg39, and amide group of Gly33. Next to the sulfate, there is an extended groove running across the protein surface; this groove is lined up with several ordered water molecules. The phosphate group (AP2) of NADP+ binds to this site. Binding sulfate may have biological relevance because it was reported that phosphate can inhibit P5CR.42
Figure 7.
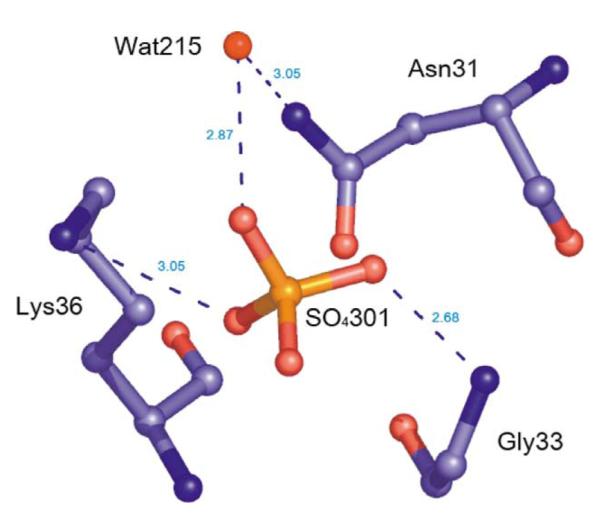
Diagram of sulfate binding in Nm-P5CR.
Proline binding
The catalytic centers of P5CRs are found at the interface of the nucleotide-binding domain of one subunit and at the part of the dimerization domain contributed by the opposite subunit. Close examination of the Fo–Fc and 2Fo–Fc density maps in the active center pockets of the structure of the Sp-P5CR/L-proline complex indicated presence of the two blobs of density, which were too large for a water molecule and were modeled as proline molecules (Figure 8(a)). One bound proline (with a B-factor of 32.8 Å2) is located in a small cavity at the bottom of the active center (Figure 8(b)). We propose that this proline represents binding of the product and provides information about binding of the substrate, P5C. The cavity is lined by the main-chain and side-chains of residues from the C-terminal domain of the symmetry-related subunit (C terminus of H10′ and N terminus of H11′ of the Sp-P5CR) (Figure 8(a),(b) and (c)). The positioning of the proline is determined by the hydrogen bonding between the carboxylate of the proline and adjacent OH groups of Thr225′ and Thr226′ and the amide group of Thr226′. The second bound proline (with a B-factor of 60.4 Å2) is positioned at the center of the active pocket. Superimposition of the structures of Sp-P5CR with L-proline and with NADP+ shows that this proline partially overlaps with the nicotinamide ring of the cofactor (Figure 8(a)). Thus, we propose that this proline acts as a competitive inhibitor, revealing the insides of the substrate inhibition mechanism of this enzyme. Inspection of the L-proline-bound structure identified a conserved, well-ordered solvent molecule at the bottom of the cavity that forms a hydrogen bond with the L-proline and could be involved in the stabilization of the substrate upon its binding and transition state during hydrate transfer. This water is surrounded by the Cys227′ and Ile231′ at one side of the cavity and hydrogen bonded with carbonyl and amide of Ser221′ and nearby carboxylate of the proline at the opposite side of the cavity. Aside from the proline carboxylate interactions, L-proline can be further stabilized by contacts between an N1 of proline and a carbonyl of Ile219′ and potentially by Ser221′.
Figure 8.

(a) Simulated annealing26 omit electron density maps (Fo–Fc) contoured at 2.5σ (blue) around L-proline molecules in the structure of Sp-P5CR complexed with L-proline. The adenosine ring of the NADP+ molecule (green) overlaps with proposed inhibitory L-proline. The NADP+ molecule is shown in the orientation as observed in the Sp-P5CR/NADP+complex. (b) Diagram of the active site L-proline binding in Sp-P5CR. (c) Close-up view of the active center of superimposed NADP and proline-bound structures showing the relative positioning between hydride donor (C4-NADPH) and acceptor (C5).
