

Abstract.
Beside its pivotal role in reproduction, the pituitary hormone prolactin (PRL) has been attributed an immunomodulatory function. Here we report that cAMP is an important stimulator of PRL transcription in primary human T lymphocytes. Inhibition of both protein kinase A (PKA) and p38 MAPK partially abrogated cAMP-induced PRL expression. In addition, cAMP-induced phosphorylation of p38 was shown to occur independently of PKA and could be mimicked by a methylated cAMP analogue which specifically activates the recently discovered cAMP receptor EPAC (exchange protein directly activated by cAMP). Our findings suggest that cAMP induces PRL expression in T lymphocytes via cooperation of at least two different signaling pathways: a PKA-dependent pathway leading to the phosphorylation of cAMP response element-binding protein, and a PKA-independent pathway leading to p38 phosphorylation.
Key words. human, T cell, gene regulation, neuroimmunology, EPAC, cAMP, PKA
The immune system is one of the targets of the polypeptide hormone prolactin (PRL). Although PRL is predominantly produced by the pituitary, the hormone is also synthesized extrapituitarily by other tissues [1, 2]. In the immune system, expression of the PRL gene has been demonstrated in human mononuclear cells [3–5] and was mainly associated with the T lymphocyte population [4]. In addition, high-affinity PRL receptors are expressed on B lymphocytes, T lymphocytes, natural killer (NK) cells and monocytes [4], suggesting the hormone may act in an autocrine or paracrine, cytokine-like, fashion in the immune system. There is indeed compelling evidence for in vitro effects of PRL on immune cell function. For example, PRL stimulates inducible nitric oxide synthetase production [6] and immunoglobulin release [7] in human leukocytes and it has anti-apoptotic properties in Nb2 rat lymphoma cells [8] and dexamethasone-treated thymocytes [9]. PRL was recently demonstrated to stimulate the maturation of rat [10] and human [11] dendritic cells. PRL-treated rat dendritic cells produced increased amounts of interleukin (IL)-12, tumor necrosis factor (TNF)-alpha and IL-1beta [10]. In addition, several groups have reported stimulatory effects of PRL on interferon (IFN)-gamma secretion [12–15].
The importance of T lymphocyte-derived PRL is indicated by the observation that PRL is an autocrine growth factor for human mononuclear cells [3, 16] as well as for the human leukemic T cell line Jurkat [17]. Hyperprolactinemia, correlating with disease activity, has been described in autoimmune conditions such as systemic lupus erythematosus [18] and rheumatoid arthritis [19, 20]. A local or endocrine role for T lymphocyte-derived PRL in systemic lupus erythematosus is suggested by enhanced PRL production in T cells from patients compared to normal controls [21, 22]. Furthermore, in patients with rheumatoid arthritis, PRL, produced by synovium-infiltrating T-lymphocytes, causes aberrant synovial cell function and might thus influence disease progression [23].
Due to the use of an alternative promoter, located 5.8 kb upstream of the pituitary PRL promoter, by extrapituitary PRL sources [24, 25], leukocyte PRL expression is regulated by different signaling pathways and different hormones, cytokines or neuropeptides when compared to pituitary PRL expression [2]. We and others have shown that cAMP is an important stimulator of PRL expression in leukocytes. Indeed, cAMP stimulates PRL expression in the T leukemic cell line Jurkat [26–28], in the eosinophilic leukaemia cell line Eol-1 [27, 29] and in human peripheral blood mononuclear cells (PBMCs) [27].
In T lymphocytes, elevation of intracellular cAMP, induced by agents such as prostaglandin E2 (PGE2), cholera toxin, forskolin or cAMP analogues, inhibits IL-2 and IL-2 receptor expression, thereby blocking cell cycle progression and proliferation [30–35]. Furthermore, cAMP inhibits the expression of Th1 cytokines, whereas it stimulates IL-5 expression by Th2 cells [34, 36, 37]. The classical view is that cAMP exerts its effects via activation of the cAMP-dependent protein kinase (PKA), which subsequently phosphorylates downstream effectors such as CREB [38]. However, PKA-independent actions of cAMP have been described in several cell types and evidence for the importance of these PKA-independent pathways in the immunomodulatory effects of cAMP is emerging. For example, the effects of cAMP on human T cell proliferation [39] and on IL-5 production by human T lymphocytes [40] were reported to be PKA-independent. In addition, we recently showed that in the myeloid leukemic cell line Eol-1, PKA independent pathways, in addition to the classical pathway involving activation of PKA and CREB, contribute to cAMP-induced PRL expression [29]. Whereas the alternative cAMP receptors in these studies remain to be identified, possible candidates could be the recently identified guanine exchange proteins directly activated by cAMP (EPAC1 and EPAC2) [41, 42]. Indeed, a recent study indicates that stimulation of EPAC1 inhibits the bactericidal activity of alveolar macrophages, implying that the protein is involved in regulating the activation of immune cells [43].
Since T lymphocyte-derived PRL has been implicated in normal and pathological immune responses, and cAMP-induced gene expression in these cells has been relatively unexplored, we addressed the mechanisms used by cAMP to induce PRL expression in human T lymphocytes. We found that cAMP stimulates PRL expression in part through PKA, and partially through PKA-independent activation of p38. We provide evidence for the expression of EPAC1, which may mediate the p38-activating effects of cAMP in human T lymphocytes.
