

Abstract
The nonsteroidal anti-inflamatory drug (NSAID) flurbiprofen is a selective amyloid lowering agent (SALA) which has been studied clinically in Alzheimer’s disease. HCT-1026 is an ester prodrug of flurbiprofen incorporating a nitrate carrier moiety that in vivo provides NO bioactivity and an improved safety profile. In vitro, HCT-1026 retained the COX inhibitory and NSAID activity of flurbiprofen, but at concentrations at which levels of Aβ1–42 were lowered by flurbiprofen, Aβ1–42 levels were elevated 200% by HCT-1026. Conversely, at lower concentrations, HCT-1026 behaved as a SALA with greater potency than flurbiprofen. The difference in concentration responses between flurbiprofen and HCT-1026 in vitro suggests different cellular targets; and in no case did a combination of nitrate drug with flurbiprofen provide similar actions. In vivo, HCT-1026 was observed to reverse cognitive deficits induced by scopolamine in two behavioral assays; activity that was also shown by a classical nitrate drug, but not by flurbiprofen. The ability to restore aversive memory and spatial working and reference memory after cholinergic blockade has been demonstrated by other agents that stimulate NO/cGMP signaling. These observations add positively to the preclinical profile of HCT-1026 and NO chimeras in Alzheimer’s disease.
Keywords: amyloid, Alzheimer‘s disease, NSAID, nitrate
Introduction
Alzheimer’s disease (AD), a neurodegenerative disorder characterized by a progressive and global deterioration in mental function, is the most common cause of dementia in older individuals. The pathology of AD is characterized by formation of plaques and tangles, inflammation, and loss of neurons and synapses. Plaques are formed predominantly of amyloid β (Aβ) peptide deposits. The disease progresses through a number of stages involving progressive impairment of memory and cognition, early AD being associated with synaptic dysfunction that itself may be caused by Aβ (Masliah 1995, Cullen et al. 1997, Vitolo et al. 2002, Walsh et al. 2002). AD is characterized by disruption of excitatory amino acid and cholinergic neurotransmission, and the only current FDA-approved drugs for therapy of mild-to-moderate AD are the acetylcholinesterase inhibitors that are viewed as inadequate, symptomatic treatments (Bartus et al. 1982, Francis et al. 1999).
The major feature of AD neuropathology is the formation of deposits of the 42- and 40-amino acid forms of Aβ (Aβ1–42 and Aβ1–40), derived from amyloid precursor protein (APP). The amyloid cascade hypothesis considers APP metabolism, amyloid deposits and plaques as the causal factors in AD (Hardy & Selkoe 2002). Therefore, lowering of levels of amyloid peptide, in particular the more toxic Aβ1–42, provides a major focus for drug discovery in AD (Kim et al. 2007). Modulation of amyloidogenesis, avoiding inhibition of secretases, which may be detrimental to normal physiology, represents an attractive therapeutic mechanism (Kowalska & Badellino 1994, Schenk et al. 1999, Mattson et al. 1999). A subset of nonsteroidal anti-inflammatory drugs (NSAIDs), including flurbiprofen, were shown to act as selective amyloid lowering agents (SALAs), reducing the levels of Aβ1–42 and the Aβ1–42/Aβ1–40 ratio (Morihara et al. 2002, Eriksen et al. 2003, Weggen et al. 2001). Since AD is one of many neurodegenerative disorders which display signs of neuroinflammation (Ting et al. 2007), the anti-inflammatory activity of NSAIDs combined with Aβ modulation was seen as promising for AD therapy.
The potential of NSAIDs as AD therapeutics is supported by epidemiological studies that have reported a decreased risk of developing AD after chronic treatment with NSAIDs (Rich et al. 1995, Stewart et al. 1997, in t’ Veld et al. 2001). However, a serious impediment to chronic NSAID therapy, particularly in the elderly, is NSAID gastrotoxicity, linked to lowering of prostaglandin levels, which leads to several thousand deaths in the USA each year (Vane et al. 1998). The R-enantiomer of flurbiprofen does not inhibit cyclooxygenase (COX), which is the initiator of gastrotoxicity, but R-flurbiprofen does retain SALA activity (Eriksen et al. 2003). Therefore, R-flurbiprofen was studied in phase 3 clinical trials as a gastric-sparing therapeutic agent for AD. In 2008, this trial was reported as a failure due to lack of efficacy, linked to poor bioavailability.
