

Abstract
Human gene expression traits have been shown to be dependent on gender, age and time of day in blood and other tissues. However, other factors that may impact gene expression have not been systematically explored. For example, in studies linking blood gene expression to obesity related traits, whether the fasted or fed state will be the most informative is an open question. Here, we employed a two-arm cross-over design to perform a genome-wide survey of gene expression in human peripheral blood to address explicitly this type of question. We were able to distinguish expression changes due to individual and time-specific effects from those due to food intake. We demonstrate that the transcriptional response to food intake is robust by constructing a classifier from the gene expression traits with >90% accuracy classifying individuals as being in the fasted or fed state. Gene expression traits that were best able to discriminate the fasted and fed states were more heritable and achieved greater coherence with respect to pathways associated with metabolic traits. The connectivity structure among gene expression traits was explored in the context of coexpression networks. Changes in the connectivity structure were observed between the fasted and fed states. We demonstrate that differential expression and differential connectivity are two complementary ways to characterize changes between fasted and fed states. Both gene sets were significantly enriched for genes associated with obesity related traits. Our results suggest that the pair of fasted/fed blood expression profiles provide more comprehensive information about an individual's metabolic states.
INTRODUCTION
The accessibility of blood-derived samples for monitoring molecular phenotypes has the potential to provide a rich source of information to biomedical researchers regarding molecular processes associated with disease and drug response. The analysis of patterns of gene expression from human blood derived samples has been explored in a large number of studies with goals as far reaching as assessing the extent of individual specific variation (1–3) identifying responses to environmental exposures (4–6), determining disease status (7–12), elucidating response to clinical treatment (13) and comparing gene expression variation between different tissues (14,15). Through many of these studies, gene expression traits from human peripheral blood tissue have been found to vary significantly by age, sex, time of day and disease status (2,3,7,8,15). The primary aim of many of these studies is the identification of biomarkers for disease and drug response: genes whose activity can be measured and used to inform on the cellular processes occurring in a given physiological state. However, most studies have not appropriately controlled for many of the covariates known to influence blood expression traits, including age, sex, time of day, fasting/feeding status, blood cell count or as yet to be determined factors. In addition, assessing the most informative state in which to sample human blood for molecular phenotypes associated with a given clinical trait of interest has not been adequately studied for any disease.
An important environmental condition for monitoring molecular phenotypes and characterizing their association with metabolic traits is feeding status. Portions of the gene network that are associated with metabolic traits like obesity may be most active in response to feeding, rather than in the fasted state (the most common condition under which clinical samples for molecular phenotyping are collected). Because the molecular networks that underlie biological systems are highly modular systems exhibiting a plasticity that allows them to adapt to a vast array of conditions (16,17), it underscores the importance of finding the appropriate condition to study the molecular network properties. In fact, several recent studies have confirmed the plasticity of biological networks by demonstrating rapid rewiring of the network in response to different environmental stresses (18–20). Some success has been achieved in studying fasting and feeding response in rodents. In livers of normal mice Foxa2 has been shown to regulate lipid metabolism and ketogenesis in the fasted state where Foxa2 is translocated into the nucleus, but not in the fed state where Foxa2 is mostly excluded from the nucleus (21). Other studies have shown that circadian clock genes in murine heart tissue are regulated differently under fasting and feeding conditions (22,23). We have also shown that some networks predicted to be strongly causal for metabolic traits are profoundly affected in response to different diets (e.g. high-fat diet versus a normal chow diet in mouse) (24).
Although much has been published on the effects of fasting and feeding on plasma protein and metabolite levels in humans (e.g. plasma leptin and glucose levels and a number of single or few gene responses to fasting and feeding have been reported) (25), there has not been a systematic human study focused on the genome-wide characterization of differences in response to fasting and feeding over multiple time points at the gene expression level. Here, we performed an extensive genome-wide survey of gene expression in human peripheral blood tissue to identify patterns of gene expression associated with age, time of day, response to food intake and variation in clinical traits related to obesity and metabolism more generally. By employing a standard two-arm cross-over design, we were able to distinguish expression changes due to individual and time-specific effects from those due to food intake. We demonstrate that the transcriptional response to food intake is robust by constructing a classifier from the gene expression traits that achieves greater than 90% accuracy to discriminate between the fasting and fed states. Gene expression traits that were best able to discriminate the fasting and fed states were also found to be more heritable, to give rise to stronger linkage signals, and to exhibit greater coherence with respect to pathways associated with metabolic traits, suggesting signaling in the transcriptional network induced by feeding is an important state to consider in studies of clinical traits related to metabolic diseases like obesity. We explored the connectivity structure among gene expression traits in the context of coexpression networks and found clear changes to this structure over time and between the fasted and fed states, providing insights into the pathways and mechanisms involved in the response to food intake.
RESULTS
To eliminate sex-specific effects on gene expression and to reduce expression variation due to age, disease, ethnicity and environment, we recruited 40 apparently healthy Caucasian males from the greater Reykjavik area in Iceland who were non-smokers, not on any medication, and within the 29–50-year-old age range. A series of clinical measurements corresponding to biometric, obesity, diabetes and cardiovascular related traits were collected for each participant (Supplementary Material, Table S1). In addition, each participant completed a survey to collect additional information on covariates that had the potential to influence obesity related traits, including characterizing normal diet, exercise, drug use (including prescription drugs and alcohol use) and vitamin use. To control for inter-individual variation as well as temporal variation, we employed a two-arm (fasting arm and feeding arm), two-period, randomized cross-over design for this study. Each individual participated in both the fasting and feeding arms of the study as depicted in Figure 1.
Figure 1.

Experimental design. A two-arm two-period crossover design was employed. Forty individuals participated in both the fasting and fed arms of the study. In each arm, blood was collected at seven time points. Individuals began each arm in the fasting state. Individuals in the fed arm consumed a meal between the second and third time points.
