

Abstract

A mild and general route for preparing dieneamides is described. Ni imidazolylidene complexes were used to mediate cycloadditive couplings between enynes and isocyanates. Dienamides were prepared in excellent yields and with good E:Z selectivity. These dieneamides can be further manipulated through oxidative cyclization methods. When a terminal enyne is employed, cyclization affords a lactam rather than a dienamide.
A primary interest of our group is the development of efficient cycloadditions that afford heterocycles and carbocycles.1–7 We have found that Ni/NHC complexes effectively catalyze the cycloaddition of diynes with CO2,2 isocyanates,3 carbonyls,4 and nitriles.5 These reactions afford pyrones, pyridones, pyrimidinones, pyrans, and pyridines in high yields. In addition, the same Ni/NHC system also mediates the rearrangement of vinyl cyclopropanes6 and cyclopropylen-ynes.7
The efficacy of our Ni/NHC catalyzed cycloaddition reactions that couple diynes/isocyanates3 and enynes/carbonyls4 prompted us to investigate the Ni/NHC catalyzed cycloaddition of enynes and isocyanates. To date, only one catalytic system, which utilizes Rh catalysts, effectively cyclizes an olefin, an alkyne, and an isocyanate. This Rh catalyst was used to couple an alkenyl-isocyanate with an alkyne in the synthesis of Lasubine alkaloids.8 Herein, we report our investigations involving the Ni catalyzed reactions between enynes and isocyanates to afford dieneamides.
Despite the precedent that enynes and isocyanates were both viable substrates individually in the Ni catalyzed cycloaddition reaction, it was unclear whether they would react with each other in a productive manner. However, success in Ni catalyzed reductive couplings between alkenes and alkyl-substituted isocyanates9 suggested enynes and isocyanates would be reactive under Ni catalyzed cycloaddition reaction conditions.
Our initial efforts revolved around evaulating a variety of conditions that would yield an isolable product. As shown in Table 1, a variety of phosphines and NHCs10 were evaluated as prospective ligands to determine the potential reactivity between enyne (1a) and cyclohexyl isocyanate (2a, Equation 1). Attention was directed toward minimizing alkyne cyclotrimerization, a known side-reaction.11 In general, poor conversions of enyne 1a were observed when either monodentate or bidentate phosphines were employed (entries 2–7). A slight increase in the conversion was observed when the reaction was run with bulky NHCs such as ItBu and IMes (entries 8–9). However, in all cases (entries 2–9), no detectable coupling product was observed. In contrast, when bulkier NHC ligands such as SIPr and IPr were employed, a distinct coupling product was isolated in good yields (entries 10–11). As expected, negligible coupling was observed when Ni(COD)2 was used in the absence of an additional donor ligand (entry 1).
Table 1.
Ni-catalyzed cycloaddition of enyne 1 and CyNCO (2a)a
| entry | L | % conversion of 1b | % yield of 3b |
|---|---|---|---|
| 1 | none | 10 | nd |
| 2 | P(n-Bu)3 | 31 | ndc |
| 3 | PPh3 | 17 | nd |
| 4 | P(p-Tol)3 | 31 | nd |
| 5 | DPPF | 7 | nd |
| 6 | BINAP | 5 | nd |
| 7 | biphenP(t-Bu)2 | 17 | nd |
| 8 | ItBu | 14 | nd |
| 9 | IMes | 33 | nd |
| 10 | SIPr | 90 | 78 |
| 11 | IPr | 100 | 80 |
Reaction conditions: 5 mol % Ni(COD)2, 10 mol % L, 0.1 M 1a, 0.11 M 2a, toluene, room temperature, 17 h.