An alternative approach to a gastric-sparing NSAID has been to incorporate a gastroprotective organic nitrate moiety in a so-called NO-donating NSAID (NO-NSAID) to counteract the effects of NSAID-induced inhibition of prostaglandin synthesis (MacNaughton et al. 1989). NO-NSAIDs, originally targeted at arthritis and pain, are well studied NSAID prodrugs, one being the NO-flurbiprofen, HCT-1026 (Wallace et al. 1994, Somasundaram et al. 1997). The purpose of this study was to compare HCT-1026 with flurbiprofen, and combinations of flurbiprofen with a clinical nitrate, with regard to important properties of potential therapeutic value in AD, including selective modulation of Aβ peptides, anti-inflammatory activity, neuroprotection, and cognition enhancement in simple animal behavioral models. The new data add positively to the preclinical profile of HCT-1026 and support further development of anti-inflammatory nitrates.
Methods
Materials
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless stated otherwise. All cell culture supplies, murine biotinylated 4G8 and 6E10 antibodies, human amyloid beta ELISA kit, and streptavidin Dynabeads T1 were purchased from Invitrogen (Carlsbad, CA). HCT-1026 was synthesized according to literature procedures (Bolla et al. 2005).
Cell culture and treatments
The murine neuroblastoma N2a cells obtained from American Type Culture Collection (ATCC, Manassas, VA) were cultured in 1:1 DMEM and OPTI-MEM supplemented with 5% fetal bovine serum, 100U/mL penicillin, and 100U/mL streptomycin. N2a cells stably transfected with the Swedish mutant of human APP (N2a/APPsw a kind gift of Dr. Gopal Thinakaran, University of Chicago) were additionally supplemented with 200 μg/mL G418 which was omitted during all drug treatments. Cells were maintained at 37 °C and 5% CO2. RAW 264.7 mouse macrophage-likecells, provided by ATCC or Dr. J. Cook (University of Illinois at Chicago, Chicago, IL), were maintained in DMEM, supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum and incubated in 5% CO2 at 37°C. For co-culture experiments, N2a/WT cells were plated in 6 well plates at a density of 10 × 104 cells/well. RAW cells were plated at a concentration of 20 × 104 cells/insert on 0.4 μm pore inserts fitted into 6 well plates. After 24 h, the N2a media was replaced with 3 mL fresh DMEM media supplemented with 0.2% FBS and the inserts containing RAW cells were transferred to the N2a 6 well plates. The co-cultured cells were treated with DMSO as vehicle control or different concentrations of flurbiprofen or HCT-1026 for 1 h prior to addition of 1 μg/mL LPS. The inserts were removed 48 h after treatment and the MTT assay was performed on N2a cells to determine cell viability. Rat primary cortical glial cultures were established from cortices of newborn rats (1–2 days old). The cells were grown in minimum essential medium (DMEM) supplemented with 10% fetal calf serum, amino acids, vitamins, D-glucose (5 mmol/L), penicillin (100 IU/mL) and streptomycin (100 μg/mL). Cells were ready to use at day 14.
Amyloid beta measurement from N2a/APPsw cell supernatant
N2a/APPsw cells were plated 24 h before the experiment at a density of 25 × 104 cell/well in a 24 well plate. Cells were washed with PBS (50mM, pH 7.4) before the addition of 500 μL of DMEM supplemented with 0.2% FBS followed by drug treatment for a period of 24 h. Conditioned media was collected, followed by the addition of NaN3 (0.01% final concentration) and a mixture of protease inhibitors (10mM phenanthroline and P2714 protease inhibitor cocktail from Sigma containing AEBSF, aprotinin, bestatin, E-64, leupeptin, and EDTA), and centrifuged for 2 min at a speed of 10,000g. Aβ1–42 levels were determined by sandwich ELISA using a human Aβ1–42 ELISA kit and following the supplied protocol. For immunoprecipitation, cells were treated as described above, but plated in a 6 well plate at a density of 100 × 104 cell/well in 2 mL media. 24 h after treatment, 1 mL of the conditioned media was collected, followed by the addition of NaN3 and protease inhibitors and spiked with Aβ1–43 at approximately 1ng/mL final concentration as an internal standard before performing immunoprecipitation. Following immunoprecipitation the samples were analyzed in a MALDI-TOF instrument for the purpose of measuring the relative abundance of Aβ peptides.