For the first period of the study, individuals were randomly assigned to one of two arms: the fasted arm, in which individuals came in fasted from the night before and continued to fast throughout the rest of the day, or the fed arm, in which individuals came in fasted and then consumed a standard meal in the morning, but then fasted throughout the rest of the day. In each arm of the study peripheral blood was collected from each of the 40 individuals at seven time points throughout the day, between the hours of 8 a.m. and 3 p.m. (Fig. 1). The second period of the study occurred 1 week later, on the same day of the week as the first period, and was carried out in the same manner as the first, but with participants crossing over into the alternate fasting/fed treatment arms. In total, 560 peripheral blood samples were collected over the course of the study (14 per individual). RNA from all but 12 of the samples were successfully processed and hybridized to custom microarrays manufactured by Agilent Technologies (Palo Alto, CA, USA) targeting 23 653 human transcripts corresponding to 20 443 genes. A number of covariates have previously been associated with gene expression traits, including age, sex, blood cell count and time of day samples were collected (15). The covariance was removed by linear regression as previously described (15), and the residuals were used in the following analysis.
Significant inter- and intra-individual variation in the blood transcriptional network
We performed a standard two-way ANOVA analysis on each gene expression trait over the fasting arm for each time period of the study, taking gene expression as the dependent variable and the individual and time as the independent factors in the analysis. Examination of the fasting arm allowed us to measure individual-specific variation and temporal variation without introducing the confounding factor of feeding response that occurs in the fed arms. Out of 23 653 expression traits profiled, 20 428 had a significant individual specific component (Supplementary Material, Table S2), whereas 8197 genes were found to have a significant time component (Supplementary Material, Table S2), at a significance threshold of 0.01. This threshold corresponds to a false discovery rate (FDR) (26) of 0.94% for the individual factor and 2.6% for the time factor, determined by permuting the individual and time point ids with respect to the gene expression traits 1000 times and then dividing the average number of counts over the 1000 runs by the count obtained from the observed data.
These data highlight a striking gene expression signature that is individual specific, likely reflecting strong genetic and environmental effects specific to each individual. The significant inter-individual variations are further highlighted by two-dimensional, agglomerative hierarchical clustering of the blood expression data, where samples from the same individual were almost always seen to cluster most tightly together (Fig. 2). However, there is also a strong temporal variation component to the gene expression traits, where, interestingly, 7996 of the 8197 genes with a significant time component overlap the 20 428 genes with a significant individual component (P < 10−323 as determined by the Fisher Exact Test), indicating that many genes have strong individual and temporal effects. Detection of a stronger temporal component was achieved by considering expression measures from all individuals over both arms and time periods. A three-way ANOVA was carried out using arm, individual and time as factors, resulting in 19 922 and 11 206 (Supplementary Material, Table S2) gene expression traits with significant individual and time components (11 003 in the overlap), respectively, at the 0.01 significance level (corresponding to FDRs of 2.0 and 2.3%, respectively), again indicating that a significant portion of the time specific genes are also individual specific. Of the 20 428 genes identified in the fasting arms, 19 434 overlap the 19 922 transcripts identified over all arms and time periods, indicating that the individual specific response is consistent between the fasting and fed states. The percentage of transcripts that had an individual specific component in our data set (81.9%) was similar to previous reports, but was on a more comprehensive set of genes that were not a priori filtered for expression or differential gene expression (27).
Figure 2.

Two-dimensional Hierarchical clustering. In this figure the gene expression values for the 1421 most differentially expressed genes from all 548 samples are clustered along the y-axis, and individuals are coded by color. About 542 (98.9%) of the 548 samples cluster with at least one other sample from the same individual, and for 35 (87.5%) out of 40 individuals, all samples from the particular individual cluster together. This highlights the extremely large individual specific effect seen in these data.
The 11 003 genes identified over both arms and time periods of the study population with a significant time and individual component were searched for enrichment of genes belonging to the Gene Ontology (GO) Biological Process categories (Supplementary Material, Table S3). The induction of apoptosis is the most significant enriched biological process (enrichment P-value = 4.7 × 10−9), which is well-known to be under circadian control (28). The cell activation, macromolecule localization and hemopoiesis biological process categories were also enriched, which are all consistent with previous findings (29–31). A number of individual genes were noteworthy within this set as well. For example, the nuclear receptor RXRA is involved in a number of biological processes including induction of apoptosis, cell activation and hemopoiesis, but then is also involved in circadian rhythm, where it is known to interact with the circadian CLOCK gene (32). CREB1, a transcription factor involved in anti-apoptosis, hemopoiesis and immune response, is also under circadian control (33). Despite the presence of these genes under circadian control, the biological process circadian rhythm was not enriched in this set. However, of the 12 core circadian rhythm genes annotated in the KEGG database (34) and represented on the microarray used in this study, eight overlapped with our set of 11 003 genes (P ∼ 0.019 as determined by the Fisher Exact Test). CLOCK, a well-known diurnal gene, was not found to vary by time in our dataset, although perhaps this should not be surprising given it has not been found to be diurnally regulated in many other studies of human circadian rhythm (35,36). We may not have been powered to see changes in CLOCK, or because expression was only measured during the daytime rather than over a 24 h period the diurnal variation for this gene may not have been represented in our study. Further, it is possible that CLOCK is regulated by time only in certain tissues like brain. The set of 11 003 genes with a time-dependent signature represents one of the largest set of diurnally regulated genes identified to date in blood or any other human tissue.
Given the extensive clinical measures scored in the present study, we explored associations between each of the clinical measures and the gene expression data. Table 1 summarizes the extent of correlation between gene expression and clinical trait values over all time points and states. Gene expression was measured at all seven time points and in both the fasted and fed states, although the clinical parameters were measured only at time point 1 in each arm of the study. Interestingly, clinical traits related to blood pressure showed the greatest number of correlated genes in the blood (Table 1). The genes that correlate with diastolic blood pressure are enriched for GO biological processes DNA replication and the response to DNA damage (P-values = 0.0004 and 0.004, respectively). The genes correlated with systolic blood pressure are enriched for GO biological processes rRNA metabolic processes and the response to reactive oxygen species (P-values = 0.002 and 0.003, respectively). Even though these P-values are not significant after the multiple testing correction, these results are consistent with previous reports showing that blood pressure is related to oxidative events and DNA damage (37). In addition, many of the clinical traits related to obesity, including hip and waist circumferences, were correlated with many genes in this dataset, even though these individuals were generally healthy (no self-reported disease and median BMI = 24.5). A number of expression traits were significantly correlated with plasma concentration of glucose and triglycerides as well as alanine aminotransferase (ALT), which can be an indicator of muscle injury, liver damage or liver disease. Therefore, these data suggest that blood gene expression traits may be related to many clinical measures that are routinely recorded and analyzed, providing a potentially rich source of molecular phenotypes to explore in the context of disease.