Materials and methods
Reagents. RMPI-1640 (with glutamax) was purchased from Life Technologies (Merelbeke, Belgium). BSA, H89, 8-(4-chloro-phenyl-thio)-cAMP (cptcAMP) and 8-(4-chloro-phenyl-thio)-2-O-methyl cAMP (Me-cptcAMP) were purchased from Sigma-Aldrich (St. Louis, M.) SB203580 was from Biomol (Plymouth, P.). Except for cptcAMP and Me-cptcAMP, which were dissolved in LPS-free water (Baxter, Lessines, Belgium), all inhibitors were dissolved in DMSO. The rabbit antibodies against Pp38, ERK1/2, P-JNK, JNK, P-CREB and CREB, and the mouse monoclonal against P-ERK1/2 were obtained from Cell Signaling (Beverly, Mass.). P-CREB antibody detects CREB only when phosphorylated at ser133 and this antibody also detects the phosphorylated form of CREB-related protein ATF-1. Rabbit antibodies against p38 were from Santa Cruz Biotechnology (Santa Cruz, Calif.). Peroxidase-conjugated donkey anti-rabbit and sheep antimouse IgGs were obtained from Amersham Pharmacia Biotech (Roosendaal, The Netherlands).
Cell preparation and cell culture. Jurkat cells were obtained from the European Collection of Cell Cultures (Salisbury, UK). T lymphocytes were isolated from buffy coats obtained from normal donors. First, PBMCs were isolated by centrifugation on Ficoll-Isopaque (Pharmacia & Upjohn, Uppsala, Sweden) density gradients (1.077 g/ml) at 1000 g for 20 min. Subsequently, T lymphocytes were isolated by sheep red blood cell rosetting [44]. Purity of the T lymphocyte fraction was always over 90% as assessed by flow cytometric analysis. T lymphocytes were resuspended in RPMI-1640 supplemented with 1% BSA, at a concentration of 5×106/ml. To measure the effects of cAMP analogues on PRL mRNA expression, cells were cultured for 6 h in a humidified 5% CO2 atmosphere at 37 °C. To address cAMP-induced protein phosphorylation, cells were cultured for 20 min in 2-ml reaction tubes (Eppendorf, Hamburg, Germany) in a water bath at 37 °C.
RT-PCR. Isolation of total RNA and reverse transcription were performed as described before [45]. Briefly, for PRL mRNA detection, a real-time cDNA amplification was performed using the Applied Biosystems (Nieuwerkerk a/d IJssel, The Netherlands) Assay-On-Demand for human prolactin (hs 00168730_m1) and a five-point standard curve (0.02–200 ng total RNA from Jurkat cells). This assay uses a specific TaqMan MGB probe with an FAM reporter dye at the 5′ end and a nonfluorescent quencher at the 3′ end. Fluorescence was monitored using the Taqman 7700 Sequence detector (Applied Biosystems). For detection of EPAC1 and EPAC2 transcripts, cDNA samples were subjected to 35 rounds of PCR cycling (94 °C for 1 min, 50°C for 2 min, 72 °C for 45 s) using the following primer sets: for EPAC1: CTTCCTCCAGAAACTCTCAG (sense) and TCAGCTCATGCGCTTCCTG (antisense); for EPAC2: CTCATTGAACCTCACGTTCC (sense) and AGTCATCTCCTTCATGCAGG (antisense).
Western blotting. Preparation of cellular extracts and Western blotting were performed as described earlier [28]. All primary antibodies were used at a 1:1000 dilution. Before reprobing, blots were stripped by washing in ddH20 for 10 min, followed by a 5-min incubation in 0.2 M NaOH and another washing in ddH20.
Statistics. Statistical differences between groups were determined by ANOVA, followed by Tukey’s post-test. Data represented are means ± SD of quadruplicate incubations of cells from one donor. The presented experiments are representative of three (figs. 1, 2, 3 and 5) or eight (fig. 4) independent experiments using different donors.
Fig. 1.
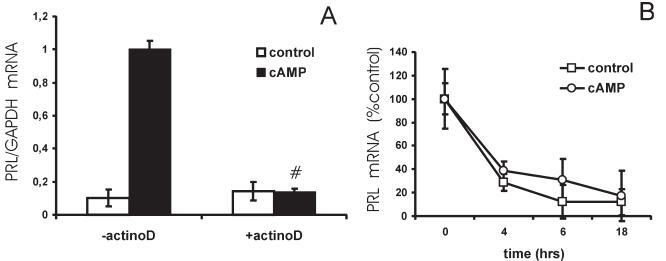
The effect of cAMP on PRL expression in primary T lymphocytes requires de novo mRNA synthesis. (A) T lymphocytes were preincubated for 1 h with vehicle or the transcriptional inhibitor actinomycin D (5 µg/ml) and next stimulated with vehicle or cptcAMP (250 µM) for 6 h. PRL mRNA levels were quantified by real-time PCR and normalized to GAPDH mRNA expression. # Significantly different from control without actinomycin D (p < 0.01). (B) Decay of PRL mRNA levels as measured by real-time PCR. T lymphocytes were preincubated for 1 h with actinomycin D (5 µg/ml) and next stimulated with vehicle or cptcAMP (250 µM) for 0, 4, 6 and 18 h. PRL mRNA levels are represented as percentage of control (actinomycin D 5 µg/ml, no cptcAMP at time 0). The results in this figure are obtained from the same donor.