Immunoprecipitation and MALDI/TOF analysis
IP-MALDI-TOF quantification was performed as described previously in detail (Abdul-Hay, 2009). Briefly, immunoprecipitation of Aβ was performed by the addition of a mixture of biotinylated 4G8 and 6E10 antibodies (Signet, MA) followed by the addition of T1 streptavidin coated magnabeads. Beads were washed with NH4CO3 (10 mM, pH 8.0) and the Aβ peptides were eluted prior to addition of the matrix solution (alpha-cyano-4-hydroxycinnamic acid in 1:1 acetonitrile and water). Analysis was performed using a MALDI-TOF instrument (Applied Biosystems) in linear positive mode at 1950 shot per spectrum. For each individual experiment, peak heights were normalized to the peak height of the Aβ1–43 standard; these normalized values were then expressed relative to the DMSO vehicle control in each set of experiments.
Griess assay
Assay of nitrite production as a measure of endogenous iNOS activation in RAW 264.7 cells was performed as described previously in detail (Hagos et al. 2008). Cells were plated at a concentration of 25 × 104 cells/well in a 24-well plate and incubated at 37°C for 24 h. The medium was changed, and the cells were drug treated, followed 30 min later by the addition of LPS (Sigma-Aldrich). After 24 h, 100 μL of the supernatant was removed and incubated with the Griess reagent (100 μL) for 30 min at room temperature in the dark. The absorbance was measured at 530 nm and calibrated using a standard curve constructed with sodium nitrite (0–100 μM) in culture media. Nitrite release from nitrate drugs was measured in RAW cell culture without LPS addition and subtracted from the measurements obtained in induced cells to quantify endogenous cellular nitrite production. Drug concentrations were selected to be non-toxic based upon cell viability assays (data not shown). For primary cell cultures, the Griess assay was used as described previously (Bhat et al. 2002): after pre-incubation with drugs or vehicle for 1 h, cells were incubated for a further 48 h with LPS (1μg/mL), or LPS (1μg/mL) + IFNγ (100 U/mL); aliquots of culture supernatant were mixed with an equal volume of Griess reagent as described above.
Prostaglandin analysis
After plating for 24 h, RAW 264.7 cells were treated with flurbiprofen and HCT-1026 at different concentrations, and 1 μg/mL LPS was added 30 min later. After a further 24 h, media were collected and immediately treated with 1 N citric acid (40 μL) and 10% butylated hydroxytoluene (5 μL). Before extraction, 20 ul of PGE2-d4 (100 ng/mL) was added to each sample as an internal standard. Prostaglandins were extracted from cell suspensions using 2 mL of hexane/ethyl acetate (1:1 (v/v)). The extraction step was repeated twice, and the organic phases were evaporated to dryness under a stream of nitrogen at room temperature. All extraction procedures were performed under low light and low temperature conditions. Samples were reconstituted in 200 μL of methanol before liquid chromatography/tandem mass spectrometry (LC/MS) analysis as described previously (Yang et al. 2002). LC/MS was performed using an API 3000 mass spectrometer (Applied Biosystems, Foster City, CA) equipped with HPLC (Shimadzu, Kyoto, Japan) separating on a Luna 3-μm, phenylhexyl, 2 × 150-mm analytical column (Phenomenex, Torrance, CA) using a methanol/ammonium acetate (10 mM; pH 8.5) gradient at a flow rate of 400 μL/min.
MTT assay
The MTT assay was performed by incubating cells in a culture media containing 0.5 mg/mL MTT solution (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) for 4 h, followed by washing with PBS and solubilization of the purple formazan crystals in DMSO. The plate was shaken on a plate rocker for 30 min then the absorbance was measured at 570 nm using 630 nm as the reference wave length on a Dynex MRX II microplate spectrophotometer.