Table 1.
The number of genes significantly correlated with each of the clinical traits (in any state, at any time point) at the P-value < 0.0001 significance level
| Clinical trait | # Correlated genes | FDR |
|---|---|---|
| Diastolic blood pressure | 3329 | 0.0138008 |
| Systolic blood pressure | 896 | 0.0499203 |
| Max hip circumference | 495 | 0.0886869 |
| Max waist circumference | 383 | 0.113092 |
| Alanine aminotransferase (ALT) | 191 | 0.234705 |
| Glucose | 160 | 0.271875 |
| Triglercerides | 132 | 0.335065 |
Expression traits associated with response to feeding
To explore the expression response to feeding as compared with fasting in a more rigorous way, we employed a mixed effects model to examine fasting and fed specific gene expression patterns over time, essentially allowing for each individual's expression behavior over time to differ from the others (see Method section for detail). Genes responding to feeding are listed in Supplementary Material, Table S2.
To compare this model to the standard ANOVA model, we constructed a Receiver Operating Characteristics curve to display the relative sensitivity and specificity of the two models, as shown in Figure 3. It is clear from the figure that the mixed effects model performs better in detecting expression changes in response to feeding than the standard ANOVA model. For example, the mixed effects model detected 10 418 significant genes at the P-value < 0.01 significance level (FDR = 0.86%, Supplementary Material, Table S2) although the standard ANOVA model detected 5182 genes at the same P-value threshold, albeit with a higher FDR (FDR = 1.9%, Supplementary Material, Table S2).
Figure 3.

Receiver operating characteristics curve showing the sensitivity and specificity of the mixed-effect model as compared with standard ANOVA models. As shown, the mixed-effect model is more sensitive in detecting true fast/fed specific genes and also results in a lower false positive rate as compared with the standard ANOVA models.
We then explored whether the 10 418 genes were enriched for genes that were correlated with obesity in either the fasted or fed states. Here, we chose to focus on time point 3, just after the meal, and used the same trait-gene correlation significance threshold of P < 0.0001 as above and an enrichment P-value threshold of <0.05 after multiple testing correction. It is interesting that in the fed state these genes were enriched for genes correlated with glucose (fasted state P = 1, fed state P = 0.019), maximum hip circumference (fasted state P = 0.53, fed state P = 0.021) and Diastolic blood pressure (fasted state P = 0.94, fed state P = 2.22 × 10−6), although in the fasted state, these genes were not enriched for correlation to any of the measured clinical traits. It is of particular note that glucose, which was measured in the fasted state, was more correlated to gene expression variation in the fed state, whereas biometric traits (such as maximum hip circumference) were more correlated with gene expression traits in response to feeding. This pattern demonstrates that at a genome-wide level, genes are more correlated with different clinical traits in different states, suggesting that the state in which blood is profiled should be considered in the context of the specific phenotypes (e.g. obesity) under investigation.
With such a striking gene expression signature associated with fasting/feeding status, we hypothesized that a robust classifier to distinguish the fasting state from the feeding states could be derived from the gene expression data. To test this, we constructed a classifier using the Elastic Net method to classify individuals as fasted or fed based on the gene expression traits (38). For classification purposes, individuals in the fed arm at time points one and two were assigned to the fasted group, since they were in the fasted state at these time points. Applying the variable selection procedure of the Elastic Net algorithm, we identified 176 genes (Supplementary Material, Table S4) to use as input into the Elastic Net classification procedure. Using 10-fold cross validation (see Methods) the mean classification accuracy rates in the training and testing sets were 99.3 and 94.0%, respectively. These classification rates far exceeded the classification accuracy of 54.6% achieved in the permuted data sets, which is close to the 50% classification accuracy we would expect by chance.
To prospectively test the predictive power of our classifier, we applied it to an independent human cohort comprised of 1002 Icelandic individuals, where peripheral blood was collected in the fasted state on each individual at a single time point (15). Each of the 1002 samples was hybridized against the same reference pool used in this study. The classification accuracy of the classifier in this independent dataset was 82.3% (825/1002), still significantly above the 50% we would have expected by chance. These results suggest that the gene expression response to fasting and fed status is robust enough to classify and predict accurately in independent datasets. Although the reduced accuracy in the independent test set is to be expected given those expression profiles making up the independent dataset were derived from a population with different age, sex and metabolic trait characteristics, we cannot exclude the possibility that some fraction of the 1002 individuals in the independent dataset did not adhere to the overnight fasting requirement before their blood draw.
To investigate the biological significance of the 176 genes found to classify fast/fed status in the full dataset, these genes were rank ordered according to their prediction ability and then characterized in the previously described cohort, consisting of 1002 Icelandic individuals from 209 extended pedigrees (15). Of the 176 genes, 78.4% (138) were detected as heritable at a FDR of 0.01. This is a significantly higher percentage than the 33.9% of genes overall that were found to be heritable after adjusting for age, gender and blood cell counts (the enrichment P-value = 1.35 × 10−11). Further, of the 176 genes, 44 (25%) had a cis-eQTL with FDR < 0.01, again significantly greater than the 6.6% of all genes for which cis-eQTL were detected. In addition, we tested these genes for cis-associations in the founders of the Icelandic cohorts as previously described (15) and found that 29.5% (52) of the 176 genes had a significant cis-association with FDR < 0.01, compared with 8.5% over all genes on the chip for which cis-associations had been detected. These findings suggest that the response to feeding has a significant genetic component.