Fig. 2.
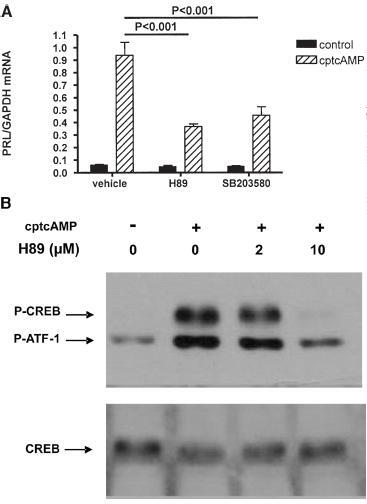
Effect of inhibition of PKA on cAMP-induced PRL mRNA expression and CREB phosphorylation in human T lymphocytes. (A) T lymphocytes were pretreated for 2 h with vehicle, H89 (10 µM) or SB203580 (50 µM), before stimulation with 250 µM cptcAMP for 6 h. PRL mRNA levels were quantified by real-time PCR and normalized to GAPDH mRNA expression. Control conditions are always significantly different from corresponding cptcAMP-stimulated conditions (p<0.01). (B) T lymphocytes were preincubated for 2 h with increasing doses of H89, before stimulation with cptcAMP (250 µM) for 20 min. P-CREB and total CREB were monitored by western blotting. The results in the figure are obtained from the same donor.
Fig. 3.
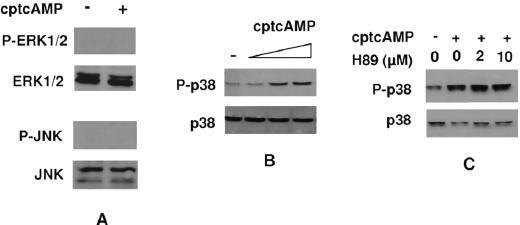
Effect of cAMP on MAPK activation in human T lymphocytes and role of PKA in this effect. Cells were stimulated for 20 min with the indicated doses of cptcAMP. MAPK activation was assessed by Western blotting. (A) P-ERK, ERK, P-JNK and JNK in T lymphocytes stimulated with 250 µM of cptcAMP. (B) P-p38 and total p38 in T lymphocytes stimulated with increasing doses (25, 250 and 500 µM) of cptcAMP. (C) P-p38 and total p38 in T lymphocytes preincubated for 2 h with increasing doses of H89, before stimulation with cptcAMP (250 mM). The results in A-C are obtained from three different donors. The results in B are obtained from the same donor as in figure 5A.
Fig. 5.
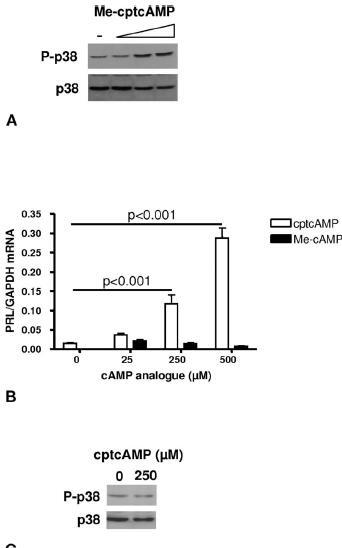
Role of EPAC in the effects of cAMP on PRL expression in human T lymphocytes. (A) T cells were stimulated for 20 min with increasing doses (25, 250 and 500 µM) of Me-cptcAMP. Activation of p38 was assessed by Western blotting. (B) T lymphocytes were stimulated for 6 h with increasing doses of cptcAMP or MecptcAMP. PRL mRNA levels were quantified by real-time PCR and normalized to GAPDH mRNA expression. (C) P-p38 and total p38 as assessed by Western blotting in Jurkat cells stimulated for 20 min with vehicle or 250 µM of cptcAMP. The results in A and B are obtained from two different donors. The results in A are obtained from the same donor as figure 3B.
Fig. 4.
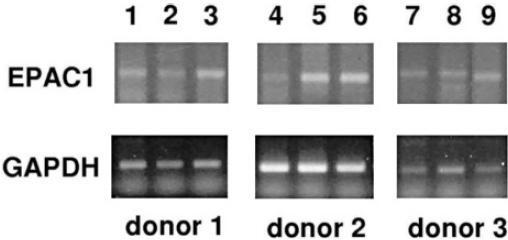
Expression of EPAC1 in primary T lymphocytes. Detection of EPAC1 mRNA by RT-PCR in human PBMC, T cell and non-T cell samples. EPAC1 expression was detected in T cells from all eight donors tested. Results are shown for three representative donors. Lanes 1, 4, 7: PBMC; lanes 2, 5, 8: T cells; lanes 3, 6, 9: non-T cells.