Step-through passive avoidance task
STPA was performed on male C57BL/6 mice (Charles River’s Laboratories) of 8–10 weeks of age and weighing 22–27 g. The mice were kept under standardized laboratory conditions (temperature 21 ± 1 °C, humidity 50–60%) with free access to food and tap water in a room with a natural light–dark cycle. The avoidance experiment was divided into 3 phases: habituation, training and retention phase. The apparatus used consisted of a box divided into 2 compartments, an illuminated compartment adjacent to a dark compartment, the floor consisted of an electric grid controlled by a switch that can deliver an electric shock only to the dark compartment. A doorway was located at floor level in the center of the connecting wall; the door closed once the mouse entered the dark compartment. The habituation phase was performed with no shock, by individually placing the mice in the light compartment; the door was closed once the mouse entered the dark compartment; the mouse left for 15 s before being returned to its cage. The training phase, performed 2 h after habituation consisted of individually placing animals in the illuminated compartment, with an electric shock (0.6 mA for 2 s) delivered immediately after the mouse entered the dark compartment, accompanied by the door closure. Trials were repeated for a maximum of 5 trials or until the mouse remained in the light compartment for 300 s in one trial. The retention test was conducted 48 h after training, consisting of a single repeat of the training protocol without foot shock: The retention trial ended when the mouse entered the dark box or 300 s had elapsed. Scopolamine (1mg/kg) dissolved in physiological saline was administered by i.p. injection 30 min before the training phase and drugs dissolved in 25% DMSO were administered by i.p. injection 20 min before the training.
Radial water maze task
The effect of HCT-1026 on spatial working and reference memory was assayed in a six arm radial water maze (RWM) using a scopolamine-induced cognitive deficit in male Long-Evans rats (200–250 g, Charles River). The RWM contained six swim paths extending out of an open central area with an escape platform located at the end of one arm. For two consecutive days, mice were trained for 16 one-minute trials grouped into 4 blocks (Day 1: blocks 1 & 2; Day 2: blocks 3 & 4; and 4 trials within each block with 15 sec inter-trial interval and 5 min interblock interval) with the hidden platform. Animals were released with their heads pointed toward the wall of the water pool in one arm. Entry into an incorrect arm was scored as an error. The total number of errors made in finding the hidden platform in each trail was counted. If the animal failed to reach the platform after 60 s, it was placed onto the platform for 10 s. Scopolamine (1 mg/kg, i.p.) was administered 30 min and drug (4.5 μmol/kg) or saline 20 min before training was conducted on each day. For each group of animals (N = 8), the mean (SE) number of errors was reported.
Statistical analysis
Results were analyzed by one-way ANOVA with Dunnett’s or Tukey’s post test as appropriate, using GraphPad Prism software version 5 (San Diego, California). For the Radial Water Maze task, the between group difference (p = 0.02) was assessed using a repeated-measure generalized linear model.
RESULTS
Anti-inflammatory activity
To compare the effect of flurbiprofen and HCT-1026 on the key target of NSAIDs, cyclooxygenase (COX), prostaglandin production was measured in RAW cells, shown previously to undergo induction of COX-2 when activated by LPS (Hagos et al. 2008). Pre-treatment with both drugs led to inhibition of LPS-induced PGD2 synthesis, as measured by LC/MS using an isotopically labeled standard (Fig. 1A). The RAW 264.7 cell line is routinely used to examine the ability of agents to inhibit cellular inflammatory response. LPS treatment reliably induces expression of iNOS, the activity of which is assessed by measuring the levels of inorganic nitrite, the major product of NO oxidative metabolism, which is readily quantified using the Griess assay. Nitrate drugs undergo metabolic denitration to directly yield NO2−, the concentration of which must be measured and subtracted to yield endogenous NO2− production. We have previously reported a complete analysis of denitration in induced and non-induced RAW cells (Hagos et al. 2008): simple aliphatic nitrates such as ISMN and HCT-1026 release only 1–3 % of the theoretical yield of NO2− in 24 h incubations. Flurbiprofen itself was observed to be a weak inhibitor of iNOS activity (IC50 > 100 μM), whereas HCT-1026 was at least 10 fold more potent (Fig. 1B). The classical mononitrate, ISMN, showed no significant activity towards inhibition of iNOS activity, and in combination with flurbiprofen was no more efficacious than flurbiprofen alone; furthermore, the combination was significantly less efficacious than the mononitrate HCT-1026 (Fig. 1C).
Fig. 1.