We also sought to explore whether there was increased biological coherence and increased association to obesity traits represented in the set of 176 genes used to construct the classifier. We first tested for enrichment of GO Biological Process categories in this gene set. All enrichment P-values reported are corrected for multiple testing. Table 2 lists a number of enriched biological process categories, including response to wounding (P-value = 6.22 × 10−6) and inflammatory response (P-value = 6.6 × 10−5). However, these enriched biological categories should not come as a surprise given the tissue under study. Next we looked at the pair-wise correlations between the 176 gene expression traits and the clinical traits in both the fasted and fed states. The primary aim here was to determine whether these genes were enriched for correlating with clinical traits/biomarkers associated with obesity. To measure this, we again restricted attention to the expression traits generated at time point 3. Using an enrichment P-value threshold of 0.05, we found that the set of 176 genes was significantly enrichment for genes that were correlated with diastolic blood pressure in both the fasted and fed states (fasted P ∼ 0.0017, fed P ∼ 0.024), and with plasma insulin levels in the fasted state only (fasted P ∼ 0.015, fed P ∼ 1), indicating that gene expression changes under the different nutritional states are tightly connected to obesity associated traits.
Table 2.
Functional category enrichment of 176 fasting/feeding classification genes
| GO biological process | Size of GO term | Overlap | P-value |
|---|---|---|---|
| Response to wounding | 1038 | 31 | 6.22E−06 |
| Inflammatory response | 721 | 24 | 6.60E−05 |
| Defense response | 1136 | 30 | 0.000189 |
| G-protein coupled receptor protein signaling pathway | 1054 | 27 | 0.001684 |
| Regulation of immune system process | 453 | 16 | 0.007481 |
| Protein kinase cascade | 1225 | 28 | 0.009481 |
| Regulation of signal transduction | 1104 | 25 | 0.039085 |
The classification genes were tested for enrichment to the various Gene Ontology Biological Process functional categories. Statistical significance was assessed using the hypergeometric function and was corrected for multiple testing.
Time and fasting/feeding dependent changes
Characterizing expression changes between fasting and feeding states is one way to capture a portion of the transcription response associated with feeding. However, there are also genes whose expression between the fasting and feeding states may not change significantly, but their connectivity (the number of genes to which a given gene is connected in the transcriptional network) may change. Most microarray studies focus on differential expression (DE), with very few of focusing on differential connectivity (DC) (39). Here, we compare genes with significant expression changes to genes with significant connectivity changes between the fasting and fed states, given these changes in connectivity may provide additional insights into the pathways and biochemical networks relating to food intake, metabolism, and ultimately, metabolic diseases such as obesity and diabetes. We defined DE between the fasting and fed states for each time point using paired t-test with P-value cutoff of 0.01. We defined a DC measure as the log ratio of each gene's connectivity in the fasting and fed states. We assumed under the null hypothesis that this log-ratio measure was normally distributed, and then called a gene differentially connected if the P-value of the change was less than 0.01 (see Method section for details). The numbers of differentially expressed and differentially connected genes at each time point are shown in Figure 4. The overlap between differentially expressed and differentially connected genes is surprisingly small. It is interesting to note that the time points with high numbers of differentially connected genes correspond to the time points with a large number changes of differentially expressed genes (e.g. see time points 3 and 5). At time points 4 and 6, the numbers of differentially expressed genes were similar to the previous time point, and the differentially expressed genes significantly overlapped with those from the previous time points (Fisher Exact Test P-values=1.33 × 10−128 and <1 × 10−326, respectively, for the significance of the overlap), which suggests that time points 4 and 6 are in semi-static states. The numbers of DC genes at time points 4 and 6 are small. These results suggest that differentially connected genes reflect the dynamic process of changes in the biological system, whereas differentially expressed genes reflect the end result of biological system changes. When differentially expressed and differentially connected genes are compared with GO biological processes, DE genes at time point 3 were enriched for innate immune response (P-value = 2.37 × 10−5), for negative regulation of transcription (P-value = 0.047) at time point 4, for induction of apoptosis and negative regulation of RNA metabolic process (P-values = 0.011 and 0.0049, respectively) at time point 5, and for induction of apoptosis and response to biotic stimulus (P-values = 1.7 × 10−5 and 8.33 × 10−6, respectively) at time point 7. DC genes at time point 3 were enriched for genes in tissue development (P-value = 0.0044). When these genes were compared with genes correlated with clinical traits in the Icelandic Family Blood (IFB) study (15), DE and DC genes were enriched for different sets of clinical traits as shown in Supplementary Material, Table S5. DE genes at different time points were consistently enriched for genes correlated with BMI, weight, plasma insulin levels and plasma leptin levels; whereas DC genes at time point 3 were significantly enriched for genes correlated with body fat mass, hip size and plasma glucose levels (P-values = 4.2 × 10−83, 1.1 × 10−70 and 4.4 × 10−16, respectively).
Figure 4.

Numbers of differentially expressed (DE) genes and differential connectivity (DC) genes across time. At time point 3 right after the meal, there are large numbers of DE and DC genes. Large numbers of DC genes at time points 3 and 5 corresponds to large number changes of DE genes. This indicates that DE and DC genes reflect different types of biological system change.
Gene-gene coexpression networks in the fasted and fed states
The connectivity structure between gene expression traits can be characterized using coexpression networks as we have previously described (24). Coexpression networks provide a genome-wide view of the connectivity structure among genes. These networks can be viewed as topological overlap maps, in which the modular structure of the networks becomes readily apparent (40). We systematically compared modules detected in the coexpression networks in fasting and fed states with respect to differentially expressed or connected genes.