Results
cAMP stimulates PRL transcription in primary human T lymphocytes. We previously showed that cAMP induces PRL mRNA expression in freshly isolated human PBMCs and in the human T leukemic cell line Jurkat [27]. To assess whether cAMP induces PRL expression in primary T lymphocytes, we investigated the effect of cAMP on PRL mRNA levels in purified primary human T lymphocytes. We found that the long-acting cAMP analogue cptcAMP stimulated PRL mRNA levels in primary T lymphocytes by 12.9 ± 11.2-fold. Whereas PRL mRNA levels were positively affected by cAMP in all investigated donors (n=12), the size of the response to cAMP was very much donor dependent, with a 34.7-fold induction in the best responder versus only a 3.6-fold induction in the weakest responder. As shown in figure 1A, preincubation of primary T lymphocytes with the transcriptional inhibitor actinomycin D completely blocked the effect of cptcAMP on PRL mRNA expression. In addition, cptcAMP did not affect the rate of decay of the PRL message in T cells treated with actinomycin D (fig. 1B), indicating that cAMP enhances PRL expression in primary T lymphocytes by stimulating transcription.
cAMP-induced PRL expression is partially mediated via PKA in primary human T lymphocytes. PKA is the best-known effector of cAMP signaling. We therefore investigated the effect of PKA inhibition, using H89, on cAMP-induced PRL expression in primary T lymphocytes. Figure 2A shows that, at maximal subcytotoxic doses (10 µM), H89 only partially blocked cptcAMP-induced PRL expression, whereas it completely abolished cptcAMP-induced phosphorylation of the principal PKA target, CREB (fig. 2B).
Role of MAPKs in cAMP-induced PRL expression. In leukocytes and other cell types, cAMP has been shown to mediate some of its effects via activation or inhibition of MAPK pathways [46]. Therefore, since inhibition of PKA did not completely block cAMP-induced expression, we addressed the effect of cptcAMP on activation of p38, ERK and JNK. We found that ERK and JNK phosphorylation were undetectable both in unstimulated and cptcAMP-stimulated T lymphocytes (fig. 3A), but could be induced by 12-O-tetradecanoyl-phorbol-13-acetate (TPA) and anisomycin respectively (data not shown). However, cptcAMP dose dependently stimulated p38 phosphorylation (fig. 3B). To address the role of PKA in the effect of cAMP on p38 activation, we investigated the effect of H89 on the phosphorylation of p38. As shown in figure 3C, H89 did not affect cptcAMP-induced p38 phosphorylation, indicating cptcAMP phosphorylates p38 in a PKA-independent manner.
The role of the p38 MAPK in the effect of cptcAMP on PRL expression in T lymphocytes was indicated by the finding that SB203580, a specific p38 inhibitor, partially abolished the effect of cptcAMP on PRL expression (fig. 2A).
Role of EPAC in PRL expression in human T lymphocytes. Whereas PKA is the best-known mediator of cAMP effects, recently, the activation of alternative, PKA-independent, signaling routes through EPAC by cAMP have been described. We therefore addressed the expression of the novel cAMP receptors EPAC1 and EPAC2 by RT-PCR in primary T lymphocytes of eight donors. EPAC1 transcripts were amplified from cDNA samples obtained from PBMCs and T cells (as well as from the residual non-T cell fraction) from all donors (n=8) (fig. 4). The PCR product identity was confirmed by DNA sequencing. EPAC2 transcripts were undetectable in all samples (data not shown). Using a methylated cptcAMP analogue, which is unable to activate PKA, but instead specifically activates EPAC [47], we investigated the role of EPAC in regulating PRL expression in T cells. As shown in figure 5A, Me-cptcAMP stimulated p38 activity in T lymphocytes, whereas it did not affect CREB phosphorylation (data not shown). However, unlike cptcAMP, which dose dependently stimulated PRL mRNA levels in human T lymphocytes, Me-cptcAMP had no effect on PRL expression (fig. 5B). We previously demonstrated that the stimulatory effect of cptcAMP on PRL expression in the human T leukemic cell line Jurkat is mediated by PKA only [28]. Indeed, as depicted in figure 5C, in these cells, cptcAMP did not induce p38 phosphorylation.
Discussion
We and others previously showed that cAMP and factors signaling via cAMP, such as PGE2, stimulate PRL expression through the extrapituitary promoter in human PBMCs and in the human T leukemic cell line Jurkat [26–28]. Here we show that cAMP is also a potent inducer of PRL transcription in primary T lymphocytes. Although cAMP increased PRL expression in all 12 donors tested, there was a large variation in the strength of the response. Since in vitro stimulation of PRL expression by phytohemagglutinin has been shown to relate to a single nucleotide polymorphisms at −1149 of the lymphoid promoter [48], the possibility exists that this polymorphism causes a variation in the strength of the cAMP effect on PRL expression. Interestingly, this polymorphism was prevalent in patients with systemic lupus erythematosus. PGE2 and other factors that signal through cAMP, such as catecholamines, usually inhibit Th1 responses [49, 50], whereas PRL predominantly stimulates expression of Th1 cytokines [10, 12–15, 51]. Since endogenous PRL has especially been shown to have immunomodulatory effects in animals subjected to stress [52, 53], one can speculate that induction of PRL expression in the immune system serves as a feedback mechanism to restrict the effect of stress hormones on the immune system. This hypothesis could be tested by assessing the effects of catecholamines in the absence and in the presence of inhibiting antibodies against PRL. Indeed, autocrine or paracrine effects of small amounts of PRL secreted by PBMCs have been shown in several studies [3, 54].