Anti-inflammatory activity of flurbiprofen and HCT-1026 in RAW 264.7 cell culture. (A) RAW cells were treated with different concentration of flurbiprofen (open circles; IC50 = 7.2 nM) or HCT-1026 (closed circles; IC50 = 40.6 nM) after induction of COX2 by 1 μg/mL of LPS. PGD2 was measured by LC/MS after 24 h, using deuterated PGD2 as an internal standard. Data is normalized to the amount of PGD2 released by vehicle treated, induced cells. (B) iNOS activity, as measured by Griess assay for NO2− in culture supernatants of LPS-induced RAW cells. Cells were pretreated for 30 min with 1, 10, or 100 μM of flurbiprofen (open circles) or HCT-1026 (closed circles) followed by LPS induction and measurement of NO2− production at 24 h. The data were normalized to DMSO vehicle control experiments in LPS-induced cells. (C) The anti-inflammatory activity of HCT-1026 was compared to that of a combination of its constituent parts: flurbiprofen + organic nitrate (ISMN). Cells were pretreated for 30 min with 100 μM of drugs followed by LPS induction and measurement of NO2− production at 24 h. The data were normalized to DMSO vehicle control experiments in LPS-induced cells. For all nitrate drugs, low background NO2− production (1–3 μM) was measured in non-induced cells and subtracted from the measurements made in induced RAW cells. Treated groups were compared to control group using ANOVA with Dunnett’s post test (***, P < 0.001). The data show mean and s.e.m. from at least 3 separate experiments performed using separate cell passages.
Observations on anti-inflammatory activity were extended to primary astrocyte cultures treated with LPS/IFNγ, measuring NO2− after 48 h incubation, and quantifying pro-inflammatory cytokines (Fig. 2). Both flurbiprofen and HCT-1026 were observed to possess anti-inflammatory activity in this system without significant differences in efficacy. In one previous study, both flurbiprofen and HCT-1026 were reported to inhibit peripheral iNOS expression in vivo in response to LPS, whereas in other studies the nitro-derivative was reported as considerably more potent (Mariotto et al. 1995).
Fig 2.

Anti-inflammatory activity of flurbiprofen and HCT-1026 in primary rat cortical astrocyte cultures. (A) iNOS activity as measured by Griess assay for NO2− in culture supernatants of primary astrocytes. Cells were pretreated 1 h with 100 μM flurbiprofen or HCT-1026 followed by induction by LPS (1 μg/ml)/IFN-γ (100 U/ml) and measurement of NO2− production at 48 h. (B) TNF-α & IL-6 production was measured by ELISA 48 h after induction by LPS/IFN-γ. Treated groups were compared to LPS/IFN-γ control group using ANOVA with Tukey’s post test (*, P <0.05; **, P < 0.001).
Neuroprotection in response to an inflammatory stimulus
A co-culture system was used in which N2a cell viability was assessed after co-culture for 24 h with RAW 264.7 cells. Anti-inflammatory drug treatment was initiated immediately after addition of RAW cell inserts to wells containing N2a cells. Neuronal cell death was observed in co-culture, but was significantly increased by LPS treatment (Fig. 3). Both flurbiprofen and HCT-1026 protected N2a cells in this system, providing neuroprotection in a concentration dependent manner.
Fig 3.

Neuroprotective effect of flurbiprofen and HCT-1026 on N2a cells in co-culture with RAW 264.7 cells. N2a cells were cultured on the bottom of 24 well plates, while RAW cells were cultured on inserts within the 24 well plates. Addition of 1 μg/ml LPS to the co-culture significantly increased the cytotoxicity of RAW cells towards N2a cells as measured 48 h later by the MTT assay. The co-cultures were pretreated for 30 min with 1, 10, or 100 μM of flurbiprofen or HCT-1026 followed by LPS induction (1 μg/ml). Pretreatment with flurbiprofen or HCT-1026 significantly attenuated the neurotoxicity of inflammatory factors towards N2a cells in a concentration (μM) dependent manner. Data show mean and s.d. from two separate cell passages compared to RAW/LPS: p < 0.05 for all groups (* p < 0.01) by one-way ANOVA with Tukey’s post test.
Modulation of amyloid β peptides
In N2a/APPsw cell culture, flurbiprofen was observed to function as a SALA at 100 μM, but at the same concentration, HCT-1026 was observed to function as a selective amyloid raising agent (SARA), in simile with the better studied SARA, fenofibrate, also a carboxylate ester (Abdul-Hay, 2009). Again the combination of mononitrate (ISMN) with flurbiprofen was compared to the flurbiprofen mononitrate HCT-1026, demonstrating no contribution from the aliphatic nitrate (Fig. 4A). In general, SALAs do not have high potency, which possibly explains why activity at lower concentrations is not reported. Both flurbiprofen and HCT-1026 were explored at lower concentrations, revealing that at these concentrations HCT-1026 acted as a SALA reducing levels of neurotoxic Aβ1–42 produced endogenously by N2a/APPsw cell cultures (Fig. 4B). Although ELISA kits are convenient for measuring the 40 and 42 amino acid fragments of Aβ, the use of Aβ immunoprecipitation in combination with MALDI-TOF mass spectroscopy allows quantification of multiple Aβ fragments. Using this IP-MALDI-TOF assay that quantifies relative levels of Aβ fragments standardized to exogenous Aβ1–43, the SALA activity of flurbiprofen and HCT-1026 was further explored (Fig. 4C). HCT-1026 (1 μM) was observed to reduce the levels of each Aβ fragment measured.