Determined by GC using naphthalene as an internal standard.
nd = not detectable by GC.
 |
(1) |
Isolation of the major product revealed that cyclization did indeed occur. Dieneamide 3a, rather than a lactam product that would arise from an additional carbon-nitrogen bond forming event (vide infra), was isolated in 70% yield.
 |
(2) |
The combination of Ni and IPr catalyzed the coupling of enyne 1a with a variety of isocyanates (Table 2, Equation 2). Alkyl isocyanates reacted smoothly at room temperature within 1–2 hours. Furthermore, these reactions afforded dieneamides in excellent overall yields with good E:Z ratios (entries 1–3). In contrast, aryl isocyanates reacted more sluggishly and required slightly more forcing conditions.9 For example, the Ni-catalyzed coupling of enyne 1a and phenyl isocyanate proceeded at 60 °C while no reaction occurred at room temperature (entry 4). Nevertheless, dieneamide 3d was isolated in 89% yield. Both aryl isocyanates possessing electron-donating groups as well as electron-withdrawing groups were converted to their respective dienamides in 71–80% yield. Aryl isocyanates possessing electron-donating groups reacted faster than those possessing electron-withdrawing groups (entries 5–7). Sterically-hindered aryl isocyanates such as 2h and 2g were also converted to their respective dieneamides, although under higher reaction temperatures, in excellent yields, 91% and 92%, respectively (entries 8–9). No reaction was observed when TMS-NCO 2i was employed.
Table 2.
Dienamide formation from enyne 1 and isocyanates 2a-2j
| entry | E/Z products | E:Z ratioa | rxn temp | % yieldb |
|---|---|---|---|---|
| 1 | 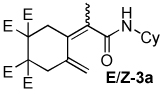 |
5:1 | rt, 1 h | 70 |
| 2 | 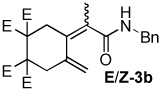 |
2:1 | rt, 2 h | 68 |
| 3 | 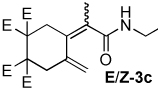 |
2:1 | 80 °C, 1 h | 75 |
| 4 | 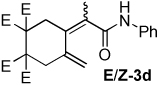 |
2:1 | 60 °C, 2 h | 79 |
| 5 | 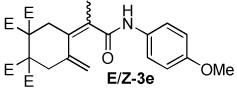 |
2:1 | 60 °C, 2 h | 74 |
| 6 | 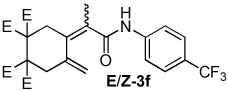 |
1.8:1 | 100 °C, 2 h | 57 |
| 7 | 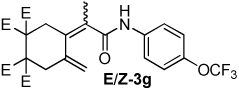 |
2:1 | 80 °C, 5 h | 66 |
| 8 | 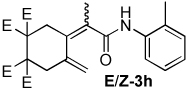 |
2:1 | 80 °C, 1 h | 71 |
| 9 | 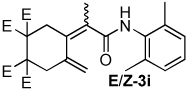 |
2:1 | 80 °C, 1.5 h | 80 |
Reaction conditions: 1 equiv 1a, 1 equiv 2,
Determined by 1H-NMR,
isolated yields, average of 2 runs
When either E or Z isomers of dieneamide 3b were resubjected to the nucleophilic IPr ligand, no isomerization was observed. In addition, when the E-isomer of 3b was resubjected to the reaction conditions, no isomerization to the Z-dieneamide was observed. However, when the Z-isomer of 3b was resubjected to the reaction conditions, a mixture of E- and Z- products was obtained (Equation 4). Thus, the Z-dieneamides are most likely the initial coupling product and undergo a Ni(0)-mediated interconversion to the more stable E-isomer over the course of the reaction.
 |
(3) |
 |
(4) |
A variety of enynes were also successfully converted to their respective enamide products (Table 3). A significant increase in enamide yield (3j and 3k) was observed when enyne 1b was used as a coupling partner in lieu of enyne 1a despite the similarity in the backbones of these two substrates (Table 3, entries 1–2 versus Table 2, entries 1 and 4, respectively). Enyne 1c afforded a 5-membered cyclic dieneamide in good yield (entries 3–4).
Table 3.