According to the classical paradigm, cAMP modulates gene transcription via activation of PKA and subsequent phosphorylation of CREB [38]. However, some of the effects of cAMP on T lymphocytes have been shown to be PKA independent. For example, in a murine Th2 cell line [55] and in human PBMCs [40], the effects of cAMP on IL-5 expression were PKA independent. Here we show that in human T lymphocytes, cAMP stimulates PRL gene expression via activation of both PKA-dependent and PKA-independent signaling pathways. The PKA inhibitor H89 completely blocked phosphorylation of CREB at Ser133, yet failed to completely inhibit cAMP-induced PRL gene transcription, suggesting that other kinases and transcription factors, beside PKA and CREB are involved in the effect of cAMP on PRL expression. Our observation that cAMP induced p38 activation, whereas a specific p38 inhibitor, SB203580, partially inhibited cAMP-induced-PRL expression in T lymphocytes, indicates p38 is one of the kinases that are activated by cAMP, in addition to PKA. Only a few studies have described effects of cAMP mediated by p38. For example, cAMP induces p38 activation in Chinese hamster ovary cells [56], in murine cardiomyocytes [57], macrophages [58] and Th2 cells [55], in rat granulosa cells [59] and in human SK-N-MC neuroblastoma cells [60]. Interestingly, whereas in most of these studies the effect of cAMP on p38 activation was PKA dependent, this was not the case in the murine Th2 cells [55]. Our results indicate that in normal human T lymphocytes, p38 is also activated independently of PKA. Whereas the signaling intermediate(s) activated by cAMP and responsible for the observed p38 activation remain to be identified, the findings that the EPAC activator Me-cptcAMP also stimulates p38 phosphorylation, and that primary T lymphocytes express EPAC1 transcripts, suggest a possible candidate could be the recently identified small guanine nucleotide exchange factor EPAC1. EPAC1 transcripts have only been detected in B lymphocytes [61] and alveolar macrophages [43] but not in other leukocyte subpopulations. Whereas Tiwari et al. [61] were unable to detect EPAC1 or EPAC2 by RT-PCR in human T lymphocytes, we did detect messages for the EPAC1 gene by RT-PCR in human T cells. Since, to date, no specific inhibitors for EPAC activity are available, proving that EPAC is indeed responsible for the effect of cAMP on p38 activation is very difficult. In addition, it has been suggested that in human T cells the cAMP-induced, but PKA-independent, pathway leading to inhibition of IL-5 does not involve EPAC [40], indicating another, unidentified, signaling protein, which is activated by Me-cptcAMP as well as by cptcAMP, could be responsible for the observed p38 activation.
Our observation that cAMP is able to stimulate some PRL expression in the presence of H89, which completely blocks CREB phosphorylation, indicates that a PKA-independent pathway is also involved in PRL expression, and that this pathway is operative in the absence of PKA activity. Indeed, inhibition of p38 also partially blocked induction of PRL expression. However, activation by Me-cptcAMP of p38 without PKA activation was not sufficient to enhance PRL expression. These observations suggest that cAMP induces a third, unidentified, route that stimulates PRL expression in human T lymphocytes. The p38 route probably acts in synergy with this unidentified route to enhance PRL transcription. In leukemic Jurkat T lymphocytes, full induction of PRL transcription has previously been demonstrated to require costimulatory signals, which can come from either the PKC-activating phorbolester TPA or the Ca2+ ionophore ionomycin [26, 27], in addition to the cAMP stimulus. These synergistic effects were not observed in normal human PBMCs, where cAMP potently induced PRL expression by itself [27]. We speculate that in human T lymphocytes, cAMP does not require a costimulus to induce PRL expression, because it induces at least one PKA-independent pathway, leading to p38 activation, in addition to the well-known PKA pathway. Interestingly, the p38 pathway was indeed not activated by cAMP in Jurkat cells (fig. 5C) but was also activated in the Eol-1 cell line [29]. In these cells, as in T lymphocytes, cAMP by itself is a potent inducer of PRL expression.
Whereas pituitary PRL overproduction can be effectively treated using dopamine agonists, leukocyte PRL expression is directed by an alternative promoter and is independent from the factors regulating pituitary PRL expression. T lymphocyte-derived PRL has been implicated in the pathophysiology of auto-immune disease and further research on the signaling routes leading to PRL expression in T lymphocytes may be of clinical importance. This paper and earlier publications from us and other groups indicate that cAMP is an important regulator of leukocyte PRL expression. Whereas our findings suggest that the PKA pathway is important in this process, at least one additional signal transmitted via p38 is required for full PRL induction in T lymphocytes. Importantly, we have detected the novel cAMP receptor EPAC1 in primary T lymphocytes and hypothesize that EPAC1 could be the signaling intermediate linking cAMP to p38.
Acknowledgements
This research was supported by the FWO Vlaanderen (G.0126.02), The Flemish Government (GOA 97-02-04) and by institutional grants from the Free University of Brussels. We would like to thank Dr. R. Hooghe for critical revision of the manuscript and E. Quartier for sequencing.