Fig. 4.

Effect of drug treatment on the level of Aβ produced by N2a/APPsw cells. (A) N2a/APPsw cells were incubated with ISMN, flurbiprofen (flurb), ISMN + flurbiprofen, or HCT-1026 (all drugs 100 μM). Levels of Aβ1–42 were measured 24 h later by ELISA and the data were normalized to DMSO vehicle treated control. The average level of Aβ1–42 in DMSO treated group was ~500 pg/mL. Treated groups were compared to control group using ANOVA with Dunnett’s post test (*, P < 0.05) (B) Concentration-response for relative levels of Aβ1–42 after treatment of N2a/APPsw cells with flurbiprofen (closed circles), or HCT-1026 (open circles). (C) Quantification of Aβ fragments (Aβ1–42, Aβ1–40, Aβ1–38, Aβ1–37) produced by N2a/APPsw cell treated with drugs or vehicle by IP-MALDI-TOF analysis; the y-axis is scaled relative to 100% for each Aβ fragment in the DMSO vehicle control group, to demonstrate the raising or lowering of peptide levels in response to drugs. The data show mean and s.e.m. from at least 4 separate experiments performed using separate cell passages.
Cognition enhancing effects of HCT-1026
The passive avoidance task measures long-term aversive memory (Venault et al. 1986). STPA was used to compare the procognitive activity of the organic nitrate, HCT-1026, with its component parts (flurbiprofen and the organic nitrate ISMN) in the presence of scopolamine blockade. The dose and delivery of HCT-1026 selected (1.6 mg/kg given by i.p. injection 20 min before training) was equimolar with that of the NO chimera nitrate, GT-1061, previously shown to be effective in a similar cognitive test. Similarly, equimolar doses of flurbiprofen and ISMN were studied by i.p. injection. Scopolamine induced a long term memory impairment in the STPA test determined 48 h after training which was reversed by treatment with HCT-1026 and ISMN, but not by flurbiprofen (Fig. 5). The STPA is normally viewed as testing hippocampal contributions to memory, however, the shock stimulus and aversive memory can be confounded by analgesic/anxiolytic drug actions. Although no such effects of HCT-1026 were apparent from general observation, the procognitive effects of HCT-1026 were further tested in the RWM task which tests spatial working and reference memory: HCT-1026 significantly reversed the cognitive deficit induced by scopolamine (Fig. 6).
Fig 5.
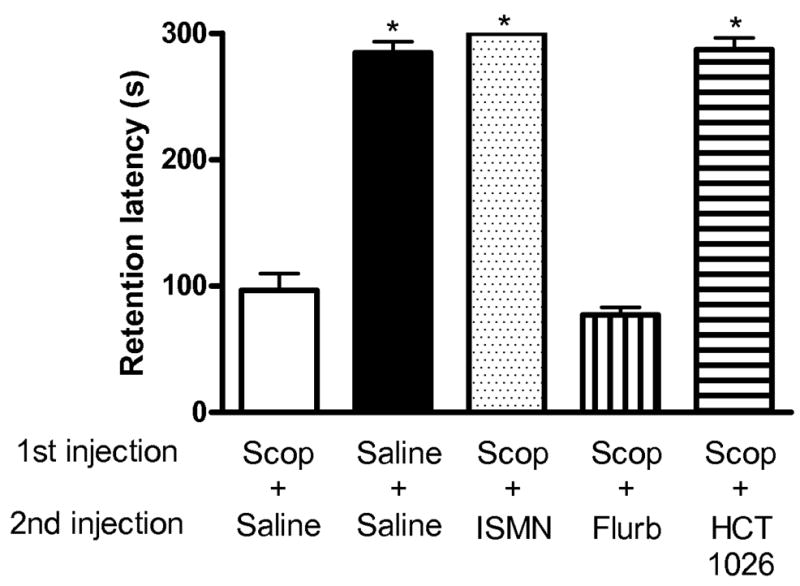
Effect of drugs on aversive memory in the STPA test. Male C57BL/6 mice received 1 mg/kg scopolamine or water by i.p. injection at t = −30 min (1st injection), 4.5 μmol/kg of drug or water i.p. at t = −20 min (2nd injection), and training was initiated at t = 0 min. Retention of aversive memory was tested 48 h later in the absence of any drugs or scopolamine. Latency to enter the dark chamber is shown at 48 h for treatment groups. Data show mean and s.e.m. (N = 8–11). Statistical significance (* p < 0.01) assessed by one-way ANOVA with Tukey post test.