P5CR catalyses oxidation of proline derivatives
We have tested P5CR enzymatic activity by using a proline dehydrogenase assay (P5CR inverse reaction). L-Proline serves as a very poor substrate for both P5CRs, suggesting that conversion of P5C to L-proline is virtually irreversible. Similar observations have been reported earlier. The end product, L-proline was shown to competitively inhibit P5CR from E. coli K-12 and H. sapiens.43,44 However, some proline derivatives (thiaproline, 3,4-dehydro-L-proline) served as good substrates, consistent with previously reported data (Figure 9).45 According to our analysis, Nm-P5CR appears to be somewhat more active than Sp-P5CR. Interestingly, L-proline inhibits the reverse reaction at a concentration of 10–20 mM. This finding is consistent with the observation of the second proline molecule in the active site of Sp-P5CR that partly overlaps with the nicotinamide ring of NADP+. Phosphate also showed some inhibitory effect at a concentration of 100 mM (data not shown), as reported.42
Figure 9.

Nm-P5CR ((a) and (b)) and Sp-P5CR ((c) and (d)) proline dehydrogenase activity. The enzymatic activity tested in the oxidation of L-thiazolidine-4-carboxylate (T4C) ((a) and (c)) and 3,4-dehydro-L-proline ((b) and (d)) in the presence of NADP+. The progress of the reaction was measured at room temperature at 340 mm. Reaction mixtures contained 100 mM Hepes (pH 8.0), 1 mM NADP+, 10 mM substrate T4C or 3,4-dehydro-L-proline and 0.5 μM of enzyme as a monomer in a total volume of 1 ml (red line). The orange line corresponds to reaction progress without enzyme, the brown line corresponds to reaction with 10 mM L-proline (no T4C or 3,4-dehydro-L-proline added), and the blue and green lines correspond to reactions containing 10 mM and 20 mM L-proline (with T4C or 3,4-dehydro-L-proline present).
The kinetic studies of cofactor affinity binding indicated that most of the P5CRs will utilize either NADPH or NADH cofactor, but generally there will be a preference for one or the other (Table 3).1,10 The structure of the complex of Sp-P5CR with NADP+ identified that Ser31 and Arg35 hydrogen bond to the 2′phosphomonoester of NADP+, adding to the stability of the cofactor/protein complex. In the structure of Nm-P5CR Asn31 and Lys36 could make similar interactions. Interaction of an arginine residue with the monophosphate of the ribose moiety of the NADP+ is considered to be a determinant of the specificity of binding NADPH over NADH.46 This residue may be an important determinant of the cofactor specificity in the P5CR family of enzymes as well. Among P5CR family members Arg35 is only moderately conserved, consistent with the data suggesting that preference for NAD and NADP vary for enzymes from different sources.
Table 3.
Km value of P5CR from various organisms, as summarized by Merrill et al.10
|
Km (mM) |
Inhibitors |
||||
|---|---|---|---|---|---|
| Source of P5CR | NADH | NADPH | Proline | NADP+ | ATP |
| Erytrocytes | 0.48 | 0.09 | − | + | − |
| Lens | 0.62 | 0.05 | − | + | + |
| Retina | 0.13 | − | + | ||
| Fibroblast | 0.41 | 0.2 | + | − | |
| LHN | 0.56 | 0.64 | + | − | |
| REH | 0.59 | 0.64 | + | + | |
| Rat liver | 0.21 | + | + | ||
| Calf liver | 0.33 | 0.69 | + | + | + |
| Yeast | 0.08 | + | |||
| Neurospora crassa | 0.53 | 0.45 | |||
| E. coli | 0.14 | 0.15 | + | + | |
Proposed catalytic mechanism
To understand the interaction of the substrate and a cofactor with the enzyme, we have compared structures of free P5CR from Nm-P5CR with structures of Sp-P5CR in complex with the products of the reaction, L-proline and the cofactor NADP+. The structures were superimposed by least-squares fit. No significant conformational changes were observed in any of the three structures, suggesting that binding of ligands closely corresponds to the lock-and-key model. The majority of residues involved in proline and NADP+ binding are conserved throughout the family (Figures 2 and 3(a)).
In the superimposed structures of complexes with L-proline and NADP+, the distance between the pro-S face of the C4 carbon of NADP+ and the C5 of L-proline is 3.3Å (Figure 8(c)), which is consistent with a direct hydride-transfer mechanism. The structural analysis suggests that during the reaction, the hydride is transferred from the NADPH pro-S face of C4 to C5 of P5C, and the proton could be potentially provided either by (1) Thr226′ or Ser221′, which are within an average distance of 3.5Å and 3.7Å from N1 of P5C, or (2) possibly a water molecule. Mutagenesis studies and crystal structures of a complex with proline analogs and NAD(P)H can be used to identify the role of these residues and should provide more insight into the mechanism of catalysis.