Footnotes
Received 21 September 2005; received after revision 31 October 2005; accepted 2 November 2005
References
- 1.Ben Jonathan N., Mershon D. L., Allen R. W., Steinwetz R. W. Extrapituitary prolactin: distribution, regulation, functions, and clinical aspects. Endocr. Rev. 1996;17:639–668. doi: 10.1210/edrv-17-6-639. [DOI] [PubMed] [Google Scholar]
- 2.R K., S G. Regulation of prolactin expression in immune cells. In: Matera L., Rapaport R., editors. Neuroimmune Biology, vol. 2, Growth and Lactogenic Hormones. Amsterdam: Elsevier; 2002. pp. 147–159. [Google Scholar]
- 3.P S., R G., W L., S V., Q L., S A., et al. Prolactin synthesized and secreted by human peripheral blood mononuclear cells: an autocrine growth factor for lymphoproliferation. Proc. Natl. Acad. Sci. USA. 1992;89:7713–7716. doi: 10.1073/pnas.89.16.7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.I P., J. J L., S A., P. A K. Expression of prolactin and its receptor in human lymphoid cells. Mol. Endocrinol. 1992;6:1023–1031. doi: 10.1210/me.6.7.1023. [DOI] [PubMed] [Google Scholar]
- 5.K. D O., D. W M., T. M T., L. Y Y.-L. Prolactin gene expression in human thymocytes. Mol. Cell. Endocrinol. 1992;87:R19–R23. doi: 10.1016/0303-7207(92)90251-Z. [DOI] [PubMed] [Google Scholar]
- 6.Z D., R H., P V., E. L H.-P. Cytokine-like effects of prolactin in human mononuclear and polymorphonuclear leukocytes. J. Neuroimmunol. 2001;120:58–66. doi: 10.1016/S0165-5728(01)00420-9. [DOI] [PubMed] [Google Scholar]
- 7.N L., A M., R S., E T. Differential effects of prolactin upon activation and differentiation of human B lymphocytes. J. Neuroimmunol. 1993;47:35–40. doi: 10.1016/0165-5728(93)90282-4. [DOI] [PubMed] [Google Scholar]
- 8.M. A L., D. J B., J. S K., J. C R., T M., A. R B. Rapid modulation of the apoptosis regulatory genes, bcl-2 and bax by prolactin in rat Nb2 lymphoma cells. Endocrinology. 1996;137:5456–5462. doi: 10.1210/en.137.12.5456. [DOI] [PubMed] [Google Scholar]
- 9.N K., O T., D. J B., N. D H., A. R B. Prolactin suppresses glucocorticoid-induced thymocyte apoptosis in vivo . Endocrinology. 2003;144:2102–2110. doi: 10.1210/en.2003-0053. [DOI] [PubMed] [Google Scholar]
- 10.P. C C., E J., R S., V. V., A. G Z. Prolactin stimulates maturation and function of rat thymic dendritic cells. J. Neuroimmunol. 2004;153:83–90. doi: 10.1016/j.jneuroim.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 11.L M., A G., M G., K V., E R., M C., et al. Individual and combined effect of granulocyte-macrophage colony-stimulating factor and prolactin on maturation of dendritic cells from blood monocytes under serum-free conditions. Immunology. 2000;100:29–36. doi: 10.1046/j.1365-2567.2000.00996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.T. C C., S Y., G C., N S., J L., N V. Enhanced yields of gamma interferon in prolactin treated human peripheral blood mononuclear cells. Proc. Soc. Exp. Biol. Med. 1994;205:89–95. doi: 10.3181/00379727-205-43683. [DOI] [PubMed] [Google Scholar]
- 13.L M., M C., G B., B F., A B. Up-modulation of interferon-gamma mediates the enhancement of spontanous cytotoxicity in prolactin-activated natural killer cells. Immunology. 1999;98:386–392. doi: 10.1046/j.1365-2567.1999.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.K. Z M. Prolactin enhances production of interferon-gamma, interleukin-12, and interleukin-10, but not of tumor necrosis factor-alpha, in a stimulus-specific manner. Cytokine. 2003;21:187–194. doi: 10.1016/S1043-4666(02)00496-9. [DOI] [PubMed] [Google Scholar]
- 15.S D., T L., H. L F., J B. Sleep associated regulation of T helper 1/T helper 2 cytokine balance in humans. Brain Behav. Immun. 2004;18:368–374. doi: 10.1016/j.bbi.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 16.D. P H., J. W H., E. W B. Inhibition of lymphocyte proliferation by antibodies to prolactin. FASEB J. 1989;3:2194–2202. doi: 10.1096/fasebj.3.10.2787766. [DOI] [PubMed] [Google Scholar]
- 17.L M., M C., M G., M C., S B., S G., et al. Prolactin is an autocrine growth factor for the Jurkat human T-leukemic cell line. J. Neuroimmunol. 1997;79:12–21. doi: 10.1016/S0165-5728(97)00096-9. [DOI] [PubMed] [Google Scholar]
- 18.J. M M., R. E P., R P., G G., D A., L B., et al. Clinical significance of serum and urine prolactin levels in lupus glomerulonephritis. Lupus. 1998;7:387–391. doi: 10.1191/096120398678920307. [DOI] [PubMed] [Google Scholar]
- 19.W. C K., G K. High levels of macrophage inflammatory protein-1alpha correlate with prolactin in female patients with active rheumatoid arthritis. Clin. Rheumatol. 1998;17:263–264. doi: 10.1007/BF01451064. [DOI] [PubMed] [Google Scholar]
- 20.B S., V F., A S., D F., M C. Serum prolactin concentrations in male patients with rheumatoid arthritis. Ann. N. Y. Acad. Sci. 2002;966:258–262. doi: 10.1111/j.1749-6632.2002.tb04224.x. [DOI] [PubMed] [Google Scholar]