For this analysis we again focused on time point 3, the first time point after the meal and the time point with the largest number of differentially connected genes. We selected for network analysis the 5000 most connected genes in each of the fasted and fed states. Of the top 5000 genes identified for each state, 1938 genes were in the overlap, resulting in a total of 8062 genes used for the network analysis. Of these 8062 genes, 48% (1640 out of 3408) were in the differentially connected gene set and 36% (713/1991) were in the differentially expressed gene set at time point 3. Figure 5 shows the connectivity map for time point 3 in both the fasted and fed states. For both Figures 5A and B, all 8062 genes are shown across the X- and Y-axes. In Figure 5A, the fasted connectivity map is displayed in the upper right half of the figure, while the fed connectivity map is shown in the lower left half of the figure. These data are ordered according to the correlation structure in the fasted dataset. In contrast, Figure 5B shows the fed data in the upper right half and the fasted data in the lower left half of the figure, and is ordered according to the fed correlation structure. We next used a previously described method (41) to identify modules of the most highly connected genes in the network. These modules were then tested for GO biological process enrichments (all reported P-values are corrected for multiple testing). Similar sets of biological processes were found to be enriched in both the fasted and fed modules (Supplementary Material, Tables S6 and S7). For example, the GO biological process RNA process is enriched in fasted modules 3 and 9 (values for enrichment are 2.81 × 10−5 and 0.004, respectively), but then is also enriched in fed module 1 (corrected P-value 3.92 × 10−5); the GO biological process ribonucleoprotein complex biogenesis and assembly is enriched in fasted module 3 (P-value = 0.014) and fed module 1 (P-value = 0.0013); the GO biological process ubiquitin cycle is enriched in fasted module 1 (P-value = 0.00097) and fed module 8 (P-value = 0.042). RNA process, especially RNA splicing and RNA localization, has previously been identified as a key process responding to fast and feeding (42). Ribosome biogenesis and ubiquitin cycle processes are also known to be regulated by fasting and feeding (43,44).
Figure 5.

Gene-gene coexpression networks of the fasted and fed states at time point 3. Shown here are two heatmaps containing the connectivity map of 8062 genes (union of 5000 most connected genes in each state at time point 3). (A) Shows the fasted state in the upper right half and the fed state in the lower left half. Genes are ordered according to the clustering in the fasted state. (B) Shows the fed state in the upper right half and the fasted state in the lower left half, and genes are ordered according to the fed data.
The network modules were then compared with genes correlated with the clinical traits (Supplementary Material, Tables S8 and S9). Fasted modules 2, 8, 9 and 10 were enriched for genes correlated with Diastolic Blood Pressure; fed modules 11, 13 and 15 were enriched for genes correlated with Diastolic Blood Pressure; and fed modules 9, 11 and 15 were also enriched for gene correlated with Systolic Blood Pressure.
Even though the fed modules were more densely connected, many fasted modules overlapped significantly with the fed modules, as shown in Supplementary Material, Figure S1. Fed module 3 is densely connected and does not significantly overlap any of the fasted network modules. This particular network module is enriched for cell–cell signaling, neurological system process and tissue development, with P-values of 0.0001, 0.017 and 0.032, respectively. As expected (because this fed network module does not overlap any of the fasted network modules), this module significantly overlaps with the set of differentially connected genes (P-value = 9.29 × 10−289), but then is under-represented in the set of differentially expressed genes (only 12 DE genes in this module, whereas 70 were expected by chance). This highlights that differences in coexpression networks provided information about a system that cannot be detected by focusing on classical differential gene expression analysis only. Genes in this module were not enriched for genes correlated with any of the clinical traits measured in this cohort. However, when the genes in this module are compared with genes correlated with clinical traits in the population-based IFB cohort (15), this module is significantly enriched for genes correlated with blood glucose concentration (P-value = 3.9 × 10−21) and hip circumference (P-value = 3.26 × 10−12). These results suggest that the dynamic transcriptional responses to feeding are associated with clinical traits, including anthropometrics and plasma status that are related to diabetes and obesity.
DISCUSSION
We employed a two-period, two-arm randomized crossover design to address the effects of feeding on the peripheral blood transcriptional network and its connection to clinical traits associated with metabolic disorders like obesity. This study design has been used extensively in clinical trials to answer specific hypotheses related to drug treatment effects. By considering feeding as a treatment we were able to address changes in gene expression levels in response to this treatment, whereas controlling for individual and temporal specific responses, as well as other confounding factors such as age and white blood cell counts. Because each individual participated in both arms of the study, we were able to measure feeding response at each time point by comparing to the matched fasting sample taken from the same individual at the same time of day. In addition, because we collected samples across seven time points in each of the two arms of the study, we were able to study temporal variation and to separate a circadian rhythm gene signature from the individual specific and feeding response signatures. In addition, we were able to study variation in the feeding response over time, where we detected a clear change in this response that was also individual specific. At systems level, we detected coexpression network structure changes over time and between the fasted and fed states, providing insights into the pathways and mechanisms involved in the response to food intake.
Because all individuals in this study were apparently healthy, non-smoking, same gender and within a relatively narrow age range, we were able to reduce biases due to these confounding factors as well as environmental specific effects due to geographic location, all of which have been shown to have a large impact on gene expression variation. Therefore, this study group is relatively homogeneous and well powered to detect the presented changes in gene expression. However, this study group may not represent well the general population, thus additional experiments would be warranted to validate whether the derived time or food response signatures still hold in a more heterogeneous population sampling.
We have identified large numbers of genes responding to time or food intake. Some of these signatures were transcripts expressed at low levels in blood. In general, when the expression level of a gene is low, the resulting experimental variation is high, requiring more samples in order to detect differential responses for such genes. This study consisted of over 500 blood samples and was therefore well powered to detect differential responses for genes expressed at low levels. Naturally, this also implies that with same number of samples, differential responses for genes expressed at high levels and with small experimental variation are most readily detected (see Supplementary Material, Fig. S2).
The most notable signature identified in this study was the individual-specific gene expression signature. Others have demonstrated that it is possible to identify a set of reporter genes that account for individual specific gene expression variation, where this set of reporters serves as an individual finger print to uniquely identify individuals (2). This has been largely confirmed in our fasting/feeding data set, given the transcriptionally most active set of genes on the array almost perfectly clustered the samples by individual in a completely unsupervised fashion. Of the 72 individual specific genes identified by Radich et al. (2), 43 were represented on the microarray used in this study and all 43 of these were detected with significant individual-specific effects in the analysis described herein. Despite this strong individual specific signature, the experimental design employed in this study specifically addressed and controlled for this type of variation. In this way, we were able to extract other smaller but very real gene expression signatures from this dataset associating with traits of interest.