While the mechanism of substrate binding and release of product awaits detailed kinetic studies, analysis of the crystal structures gives some insights regarding the possible order of substrate binding. A surface rendering of the active center of the structure of Sp-P5CR with NADP+ shows that upon binding of the coenzyme, the entrance to the active center cavity is effectively blocked, leaving only a small opening insufficient to allow P5C to pass by. This suggests that P5C may bind first followed by binding of NADPH. It also suggests that upon product formation the order is reversed and NADP+ is released first, followed by the release of L-proline.
In the dimer, the active sites are ~36Å apart and appear to operate independently. However, Sp-P5CR and other multimeric P5CRs may allow for communication between active sites through interaction within the decamer or other oligomeric forms.
The P5CR is constructed in a very modular manner, where one domain (C-terminal) provides nearly all of the constituents required for binding P5C; the second domain (N-terminal) provides the NADPH binding site, and these two domains are aligned by using a few key anchoring residues. Therefore, the catalytically functional unit of P5CR is a dimer, which can assemble into different oligomeric states by using the dimer as a building unit, depending on the cellular requirements (Figure 10).
Figure 10.

Proposed catalytic mechanism for P5CR.
Materials and Methods
Gene cloning and protein expression
The P5CR ORFs from 14 organisms (A. pernix, B. anthracis, B. subtilis, E. coli, E. faecalis, H. influenzae, H. pylori, N. meningitides, S. choleraesuis, S. flexneri, S. oneidensis, S. pneumoniae, S. pyogenes, and V. cholerae) were cloned by using procedures established earlier.47 Nine P5CR enzymes expressed well in a soluble form, one clone expressed poorly, and four did not show significant levels of expression. All proteins obtained in a soluble form were processed in the MCSG pipeline. In the following, we describe details of the cloning and expression protocols for P5CR from N. meningitides and S. pyogenes.
The ORF of P5CR (proC) was amplified by PCR from N. meningitides MC58 genomic DNA (ATCC) with KOD DNA polymerase by using conditions and reagents provided by the vendor (Novagen, Madison, WI). The gene was cloned into a pMCSG7 vector by using a modified ligation-independent cloning protocol.49,50 This process generated an expression clone of a fusion protein with an N-terminal His6-tag and a TEV protease recognition site (ENLYFQ↓S). The fusion protein was over-produced in an E. coli BL21-derivative that harbored a plasmid pMAGIC encoding three rare E. coli tRNAs (Arg (AGG/AGA) and Ile (ATA)) as described.50 This construct provides an N-terminal His6-tag separated from the gene by a TEV protease recognition sequence. By using an identical approach, the ORF of P5CR was amplified by PCR from S. pyogenes genomic DNA (ATCC), cloned into pMCSG7, and expressed in E. coli BL21/pMAGIC.
A selenomethionine (SeMet) derivative of the expressed protein was prepared as described.51 The transformed BL21 cells were grown at 37 °C in M9 medium supplemented with 0.4% (w/v) glucose, 8.5 mM NaCl, 0.1 mM CaCl2, 2 mM MgSO4, and 1% (w/v) thiamine. After the A600 reached 0.5, 0.01% (w/v) each of leucine, isoleucine, lysine, phenylalanine, threonine, and valine was added to inhibit the metabolic pathway of methionine and encourage SeMet incorporation. SeMet (60 mg) was added to one liter of culture, and 15 min later, protein expression was induced by 1 mM isopropyl-β-D-thiogalactoside (IPTG). The cells were then incubated at 20 °C overnight. The harvested cells were resuspended in lysis buffer (500 mM NaCl, 5% (v/v) glycerol, 50 mM Hepes (pH 8.0), 10 mM imidazole, and 10 mM 2-mercaptoethanol) and stored at −20 °C.
Protein purification and crystallization
Both native and SeMet proteins were purified according to a standard protocol.49 The harvested cells were resuspended in lysis buffer and lysozyme was added at a final concentration of 1 mg/ml, as was 100 ml of protease inhibitor (Sigma, P8849) per 2 g of wet cells. This mixture was kept on ice for 20 min and then sonicated. The lysate was clarified by centrifugation at 36,000g for 1 h and filtration with a 0.44 μm membrane. The clarified lysate was applied to a 5 ml HiTrap Ni-NTA column (GE Health Systems) on an AKTAexpress system (GE Health Systems). The His6-tagged protein was released with elution buffer (500 mM NaCl, 5% glycerol, 50 mM Hepes (pH 8.0), 250 mM imidazole, 10 mM 2-mercaptoethanol), and the fusion tag was removed by treatment with recombinant His6-tagged TEV protease (a gift from Dr D. Waugh, NCI). A Ni-NTA affinity chromatography step was performed by using a standard bench-top procedure to remove the His6-tag, uncut protein, and His6-tagged TEV protease. Proteins were concentrated by using Centricon concentrators with 5000 Mr cutoff (Amicon), flash frozen, and stored at the liquid nitrogen.