- 21.M. A G., J. F M., L. J J., M. L C., C G., S G.-U., et al. Prolactin and systemic lupus erythematosus: prolactin secretion by SLE lymphocytes and proliferative (autocrine) activity. Lupus. 1995;4:348–352. doi: 10.1177/096120339500400504. [DOI] [PubMed] [Google Scholar]
- 22.F L., A M.-C., V C., J A.-V., G Q., C C., et al. A bioactive 60-kilodalton prolactin species is preferentially secreted in cultures of mitogen-stimulated and nonstimulated peripheral blood mononuclear cells from subjects with systemic lupus erythematosus. J. Clin. Endocrinol. Metab. 1997;82:3664–3669. doi: 10.1210/jc.82.11.3664. [DOI] [PubMed] [Google Scholar]
- 23.H N., N S., A K., T A., T S. Prolactin locally produced by synovium infiltrating T lymphocytes induces excessive synovial cell functions in patients with rheumatoid arthritis. J. Rheumatol. 1999;26:1890–1900. [PubMed] [Google Scholar]
- 24.M B., J. R M., J. R D. Characterization of an up-stream promoter directing extrapituitary expression of the human prolactin gene. Mol. Endocrinol. 1994;8:635–642. doi: 10.1210/me.8.5.635. [DOI] [PubMed] [Google Scholar]
- 25.B G., R K., R T., G. E D. Nonpituitary human prolactin gene transcription is independent of Pit-1 and differentially controlled in lymphocytes and in endometrial stroma. Mol. Endocrinol. 1994;8:356–373. doi: 10.1210/me.8.3.356. [DOI] [PubMed] [Google Scholar]
- 26.G. H R., D. W R., J. R D. The human prolactin gene upstream promoter is regulated in lymphoid cells by activators of T-cells and by cAMP. Mol. Endocrinol. 1999;22:285–292. doi: 10.1677/jme.0.0220285. [DOI] [PubMed] [Google Scholar]
- 27.S G., Vanden W B., P V., E. L H.-P., R K. Regulation of prolactin expression in leukemic cell lines and peripheral blood mononuclear cells. J. Neuroimmunol. 2003;135:107–116. doi: 10.1016/S0165-5728(02)00438-1. [DOI] [PubMed] [Google Scholar]
- 28.S G., P V., B G., E. L H.-P., R K. Mechanism of prostaglandin (PG)E2-induced prolactin expression in human T cells: cooperation of two PGE2 receptor subtypes, E-prostanoid (EP) 3 and EP4, via calcium- and cyclic adenosine 5′-monophosphate-mediated signaling pathways. J. Immunol. 2004;173:5952–5962. doi: 10.4049/jimmunol.173.10.5952. [DOI] [PubMed] [Google Scholar]
- 29.S G., P V., E. L H.-P., R K. Multiple, PKA-dependent and PKA-independent, signals are involved in cAMP-induced PRL expression in the eosinophilic cell line Eol-1. Cell Signal. 2004;17:901–909. doi: 10.1016/j.cellsig.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 30.D M., C A., B F., M F. Regulation of interleukin 2 synthesis by cAMP in human T cells. J. Immunol. 1987;139:1179–1184. [PubMed] [Google Scholar]
- 31.A L., B J., R. A M. Cyclic AMP concentrations modulate both calcium flux and hydrolysis of phosphatidylinositol phosphates in mouse T lymphocytes. J. Immunol. 1988;140:936–940. [PubMed] [Google Scholar]
- 32.L. E A., R. L S., G. M K. Control of human T-lymphocyte interleukin-2 production by a cAMP-dependent pathway. Cell. Immunol. 1988;115:88–99. doi: 10.1016/0008-8749(88)90164-5. [DOI] [PubMed] [Google Scholar]
- 33.D C., E. V R. Interleukin 2 transcription factors as molecular targets of cAMP inhibition: delayed inhibition kinetics and combinatorial transcription roles. J. Exp. Med. 1994;179:931–942. doi: 10.1084/jem.179.3.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.M B., B. S F. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991;146:108–113. [PubMed] [Google Scholar]
- 35.E. D A., F P., J. P B., H Y., D. T B. Prostaglandin E2 and other cyclic AMP-elevating agents modulate IL-2 and IL-2R alpha gene expression at multiple levels. J. Immunol. 1992;148:2845–2852. [PubMed] [Google Scholar]
- 36.T. F G., S. R S., F. W F. Evidence implicating utilization of different T cell receptor-associated signaling pathways by TH1 and TH2 clones. J. Immunol. 1990;144:4110–4120. [PubMed] [Google Scholar]
- 37.E M., A. M Z., M M., N. P S., B. T H. Cholera toxin discriminates between T helper 1 and 2 cells in T cell receptor-mediated activation: role of cAMP in T cell proliferation. J. Exp. Med. 1990;172:95–103. doi: 10.1084/jem.172.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.B M., M M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell. Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 39.P. J B., M. J D., I. V H. Immunomodulatory effects of pharmacological elevation of cyclic AMP in T lymphocytes proceed via a protein kinase A independent mechanism. Immunopharmacology. 1999;41:139–146. doi: 10.1016/S0162-3109(98)00060-5. [DOI] [PubMed] [Google Scholar]
- 40.K. J S., M B., K T., M. D H., I M., P. J B., et al. Adenosine 3′,5′-cyclic monophosphate (cAMP)-dependent inhibition of IL-5 from human T lymphocytes is not mediated by the cAMP-dependent protein kinase A. J. Immunol. 2001;167:2074–2080. doi: 10.4049/jimmunol.167.4.2074. [DOI] [PubMed] [Google Scholar]