We have shown that there is a detectable gene expression response to feeding that occurs in blood derived samples and that this response is predictive with 80–90% classification accuracy. Furthermore, the genes involved in the feeding response are significantly enriched for categories related to metabolism, cell communication, and immune response, and were found to be more heritable than genes overall in a separate dataset. We have also explored the relationship between differential response to feeding and DC for characterizing transcriptional response. Our systems-level approach clearly demonstrates that the connections between genes, and the changes in these connections associated with the physiological state, may be a more informative way to measure gene expression response to disease and/or environmental stimulus. Finally, we have shown that the genes responding to the fasted or fed states are differently correlated to various metabolic traits, suggesting that the state in which blood is profiled should be seriously considered as regards the phenotype or condition under investigation. The results of this study have broader implications including studies of drug response in clinical trials. Our results suggest that that the response to feeding is an important variable that may bring us closer to dissecting the underlying causes of obesity.
MATERIALS AND METHODS
Gene expression data
Erythrocytes were lysed and all cell debris, including globins, were removed prior to RNA extraction as previously described (4). RNA from blood was processed and gene expression data was generated as previously described (45). Samples were hybridized against a reference pool containing 85 Icelandic individuals, 10 of whom also participated in the current study, to an array containing 23 653 unique substances. Background corrected intensity values for each probe in each sample were calculated, and then expression ratio values, defined as the average log [intensity of sample/intensity of reference pool], were then corrected for age, white blood cell counts, lymphocytes, and neutrophils by linear regression using the following model:
Two-dimensional, agglomerative clustering of the blood gene expression data
The extent of individual specific effects in the blood gene expression data was explored by clustering individuals over a set of differentially expressed genes (at least 1.5-fold difference in expression comparing to the reference pool and P-value <0.05 for at least 10% of the samples). 5829 genes were differentially expressed. 1421 top variated genes of 5892 was then clustered using a previously described two-dimensional, agglomerative hierarchical clustering algorithm in the experiment (individuals) and gene dimensions (Fig. 2).
Mixed-effect model to measure response to fasting and feeding
In general, time series data exhibit a strong autocorrelation between successive time points. The behavior over time can vary between individuals (referred as random effect). ANOVA model treats data from different time points independently without considering autocorrelation information. To capture both dependency across time points within individual and independency between individual, we deployed a mixed-effect model (46). For expression trait values Yijk at the time point tk for the jth person within the i period, the linear nested mixed effects model of interest is:
where β0, β1, β2, and β3 are the fixed effects for intercept, fasting/feeding status (indicator random variable taking the value 1 for feeding state and 0 for fasting state), time (numeric value) and fasting/feeding-by-time interaction, respectively. bi,j= (b0i,j, b1i,j) is the individual within period random effects vector, and εijk is the residual error. Here, we assume normal distributions εijk ∼ N (0, σ2), bi,j ∼ N (0, Σ2), where independence is assumed among the bi,j (for different i and j). The P-value of coefficient β1 indicates whether the gene expression trait responds to fasting and feeding.
Classification of individuals based on fast/fed status
To discriminate between the fasted and fed states, we constructed a classifier using the gene expression data and the Elastic Net method (38). Elastic Net has been shown to perform well in evaluating microarray expression data (38). The Elastic Net algorithm gave the best classification results compared with other procedures we tested (data not shown) and also was advantageous in that this method selected the predictor variables. The age and cell count adjusted mean log-ratio measure was used to construct the Elastic Net classifier, with fasting/feeding status treated as the dependent variable. To reduce the number of genes entered into this procedure, a filtering criterion was introduced requiring significant DE between the fasted and fed groups. To determine the classification accuracy rate we used a 10-fold cross validation strategy in which the samples were randomly divided into training groups consisting of 90% of the samples and testing groups consisting of the remaining 10% of the samples. For each of the cross validation procedure, the filtering step just described was first applied within the training data to identify genes of interest, and then this subset of genes was used to construct the classifier. We performed the 10-fold cross validation 100 times. The final classification accuracy is the average of the 100 test results. The significance of the classifier was assessed through permutation testing by permuting group assignments, and then assessing the prediction accuracy using the 10-fold cross validation strategy on the permuted data.
Functional category enrichment
All tests of functional category enrichment for particular gene sets were performed using the Gene Ontology Biological Process category. Enrichment P-values were corrected for multiple testing using the Bonferroni correction.
Differential connectivity
Two genes are defined as connected or correlated if the P-value of the expression correlation is less than 0.0001. The connectivity of a gene is defined as how many genes it connects to in a system. The difference of connectivities of fast and fed states is measured by the log ratio of their connectivities, 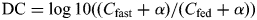 . All connectivities are offset by α to avoid large ratio changes due to small connectivities as denominators. By chance, positive and negative DC are equal likely and the distribution of DC is symmetric. When system changes are expected, there will be systematic gain or loss connectivity and the distribution of DC will be asymmetric. To determine whether a DC is significant or not, we fit the smaller half of DC distribution as a half-normal distribution, and identify a cutoff value corresponding to P-value 0.01. A DC is significant if its absolute value is greater than the cutoff.
. All connectivities are offset by α to avoid large ratio changes due to small connectivities as denominators. By chance, positive and negative DC are equal likely and the distribution of DC is symmetric. When system changes are expected, there will be systematic gain or loss connectivity and the distribution of DC will be asymmetric. To determine whether a DC is significant or not, we fit the smaller half of DC distribution as a half-normal distribution, and identify a cutoff value corresponding to P-value 0.01. A DC is significant if its absolute value is greater than the cutoff.
Gene-gene coexpression network
The correlation between genes was measured using the Pearson correlation statistic. The P-value threshold for significance was determined similar as described by Chen et al. (24), to minimize the FDR over all pair-wise gene–gene correlations. Topological overlap maps showing the connectivity between genes were calculated and plotted using the methods described by Barabasi and Oltvai (47).
SUPPLEMENTARY MATERIAL
FUNDING
Funding to pay the Open Access publication charges for this article was provided by Merck & Co., Inc.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank the 40 individuals who participated in this study, and the staff of the Clinical Research Center, deCODE Genetics, Inc., Reykjavik, Iceland, for their cooperation and assistance.