The Nm-P5CR protein was dialyzed against crystallization buffer containing 200 mM NaCl, 20 mM Hepes (pH 8.0), and 2 mM DTT and then concentrated to 130 mg/ml. Crystallization was performed by the sitting-drop vapor diffusion method at 16 °C. Index (Hampton Research) was used for the initial screening. Plate-shaped crystals were grown from 0.2 M ammonium sulfate, 0.1 M Hepes (pH 7.5), and 25% (w/v) polyethylene glycol 3350. Crystals of complex with NADP+ were obtained by adding NADP+ to the protein solution to a final concentration of 3 mM and crystallized under the same condition as the native enzyme. The crystals reached the size 0.2 mm×0.2 mm×0.15 mm within two days. The mother liquor containing 10% glycerol was used as a cryoprotectant. The crystal was picked up with a nylon loop (Hampton Research) and flash-frozen in liquid nitrogen. Both native P5CR and the complex with NADP+ crystals belong to space group P21212. The unit cell parameters for free enzyme were a=89.52Å, b=49.93Å, c=65.60Å, and α=γ=β=90°, and those for the complex are a=89.70Å, b=49.83Å, c=65.45, and α=γ=β=90°. The asymmetric unit contains one molecule with the Vm value of 2.6Å3/Da and solvent content 52%.
The Sp-P5CR protein was dialyzed against crystallization buffer containing the same formulation as stated previously and was concentrated to 125 mg/ml. Crystallization was performed by the sitting and hanging-drop method by using the vapor diffusion method at 25 °C. Large diffraction-quality crystals were obtained by mixing equal (1 μl) volumes of the protein and solution containing a mixture of the reservoir solution (3.5 M sodium formate (pH 7.0)) with either 5 mM L-proline or 10 mM NADP+ and equilibrated over 500 μl of the reservoir solution. The colorless crystals appeared typically within 24–48 h and grew to 0.3 mm×0.3 mm×0.3 mm. Sp-P5CR crystallizes in the monoclinic space group C2, with cell dimensions of a=171.82Å, b=109.96Å, c=84.16Å, α=γ=90°, and β=95.4°. The crystal asymmetric unit contains five molecules, with the Vm value of 2.88Å3/Da and solvent content 57%. Before data collection, crystals were soaked for ~10 s in a cryoprotectant consisting of 25% glycerol in the crystal mother liquor and then flash-frozen in liquid nitrogen.
Data collection and structure determination
For the ligand-free Nm-P5CR, two wavelengths of anomalous diffraction26 data near the selenium edge (peak and inflection) were collected from a SeMet-substituted protein. A data set at the selenium absorption edge was also obtained for a complex of Nm-P5CR with NADP+. Both data sets were collected on SBC-2, which is a 3×3 charged coupled device detector at 100 K in the 19ID beamline of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory. The diffraction data were processed by using the HKL2000 suite of programs.52 Data collection statistics are presented in Table 1. All procedures for MAD phasing, phase improvement by density modification, and initial protein model building were done by using the HKL2000_PH software package.52-58 At this stage, the mean figure of merit of the phase set was 0.383 for 30Å–2.2Å data and improved to 0.542 after density modification (DM).56 The autotracing modules built 201 out of 263 amino acid residues, and 110 residues were built with side-chains and 91 residues without. The initial model was rebuilt with the program COOT and O by using electron density maps based on DM-phased reflection file (COOT), or 2Fo–Fc maps (O).53,54 After each cycle of rebuilding, the model was refined by using CNS-SOLVE at the resolution range of 40Å to 1.9Å.26 For the final stage of refinement, REFMAC5 from the CCP4 suite was used with TLS refinement.27 The geometrical properties of the model were assessed by the program PROCHECK.28 The crystal of the NADP+ complex was isomorphous with the NADP-free crystal. The model of P5CR complexed with NADP+ was refined from the NADP-free structure without any phasing step by using REFMAC5.57 The NADP+ model was adopted from the HICUP server and rebuilt with program O.46,53