Substrate scope using enynes 1b-1e and isocyanates 2a and 2b
| entry | enyne | isocyanate | E, Z products | E:Z ratioa | % yieldb |
|---|---|---|---|---|---|
| 1 | 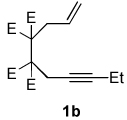 |
CyNCO | 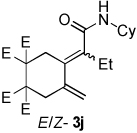 |
4:1 | 89c |
| 2a | |||||
| 2 | 1b | PhNCO | 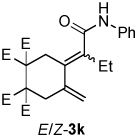 |
4:1 | 84d |
| 2b | |||||
| 3 | 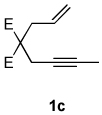 |
2a | 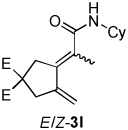 |
4:1 | 65e |
| 4 | 1c | 2b | 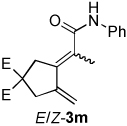 |
4:1 | 72f |
| 5 | 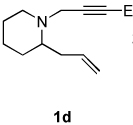 |
2a | 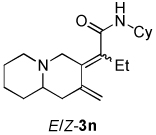 |
1.4:1 | 72f |
| 6 | 1d | 2b | 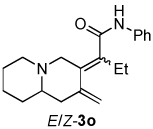 |
1.4:1 | 72f |
| 7 | 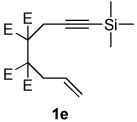 |
nd | — | — | |
| 2a/2b |
Reaction conditions: 1 equiv enyne, 1 equiv isocyanate, 0.1 M toluene,
determined by 1H-NMR,
isolated yields, average of 2 runs,
rt, 4h,
rt, 6 h,
80 °C, 4 h,
80 °C, 3 h
Dienamides having a bicyclic ring system with a nitrogen atom on the bridgehead were also prepared in good yields (entries 5–6). Somewhat surprisingly, although enyne 1e, which possesses a bulky trimethylsilyl group on the alkyne, has been used as a substrate in other Ni-catalyzed cycloddition reactions,2b this enyne undergoes cycloisomerization12 exclusively and does not afford a dienamide product (entries 7).
Two possible mechanisms for dienamide formation are depicted in Scheme 1.13 The primary difference between these two mechanisms is at which point β-hydride elimination occurs. In pathway A, intramolecular initial oxidiative coupling of the enyne and subsequent insertion of the isocyanate leads to 7-membered intermediate 5. Rather than undergoing C-N bond-forming reductive elimination,14 β-hydride elimination occurs resulting in 6. Facile reductive elimination from 6 would afford the Z-dienamide product. Alternatively, nickelacycle 5 can arise from oxidative coupling of the alkyne and the isocyanate to produce 7 prior to insertion of the pendant olefin (Pathway B, Scheme 1).12
Scheme 1.

Possible mechanisms for dieneamide formation
Interestingly, when enyne 1f was subjected to the coupling conditions with either isocyanate 2a or 2b, dienamide formation did not occur. Instead, a cyclic amide was formed as the sole product, albeit in low yields (Equation 5)
 |
(5) |
Lactams 10 and 11 likely arise from C-N bond-forming reductive elimination from a 7 membered nickelacycle such as 5b (Scheme 2). Insertion of the isocyanate into the Ni-Csp3 bond, rather than the Ni-Csp2 bond in 4 would lead to the formation of 5b (mechanism A, Scheme 1). Althernatively, nickelacycle 5b may arise from initial oxidative coupling between the olefin of the enyne and the isocyante folleowed by insertion of the pendant alkyne (mechanism B, Scheme 1). Formation of 5b may be favored over 5a when the steric interaction of the alkyne substituent and the ligand (i.e., IPr) is small. In fact, we have observed this type of sterically-driven selectivity in other Ni-catalyzed cycloaddition chemistry.2b,4
Scheme 2.

Possible mechanisms for dienamide formation
 |
(6) |
Dienemades can be conveniently converted to iminoethers.15 For example, the reaction of 3b with NBS afforded the bromo-substituted iminoether 12 in 53% isolated yield. Furthermore, when 3b was subjected to I2 in lieu of NBS, higher yields were obtained of the halo substituted iminoether. That is, the iodo-substituted iminoether 13 was obtained in 90% isolated yield.
We have demonstrated that reductive coupling of enynes and isocyanates can successfully take place using Ni(COD)2 and IPr ligand system to afford dienamides in good to excellent yields.. This catalyst system can be used to prepare dienamides which contain 5 or 6-membered rings.