- 41.de J R., F. J. T Z., M. H. G V., R. H C., A N., J W., et al. Epac is a Rap1 guaninenucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 42.H K., G. M S., S T., J. J C., P H., J. P B., et al. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. USA. 1998;95:13278–13283. doi: 10.1073/pnas.95.22.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.J A., C C., C. H S., M L., M P.-G. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J. Immunol. 2005;174:595–599. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- 44.F G.-M., R. E B. Simplified procedure for the separation of human T and non-T cells. Vox. Sang. 1977;33:5–8. doi: 10.1111/j.1423-0410.1977.tb02229.x. [DOI] [PubMed] [Google Scholar]
- 45.R K., A C. Insulin-like growth factor-I stimulates IL-10 production in human T cells. J. Leukoc. Biol. 2004;76:862–867. doi: 10.1189/jlb.0404248. [DOI] [PubMed] [Google Scholar]
- 46.P. J. S S., J. M S. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell. Biol. 2002;12:258–266. doi: 10.1016/S0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 47.J. M E., A. E C., de J R., van M T., F S., H. G G., et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 48.A S., D R., A A., A H., W T., R D., et al. Characterization of a prolactin gene polymorphism and its associations with systemic lupus erythematosus. Arthritis Rheum. 2001;44:2358–2366. doi: 10.1002/1529-0131(200110)44:10<2358::AID-ART399>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 49.M B., B. S F. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991;146:108–113. [PubMed] [Google Scholar]
- 50.I. J E., R. L W., G. P C., E. S V. The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol. Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- 51.R M., C B., I N., G M. R., E G., M G., et al. Th1-Th2 response in hyperprolactinemic mice infected with Salmonella enterica serovar Typhimurium. Eur. Cytokine Netw. 2003;14:186–191. [PubMed] [Google Scholar]
- 52.A. L D., O T., D. J B., A. R B., C. K O., N. D H. Prolactin deficiency on myelopoiesis and splenic T lymphocyte proliferation in thermally injured mice. Endocrinology. 2002;143:4147–4151. doi: 10.1210/en.2002-220515. [DOI] [PubMed] [Google Scholar]
- 53.K D., N. D H. Anterior pituitary hormones, stress, and immune system homeostasis. Bioessays. 2001;23:288–294. doi: 10.1002/1521-1878(200103)23:3<288::AID-BIES1039>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 54.K C.-R., J H., E Z., A L.-M., M. V L.-H., F B.-F. Identification of prolactin as a novel immunomodulator on the expression of co-stimulatory molecules and cytokine secretions on T and B human lymphocytes. Clin. Immunol. 2005;116:182–191. doi: 10.1016/j.clim.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 55.C C., D Z., J. M L., A R. Cyclic AMP activates p38 mitogen-activated protein kinase in Th2 cells: phosphorylation of GATA-3 and stimulation of Th2 cytokine gene expression. J. Immunol. 2000;165:5597–5605. doi: 10.4049/jimmunol.165.10.5597. [DOI] [PubMed] [Google Scholar]
- 56.M P., H. B A., S K., C C., J. P B. Thyroid-stimulating hormone and cyclic AMP activate p38 mitogen-activated protein kinase cascade: involvement of protein kinase A, rac1, and reactive oxygen species. J. Biol. Chem. 2000;22:40539–40546. doi: 10.1074/jbc.M002097200. [DOI] [PubMed] [Google Scholar]
- 57.M Z., S. J Z., B Z., B. K K., R. P X. Beta 2-adrenergic receptor-induced p38 MAPK activation is mediated by protein kinase A rather than by Gi or gbeta gamma in adult mouse cardiomyocytes. J. Biol. Chem. 2000;22:40635–40640. doi: 10.1074/jbc.M006325200. [DOI] [PubMed] [Google Scholar]
- 58.C C., Y C., Y H., K C., W L. PKA-dependent activation of PKC, p38 MAPK and IKK in macrophages: implication in the induction of inducible nitric oxide synthase and interleukin-6 by dibutyryl cAMP. Cell Signal. 2004;16:565–575. doi: 10.1016/j.cellsig.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 59.E. T M., J C., J. C J., M H.-D. Follicle stimulating hormone (FSH) activates the p38 mitogen-activated protein kinase pathway, inducing small heat shock protein phosphorylation and cell rounding in immature rat ovarian granulosa cells. Endocrinology. 1998;139:3353–3356. doi: 10.1210/en.139.7.3353. [DOI] [PubMed] [Google Scholar]
- 60.X Z., K U., H W., E F. D1 dopamine receptor agonists mediate activation of p38 mitogenactivated protein kinase and c-Jun amino-terminal kinase by a protein kinase A-dependent mechanism in SK-N-MC human neuroblastoma cells. Mol. Pharmacol. 1998;54:453–458. doi: 10.1124/mol.54.3.453. [DOI] [PubMed] [Google Scholar]
- 61.S T., K F., E. Y M., A F., D. H S., A L. Among circulating hematopoietic cells, BCLL uniquely expresses functional EPAC1, but EPAC1-mediated Rap1 activation does not account for PDE4 inhibitor-induced apoptosis. Blood. 2004;103:2661–2667. doi: 10.1182/blood-2003-06-2154. [DOI] [PubMed] [Google Scholar]