For the Sp-P5CR, a single anomalous dispersion (SAD) data set was collected on a Q315 CCD detector (ADCS) at the 19ID beamline of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory, to 2.15Å resolution from a single crystal of SeMet-labeled Sp-P5CR/NADP+ complex at the selenium-edge wavelength (Table 1). The Sp-P5CR/L-proline complex data were collected at the same wavelength to 2.20Å. All diffraction data were collected at 100 K. All data sets were processed by using the HKL2000 suite of programs.52 Data collection statistics are presented in Table 1. The structure of the NADP+ bound enzyme has been determined by using SAD phasing, density modification, and initial protein model building as implemented in HKL2000_PH software package.52-58 The initial model (~70% completed) was subsequently submitted for model building with programs ARP/wARP.59 The more completed model (85%) obtained from ARP/wARP was further extended manually by using the program COOT. The crystals of the Sp-P5CR/L-proline complex were isomorphous with the crystals of the SpP5CR/NADP+ complex. Hence, the structure of the latter was used as the starting model for refinement after removing NADP+ and solvent molecules. Cycles of manual corrections of both the models in program COOT and refinement with REFMAC with non-crystallographic symmetry (NCS) averaging restraints were repeated at the initial stage of refinement. Upon model improvement, NCS restrains were released. The final model was refined against all reflections in the resolution ranges of 50.0Å–2.15Å and 50Å–2.20Å, except for 5% of randomly selected reflections, which were used for monitoring Rfree.
The final round of refinement was carried out by using TLS refinement. The stereochemical quality of the structure models was checked with PROCHECK.28 Final refinement statistics are presented in Table 2. The sequence alignment Figure was generated by using the program Multalin and ESprint.60,61 The rest of the Figures were prepared by using programs PyMOL† and Ligplot.62
P5CR enzyme assays
The enzyme assays were performed spectroscopically as described by Charle et al.45 L-Proline dehydrogenase (the P5CR inverse reaction) activity was measured at room temperature (~22 °C) as an increase in absorbance at 340 mm by using an Agilent 8453 UV spectrophotometer. Assays were measured every 10 s for a total of 200 s. Reaction mixtures contain 100 mM Hepes (pH 8.0), 1 mM NADP+, and 10 mM substrate (thiaproline, 3,4-dehydro-L-proline, or L-proline), and the reaction was initiated by addition of enzyme at 0.5 μM in a total volume of 1 ml. Activities were calculated by using a 1 mM extinction coefficient of 6.22 for NADPH. To investigate the effect of L-proline on the enzyme activity, 10 mM and 20 mM of L-proline were included in the reaction mixture.
Size-exclusion chromatography
The molecular mass of P5CR proteins in solution was evaluated by size-exclusion chromatography on a Superdex-200 10/30 column (Pharmacia) by using crystallization buffer and calibrated with ribonuclease A (13.7 kDa), ovalbumin (43 kDa), albumin (67 kDa), aldolase (158 kDa), catalase (232 kDa), and thyroglobulin (669 kDa) as standards. The calibration curve of Kav versus log molecular mass was prepared by using the equation Kav=Ve–Vo/Vt–Vo, where Ve=elution volume for the protein, Vo = column void volume, and Vt=total bed volume.63
Protein Data Bank accession numbers
The coordinates of Nm-P5CR, Nm-P5CR+NADP+, Sp-P5CR+NADP+, and Sp-P5CR+L-proline have been deposited in the RSCB Protein Data Bank, with accession codes 1YQG, 2AG8, 2AHR, and 2AMF, respectively.
Acknowledgements
The authors are grateful to M. Cuff, J. Osipiuk, Y. Kim and M. Chodkiewicz-Nocek for helpful discussion, suggestions, and assistance with Figures generation. We also thank all members of the Structural Biology Center at Argonne National Laboratory for their help in conducting these experiments. This work was supported by National Institutes of Health grant GM62414 and by the US Department of Energy, Office of Biological and Environmental Research, under contract W-31-109-Eng-38.