Supplementary Material
Acknowledgment
We gratefully acknowledge the Camille and Henry Drefus Foundation and the NIGMS for support of this reasearch.
Footnotes
Supporting Information Available: 1H NMR, 13C NMR and IR data for all compounds in PDF format. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Zhang K, Chopade PR, Louie J. Tetrahedron Lett. 2008;49:4306. doi: 10.1016/j.tetlet.2008.04.121. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo G, Zhang K, Louie J. Tetrahedron Lett. 2008;49:6797. doi: 10.1016/j.tetlet.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Louie J, Gibby JE, Farnworth MV, Tekavec TN. J. Am. Chem. Soc. 2002;124:15188. doi: 10.1021/ja027438e. [DOI] [PubMed] [Google Scholar]; (b) Tekavec TN, Arif AM, Louie J. Tetrahedron. 2004;60:7431. [Google Scholar]
- 3.(a) Duong HA, Cross MJ, Louie J. J. Am. Chem. Soc. 2004;126:11438. doi: 10.1021/ja046477i. [DOI] [PubMed] [Google Scholar]; (b) Duong HA, Louie J. J. Organomet. Chem. 2005;690:5098. [Google Scholar]; (c) Duong HA, Louie J. Tetrahedron. 2006;62:7552. [Google Scholar]
- 4.(a) Tekavec TN, Louie J. Org. Lett. 2005;7:4037. doi: 10.1021/ol0515558. [DOI] [PubMed] [Google Scholar]; (b) Tekavec TN, Louie J. J. Org. Chem. 2008;73:2641. doi: 10.1021/jo702508w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) McCormick MM, Duong HA, Zuo G, Louie J. J. Am. Chem. Soc. 2005;127:5030. doi: 10.1021/ja0508931. [DOI] [PubMed] [Google Scholar]; (b) Tekavec TN, Zuo G, Simon K, Louie J. J. Org. Chem. 2006;71:5834. doi: 10.1021/jo0608669. [DOI] [PubMed] [Google Scholar]
- 6.Zuo G, Louie J. Angew. Chem. Int. Ed. 2004;43:2777. doi: 10.1002/anie.200353469. [DOI] [PubMed] [Google Scholar]
- 7.Zuo G, Louie J. J. Am. Chem. Soc. 2005;127:5798. doi: 10.1021/ja043253r. [DOI] [PubMed] [Google Scholar]
- 8.(a) Yu TR, Rovis T. J. Am. Chem. Soc. 2006;128:2782. doi: 10.1021/ja057803c. [DOI] [PubMed] [Google Scholar]; (b) Yu TR, Rovis T. J. Am. Chem. Soc. 2006;128:12370. doi: 10.1021/ja064868m. [DOI] [PubMed] [Google Scholar]; (c) Lee EE, Rovis T. Org. Lett. 2008;10:1231. doi: 10.1021/ol800086s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scleicher KD, Jamison T. Org. Lett. 2007;9:875. doi: 10.1021/ol063111x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.IPr=1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene; SIPr=1,3-bis(2,5-diisopropylphenyl)-4,5-dihydroimidazolin-2-ylidene; IMes=1,3-bis(2,4,6-trimethylphenyl)-imidazol-2-ylidene; ItBu=1,3-ditert-butylimidazol-2-ylidene.
- 11.(a) Saito S, Yamamoto Y. Chem. Rev. 2000;100:2901. doi: 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]; (b) Tsuda T, Morikawa S, Sumiya R, Saegusa T. J. Org. Chem. 1988;53:3140. [Google Scholar]
- 12.Tekavac T, Louie J. Tetrahedron. 2008;64:6870. doi: 10.1016/j.tet.2008.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For an insightful discussion on the mechanism of the Rh catalyzed couplings of alkenes, alkynes, and isocyanates, see reference 8.
- 14.Koo K, Hillhouse G. Organometallics. 1995;14:4421. [Google Scholar]
- 15.Wang C, Lu J, Mao G, Xi Z. J. Org. Chem. 2005;70:5150. doi: 10.1021/jo050433q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.