Conflict of Interest statement. A.L., J.Z., Y.C., K.W., J.R.L., M.R., V.E. and E.E.S. work for Merck & Co., Inc. and own Merck stocks.
REFERENCES
- 1.Boivin D.B., James F.O., Wu A., Cho-Park P.F., Xiong H., Sun Z.S. Circadian clock genes oscillate in human peripheral blood mononuclear cells. Blood. 2003;102:4143–4145. doi: 10.1182/blood-2003-03-0779. [DOI] [PubMed] [Google Scholar]
- 2.Radich J.P., Mao M., Stepaniants S., Biery M., Castle J., Ward T., Schimmack G., Kobayashi S., Carleton M., Lampe J., et al. Individual-specific variation of gene expression in peripheral blood leukocytes. Genomics. 2004;83:980–988. doi: 10.1016/j.ygeno.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 3.Whitney A.R., Diehn M., Popper S.J., Alizadeh A.A., Boldrick J.C., Relman D.A., Brown P.O. Individuality and variation in gene expression patterns in human blood. Proc. Natl. Acad. Sci. USA. 2003;100:1896–1901. doi: 10.1073/pnas.252784499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lampe J.W., Stepaniants S.B., Mao M., Radich J.P., Dai H., Linsley P.S., Friend S.H., Potter J.D. Signatures of environmental exposures using peripheral leukocyte gene expression: tobacco smoke. Cancer Epidemiol. Biomarkers Prev. 2004;13:445–453. [PubMed] [Google Scholar]
- 5.Ryder M.I., Hyun W., Loomer P., Haqq C. Alteration of gene expression profiles of peripheral mononuclear blood cells by tobacco smoke: implications for periodontal diseases. Oral Microbiol. Immunol. 2004;19:39–49. doi: 10.1046/j.0902-0055.2003.00110.x. [DOI] [PubMed] [Google Scholar]
- 6.Zieker D., Fehrenbach E., Dietzsch J., Fliegner J., Weidmann M., Nieselt K., Gebicke-Haerter P., Spanagel R., Simon P., Niess A.M., et al. cDNA-microarray analysis reveals novel candidate genes expressed in human peripheral blood following exhaustive exercise. Physiol. Genomics. 2005;23:287–294. doi: 10.1152/physiolgenomics.00096.2005. [DOI] [PubMed] [Google Scholar]
- 7.Borovecki F., Lovrecic L., Zhou J., Jeong H., Then F., Rosas H.D., Hersch S.M., Hogarth P., Bouzou B., Jensen R.V., et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc. Natl. Acad. Sci. USA. 2005;102:11023–11028. doi: 10.1073/pnas.0504921102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bull T.M., Coldren C.D., Moore M., Sotto-Santiago S.M., Pham D.V., Nana-Sinkam S.P., Voelkel N.F., Geraci M.W. Gene microarray analysis of peripheral blood cells in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2004;170:911–919. doi: 10.1164/rccm.200312-1686OC. [DOI] [PubMed] [Google Scholar]
- 9.Rus V., Chen H., Zernetkina V., Magder L.S., Mathai S., Hochberg M.C., Via C.S. Gene expression profiling in peripheral blood mononuclear cells from lupus patients with active and inactive disease. Clin. Immunol. 2004;112:231–234. doi: 10.1016/j.clim.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka T., Takada H., Nomura A., Ohga S., Shibata R., Hara T. Distinct gene expression patterns of peripheral blood cells in hyper-IgE syndrome. Clin. Exp. Immunol. 2005;140:524–531. doi: 10.1111/j.1365-2249.2005.02805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Y.S., Fan R.Q., Chen D.C., Xuan G.W. Gene expression profiling of peripheral leukocytes from patients with systemic lupus erythematosus using oligonucleotide DNA microarray. Di Yi Jun Yi Da Xue Xue Bao. 2005;25:929–934. [PubMed] [Google Scholar]
- 12.Yu S.Y., Hu Y.W., Liu X.Y., Xiong W., Zhou Z.T., Yuan Z.H. Gene expression profiles in peripheral blood mononuclear cells of SARS patients. World J. Gastroenterol. 2005;11:5037–5043. doi: 10.3748/wjg.v11.i32.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi S., Park H.S., Cheon M.S., Lee K. Expression profile analysis of human peripheral blood mononuclear cells in response to aspirin. Arch. Immunol. Ther. Exp. (Warsz) 2005;53:151–158. [PubMed] [Google Scholar]
- 14.Glatt S.J., Everall I.P., Kremen W.S., Corbeil J., Sasik R., Khanlou N., Han M., Liew C.C., Tsuang M.T. Comparative gene expression analysis of blood and brain provides concurrent validation of SELENBP1 up-regulation in schizophrenia. Proc. Natl. Acad. Sci. USA. 2005;102:15533–15538. doi: 10.1073/pnas.0507666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emilsson V., Thorleifsson G., Zhang B., Leonardson A.S., Zink F., Zhu J., Carlson S., Helgason A., Walters G.B., Gunnarsdottir S., et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 16.Schadt E.E., Sachs A., Friend S. Embracing complexity, inching closer to reality. Sci. STKE. 2005;2005:pe40. doi: 10.1126/stke.2952005pe40. [DOI] [PubMed] [Google Scholar]
- 17.Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 18.Han J.D., Bertin N., Hao T., Goldberg D.S., Berriz G.F., Zhang L.V., Dupuy D., Walhout A.J., Cusick M.E., Roth F.P., et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 19.Lee I., Date S.V., Adai A.T., Marcotte E.M. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- 20.Pinto S., Roseberry A.G., Liu H., Diano S., Shanabrough M., Cai X., Friedman J.M., Horvath T.L. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 21.Wolfrum C., Asilmaz E., Luca E., Friedman J.M., Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004;432:1027–1032. doi: 10.1038/nature03047. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi H., Oishi K., Hanai S., Ishida N. Effect of feeding on peripheral circadian rhythms and behaviour in mammals. Genes Cells. 2004;9:857–864. doi: 10.1111/j.1365-2443.2004.00769.x. [DOI] [PubMed] [Google Scholar]