Abbreviations used
- P5CR
Δ1-pyrroline-5-carboxylate reductase
- SeMet
selenomethionine
- rmsd
root-mean-square deviation
- pdb
Protein Data Bank
- ORF
open reading frame
- SAD
single anomalous dispersion
- MAD
multiple anomalous dispersion
- NCS
non-crystallo-graphic symmetry
Footnotes
DeLano, W.L., The PyMOL Molecular Graphics System http://www.pymol.org
References
- 1.Adams E, Frank L. Metabolism of proline and the hydroxyprolines. Annu. Rev. Biochem. 1980;49:1005–1061. doi: 10.1146/annurev.bi.49.070180.005041. [DOI] [PubMed] [Google Scholar]
- 2.Phang JM. The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr. Top. Cell Regul. 1985;25:91–132. doi: 10.1016/b978-0-12-152825-6.50008-4. [DOI] [PubMed] [Google Scholar]
- 3.Ambikapathy J, Marshall JS, Hocart CH, Hardham AR. The role of proline in osmoregulation in Phytophthora nicotianae. Fungal Genet. Biol. 2002;35:287–299. doi: 10.1006/fgbi.2001.1327. [DOI] [PubMed] [Google Scholar]
- 4.Csonka LN, Hanson AD. Prokaryotic osmoregulation: genetics and physiology. Annu. Rev. Microbiol. 1991;45:569–606. doi: 10.1146/annurev.mi.45.100191.003033. [DOI] [PubMed] [Google Scholar]
- 5.Whatmore AM, Chudek JA, Reed RH. The effects of osmotic upshock on the intracellular solute pools of Bacillus subtilis. J. Gen. Microbiol. 1990;136:2527–2535. doi: 10.1099/00221287-136-12-2527. [DOI] [PubMed] [Google Scholar]
- 6.Verbruggen N, Villarroel R, Van Montagu M. Osmoregulation of a pyrroline-5-carboxylate reductase gene in Arabidopsis thaliana. Plant Physiol. 1993;103:771–781. doi: 10.1104/pp.103.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deutch AH, Smith CJ, Rushlow KE, Kretschmer PJ. Escherichia coli delta 1-pyrroline-5-carboxylate reductase: gene sequence, protein overproduction and purification. Nucl. Acids Res. 1982;10:7701–7714. doi: 10.1093/nar/10.23.7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith RJ, Downing SJ, Phang JM, Lodato RF, Aoki TT. Pyrroline-5-carboxylate synthase activity in mammalian cells. Proc. Natl Acad. Sci. USA. 1980;77:5221–5225. doi: 10.1073/pnas.77.9.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohl DH, Schubert KR, Carter MB, Hagedorn CH, Shearer G. Proline metabolism in N2-fixing root nodules: energy transfer and regulation of purine synthesis. Proc. Natl Acad. Sci. USA. 1988;85:2036. doi: 10.1073/pnas.85.7.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merrill MJ, Yeh GC, Phang JM. Purified human erythrocyte pyrroline-5-carboxylate reductase. Preferential oxidation of NADPH. J. Biol. Chem. 1989;264:9352–9358. [PubMed] [Google Scholar]
- 11.Dougherty KM, Brandriss MC, Valle D. Cloning human pyrroline-5-carboxylate reductase cDNA by complementation in Saccharomyces cerevisiae. J. Biol. Chem. 1992;267:871–875. [PubMed] [Google Scholar]
- 12.Delauney AJ, Verma DP. Proline biosynthesis and osmoregulation in plants. Plant J. 1993;4:215–223. [Google Scholar]
- 13.Delauney AJ, Hu CA, Kishor PB, Verma DP. Cloning of ornithine delta-aminotransferase cDNA from Vigna aconitifolia by trans-complementation in Escherichia coli and regulation of proline biosynthesis. J. Biol. Chem. 1993;268:18673–18678. [PubMed] [Google Scholar]
- 14.Taylor CB. Proline and water deficit: ups and downs. Plant Cell. 1996;8:1221–1224. [Google Scholar]
- 15.Hanson AD, Hitz WD. Metabolic responses of mesophytes to plant water deficits. Annu. Rev. Plant Physiol. 1982;33:163–203. [Google Scholar]
- 16.Hare PD, Cress WA. Metabolic implications of stress-induced proline accumulation in plants. Plant Growth Reg. 1997;21:79–102. [Google Scholar]
- 17.Delauney AJ, Verma DP. A soybean gene encoding delta 1-pyrroline-5-carboxylate reductase was isolated by functional complementation in Escherichia coli and is found to be osmoregulated. Mol. Gen. Genet. 1990;221:299–305. doi: 10.1007/BF00259392. [DOI] [PubMed] [Google Scholar]
- 18.Misener SR, Chen C, Walker VK. Cold tolerance and proline metabolic gene expression in Drosophila melanogaster. J. Insect. Physiol. 2001;47:393–400. doi: 10.1016/s0022-1910(00)00141-4. [DOI] [PubMed] [Google Scholar]
- 19.Phang JM, Downing SJ, Yeh GC, Smith RJ, Williams JA, Hagedorn CH. Stimulation of the hexosemonophosphate-pentose pathway by pyrroline-5-carboxylate in cultured cells. J. Cell Physiol. 1982;110:255–261. doi: 10.1002/jcp.1041100306. [DOI] [PubMed] [Google Scholar]
- 20.Kohl DH, Schubert KR, Carter MB, Hagedorn CH, Shearer G. Proline metabolism in N2-fixing root nodules: energy transfer and regulation of purine synthesis. Proc. Natl Acad. Sci. USA. 1988;85:2036–2040. doi: 10.1073/pnas.85.7.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeh GC, Harris SC, Phang JM. Pyrroline-5-carboxylate reductase in human erythrocytes. J. Clin. Invest. 1981;67:1042–1046. doi: 10.1172/JCI110115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagedorn CH. Demonstration of a NADPH-linked delta 1-pyrroline-5-carboxylate-proline shuttle in a cell-free rat liver system. Biochim. Biophys. Acta. 1986;884:11–17. doi: 10.1016/0304-4165(86)90220-5. [DOI] [PubMed] [Google Scholar]
- 23.Hagedorn CH, Phang JM. Catalytic transfer of hydride ions from NADPH to oxygen by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch. Biochem. Biophys. 1986;248:166–174. doi: 10.1016/0003-9861(86)90413-3. [DOI] [PubMed] [Google Scholar]
- 24.Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61:1810–1815. [PubMed] [Google Scholar]
- 25.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 26.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallog. sect. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallog. sect. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 29.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallog. sect. D. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 30.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 31.Barycki JJ, O’Brien LK, Bratt JM, Zhang R, Sanishvili R, Strauss AW, Banaszak LJ. Biochemical characterization and crystal structure determination of human heart short chain L-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochemistry. 1999;38:5786–5798. doi: 10.1021/bi9829027. [DOI] [PubMed] [Google Scholar]
- 32.Ahn HJ, Eom SJ, Yoon HJ, Lee BI, Cho H, Suh SW. Crystal structure of class I acetohydroxy acid isomeroreductase from Pseudomonas aeruginosa. J. Mol. Biol. 2003;328:505–515. doi: 10.1016/s0022-2836(03)00264-x. [DOI] [PubMed] [Google Scholar]
- 33.Matak-Vinkovic D, Vinkovic M, Saldanha SA, Ashurst JL, von Delft F, Inoue T, et al. Crystal structure of Escherichia coli ketopantoate reductase at 1.7Å resolution and insight into the enzyme mechanism. Biochemistry. 2001;40:14493–14500. doi: 10.1021/bi011020w. [DOI] [PubMed] [Google Scholar]
- 34.Winter VJ, Cameron A, Tranter R, Sessions RB, Brady RL. Crystal structure of Plasmodium berghei lactate dehydrogenase indicates the unique structural differences of these enzymes are shared across the Plasmodium genus. Mol. Biochem. Parasitol. 2003;131:1–10. doi: 10.1016/s0166-6851(03)00170-1. [DOI] [PubMed] [Google Scholar]
- 35.Walsh MA, Otwinowski Z, Perrakis A, Anderson PM, Joachimiak A. Structure of cyanase reveals that a novel dimeric and decameric arrangement of subunits is required for formation of the enzyme active site. Structure Fold. Des. 2000;8:505–514. doi: 10.1016/s0969-2126(00)00134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, et al. Crystal structure of trp repressor/operator complex at atomic resolution. Nature. 1988;335:321–329. doi: 10.1038/335321a0. [DOI] [PubMed] [Google Scholar]
- 37.Matsuzawa T, Ishiguro I. delta 1-Pyrroline-5-carboxylate reductase from Baker’s yeast. Purification, properties and its application in the assays of L-delta 1-pyrroline-5-carboxylate and L-ornithine in tissue. Biochim. Biophys. Acta. 1980;613:318–323. doi: 10.1016/0005-2744(80)90086-8. [DOI] [PubMed] [Google Scholar]
- 38.Kenklies J, Ziehn R, Fritsche K, Pich A, Andreesen JR. Proline biosynthesis from L-ornithine in Clostridium sticklandii: purification of delta1-pyrroline-5-carboxylate reductase, and sequence and expression of the encoding gene, proC. Microbiology. 1999;145:819–826. doi: 10.1099/13500872-145-4-819. [DOI] [PubMed] [Google Scholar]
- 39.Shiono T, Kador PF, Kinoshita JJ. Purification and characterization of rat lens pyrroline-5-carboxylate reductase. Biochim. Biophys. Acta. 1986;881:72–78. doi: 10.1016/0304-4165(86)90098-x. [DOI] [PubMed] [Google Scholar]