- 23.Sakamoto K., Kadota K., Oishi K. Light-induced phase-shifting of the peripheral circadian oscillator in the hearts of food-deprived mice. Exp. Anim. 2004;53:471–474. doi: 10.1538/expanim.53.471. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y., Zhu J., Lum P.Y., Yang X., Pinto S., MacNeil D.J., Zhang C., Lamb J., Edwards S., Sieberts S.K., et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heilbronn L.K., Civitarese A.E., Bogacka I., Smith S.R., Hulver M., Ravussin E. Glucose tolerance and skeletal muscle gene expression in response to alternate day fasting. Obes. Res. 2005;13:574–581. doi: 10.1038/oby.2005.61. [DOI] [PubMed] [Google Scholar]
- 26.Grant G.R., Liu J., Stoeckert C.J., Jr A practical false discovery rate approach to identifying patterns of differential expression in microarray data. Bioinformatics. 2005;21:2684–2690. doi: 10.1093/bioinformatics/bti407. [DOI] [PubMed] [Google Scholar]
- 27.Storey J.D., Madeoy J., Strout J.L., Wurfel M., Ronald J., Akey J.M. Gene-expression variation within and among human populations. Am. J. Hum. Genet. 2007;80:502–509. doi: 10.1086/512017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu L., Pelicano H., Liu J., Huang P., Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- 29.Bourin P., Ledain A.F., Beau J., Mille D., Levi F. In-vitro circadian rhythm of murine bone marrow progenitor production. Chronobiol. Int. 2002;19:57–67. doi: 10.1081/cbi-120002677. [DOI] [PubMed] [Google Scholar]
- 30.Walters J., Skene D., Hampton S.M., Ferns G.A. Biological rhythms, endothelial health and cardiovascular disease. Med. Sci. Monit. 2003;9:RA1–8. [PubMed] [Google Scholar]
- 31.Yang S., Wang K., Valladares O., Hannenhalli S., Bucan M. Genome-wide expression profiling and bioinformatics analysis of diurnally regulated genes in the mouse prefrontal cortex. Genome Biol. 2007;8:R247. doi: 10.1186/gb-2007-8-11-r247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNamara P., Seo S.P., Rudic R.D., Sehgal A., Chakravarti D., FitzGerald G.A. Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature: a humoral mechanism to reset a peripheral clock. Cell. 2001;105:877–889. doi: 10.1016/s0092-8674(01)00401-9. [DOI] [PubMed] [Google Scholar]
- 33.Scheving L.A., Gardner W. Circadian regulation of CREB transcription factor in mouse esophagus. Am. J. Physiol. 1998;274:C1011–C1016. doi: 10.1152/ajpcell.1998.274.4.C1011. [DOI] [PubMed] [Google Scholar]
- 34.Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bjarnason G.A., Jordan R.C., Wood P.A., Li Q., Lincoln D.W., Sothern R.B., Hrushesky W.J., Ben-David Y. Circadian expression of clock genes in human oral mucosa and skin: association with specific cell-cycle phases. Am. J. Pathol. 2001;158:1793–1801. doi: 10.1016/S0002-9440(10)64135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takata M., Burioka N., Ohdo S., Takane H., Terazono H., Miyata M., Sako T., Suyama H., Fukuoka Y., Tomita K., et al. Daily expression of mRNAs for the mammalian Clock genes Per2 and clock in mouse suprachiasmatic nuclei and liver and human peripheral blood mononuclear cells. Jpn. J. Pharmacol. 2002;90:263–269. doi: 10.1254/jjp.90.263. [DOI] [PubMed] [Google Scholar]
- 37.Alexander R.W. Theodore Cooper Memorial Lecture. Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension. 1995;25:155–161. doi: 10.1161/01.hyp.25.2.155. [DOI] [PubMed] [Google Scholar]
- 38.Zou H., Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B. 2005;67:301–320. [Google Scholar]
- 39.Reverter A., Ingham A., Lehnert S.A., Tan S.H., Wang Y., Ratnakumar A., Dalrymple B.P. Simultaneous identification of differential gene expression and connectivity in inflammation, adipogenesis and cancer. Bioinformatics. 2006;22:2396–2404. doi: 10.1093/bioinformatics/btl392. [DOI] [PubMed] [Google Scholar]
- 40.Ravasz E., Somera A.L., Mongru D.A., Oltvai Z.N., Barabasi A.L. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 41.Lum P.Y., Chen Y., Zhu J., Lamb J., Melmed S., Wang S., Drake T.A., Lusis A.J., Schadt E.E. Elucidating the murine brain transcriptional network in a segregating mouse population to identify core functional modules for obesity and diabetes. J. Neurochem. 2006;97(Suppl. 1):50–62. doi: 10.1111/j.1471-4159.2006.03661.x. [DOI] [PubMed] [Google Scholar]
- 42.Amir-Ahmady B., Salati L.M. Regulation of the processing of glucose-6-phosphate dehydrogenase mRNA by nutritional status. J. Biol. Chem. 2001;276:10514–10523. doi: 10.1074/jbc.M010535200. [DOI] [PubMed] [Google Scholar]
- 43.Conde R.D., Franze-Fernandez M.T. Increased transcription and decreased degradation control and recovery of liver ribosomes after a period of protein starvation. Biochem. J. 1980;192:935–940. doi: 10.1042/bj1920935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshizawa F., Nagasawa T., Nishizawa N., Funabiki R. Protein synthesis and degradation change rapidly in response to food intake in muscle of food-deprived mice. J. Nutr. 1997;127:1156–1159. doi: 10.1093/jn/127.6.1156. [DOI] [PubMed] [Google Scholar]
- 45.Shoemaker D.D., Schadt E.E., Armour C.D., He Y.D., Garrett-Engele P., McDonagh P.D., Loerch P.M., Leonardson A., Lum P.Y., Cavet G., et al. Experimental annotation of the human genome using microarray technology. Nature. 2001;409:922–927. doi: 10.1038/35057141. [DOI] [PubMed] [Google Scholar]
- 46.Laird N.M., Ware J.H. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 47.Barabasi A.L., Oltvai Z.N. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.