- 40.Murahama M, Yoshida T, Hayashi F, Ichino T, Sanada Y, Wada K. Purification and characterization of Delta(1)-pyrroline-5-carboxylate reductase isoenzymes, indicating differential distribution in spinach (Spinacia oleracea L.) leaves. Plant Cell Physiol. 2001;42:742–750. doi: 10.1093/pcp/pce093. [DOI] [PubMed] [Google Scholar]
- 41.Bottoms CA, Smith PE, Tanner JJ. A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 2002;11:2125–2137. doi: 10.1110/ps.0213502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miler PA, Stewart CR. Pyrroline-5-carboxylic acid reductase from soybean leaves. Phytochemistry. 1976;15:1855–1857. [Google Scholar]
- 43.Rossi JJ, Vender J, Berg CM, Coleman WH. Partial purification and some properties of delta1-pyrroline-5-carboxylate reductase from Escherichia coli. J. Bacteriol. 1977;129:108–114. doi: 10.1128/jb.129.1.108-114.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valle D, Downing SJ, Phang JM. Proline inhibition of pyrroline-5-carboxylate reductase: differences in enzymes obtained from animal and tissue culture sources. Biochem. Biophys. Res. Commun. 1973;54:1418–1424. doi: 10.1016/0006-291x(73)91144-3. [DOI] [PubMed] [Google Scholar]
- 45.Deutch CE, Klarstrom JL, Link CL, Ricciardi DL. Oxidation of L-thiazolidine-4-carboxylate by delta1-pyrroline-5-carboxylate reductase in Escherichia coli. Curr. Microbiol. 2001;42:426–442. doi: 10.1007/s002840010245. [DOI] [PubMed] [Google Scholar]
- 46.Kleywegt GJ, Jones TA. Databases in protein crystallography. Acta Crystallog. sect. D. 1998;54:1119–1131. doi: 10.1107/s0907444998007100. [DOI] [PubMed] [Google Scholar]
- 47.Dieckman L, Gu M, Stols L, Donnelly MI, Collart FR. High throughput methods for gene cloning and expression. Protein Expr. Purif. 2002;25:1–7. doi: 10.1006/prep.2001.1602. [DOI] [PubMed] [Google Scholar]
- 48.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 49.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, et al. Automation of protein purification for structural genomics. J. Struct. Funct. Genoms. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang RG, Skarina T, Katz JE, Beasley S, Khachatryan A, Vyas S, et al. Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure (Camb.) 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh MA, Dementieva I, Evans G, Sanishvili R, Joachimiak A. Taking MAD to the extreme: ultrafast protein structure determination. Acta Crystallog. sect. D. 1999;55:1168–1173. doi: 10.1107/s0907444999003698. [DOI] [PubMed] [Google Scholar]
- 52.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallog. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 53.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog. sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallog. sect. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallog. sect. D. 2002;58:1772177–1772179. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 56.Cowtan KD, Main P. Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints. Acta Crystallog. sect. D. 1993;49:148–157. doi: 10.1107/S0907444992007698. [DOI] [PubMed] [Google Scholar]
- 57.The Collaborative Computational Project Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallog. sect. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 58.Terwilliger TC. SOLVE and RESOLVE automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 59.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 60.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucl. Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 62.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein–ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 63.Zhang RG, Grembecka J, Vinokour E, Collart F, Dementieva I, Minor W, Joachimiak A. Structure of Bacillus subtilis YXKO–a member of the UPF0031 family and a putative kinase. J. Struct. Biol. 2002;139:161–170. doi: 10.1016/s1047-8477(02)00532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun G, Voigt JH, Filippov IV, Marquez VE, Nicklaus MC. PROSIT: pseudo-rotational online service and interactive tool, applied to a conformational survey of nucleosides and nucleotides. J. Chem. Inf. Comput. Sci. 2004;44:1752–1762. doi: 10.1021/ci049881+. [DOI] [PubMed] [Google Scholar]