Fig 6.

Effect of HCT-1026 on spatial memory in a 6-arm RWM. Male Long-Evans rats were administered scopolamine (1mg/kg, i.p.) 30 min before and HCT-1026 (open circles; 4.5 μmol/kg) or saline (crosses) 20 min before training/testing on each day: Day 1: blocks 1 & 2; Day 2: blocks 3 & 4; 4 trials within each block; 5 min inter-block interval; each trial 60 s or less). Short-term and long-term spatial memory was measured by the total number of errors made in finding the hidden platform in the RWM in each trial. For each group of animals (N = 8), the mean and s.e. are shown: the statistical significance between groups (p = 0.02) was assessed using a repeated-measure generalized linear model.
Discussion
NSAIDs, flurbiprofen and AD
Neuroinflammation is part of the innate immunity that protects the brain from harmful stimuli. Acute local inflammatory reactions may involve limited and reversible damage, however, chronic inflammatory reactions in the CNS are believed to lead to tissue damage and to contribute to neurodegeneration in diseases such as AD (Akiyama et al. 2000). The etiology of the inflammatory reaction observed in AD is still debated, but Aβ has been implicated as a stimulus to induction of inflammation (Butterfield et al. 2002). Flurbiprofen is an NSAID that inhibits COX and the activation of monocytes (Chalmers et al. 1972, Hinz et al. 2001). Flurbiprofen is also one of a subset of NSAIDs that have been reported to act as SALAs, reducing the levels of neurotoxic Aβ1–42 and the ratio of Aβ1–42/Aβ1–40; of common NSAIDs, flurbiprofen is the most efficacious in vitro (Morihara et al. 2002, Eriksen et al. 2003, Weggen et al. 2001). The SALA activity of flurbiprofen was replicated in the present study in N2a/APPsw cell culture, although in accord with other studies, the potency was not high. Low potency combined with low bioavailability (only 1% of the drug is reported to cross the blood brain barrier (Galasko et al. 2007)) provides one rationale for the failure of R-flurbiprofen in Phase 3 clinical trials reported in 2008. Alternative explanations have been offered, and some reports argue that NSAIDs are unable to modify established AD pathology (Szekely et al. 2008), or question the epidemiological evidence of efficacy (Breitner et al. 2009).
R-flurbiprofen was selected for clinical trials because being the COX inactive isomer, gastrotoxicity was attenuated; excellent safety and lowered Aβ1–42 plasma levels were reported associated with higher drug plasma levels (Galasko et al. 2007). However, these promising phase 2 trial results were followed by the subsequent failure at an 800 mg dose (bid) in a phase 3 trial (Wilcock et al. 2008). Gastric-sparing NO-flurbiprofen represents an alternative strategy to R-flurbiprofen in AD therapy.
NO-NSAIDs, NO-flurbiprofen, HCT-1026, and NCX-2216
NO-NSAIDs are NSAID prodrugs that undergo esterase mediated hydrolysis to liberate the NSAID and an organic nitrate (Bolla et al. 2005). Wallace reported the gastroprotective actions of glyceryl trinitrate in 1989 leading to the development of NO-NSAID hybrid nitrates to ameliorate the gastrotoxicity of the parent NSAID (MacNaughton et al. 1989). The NO-flurbiprofen, HCT-1026, releases 4-hydroxybutyl nitrate and flurbiprofen. The improved safety profile of this hybrid nitrate was reported 15 years ago (Wallace et al. 1994). The organic nitrate is a source of low fluxes of NO, although the clear identification of the metabolic steps by which NO-NSAIDs produce NO has not been established (Govoni et al. 2006). NO-NSAIDs have often been reported to manifest more potent or more varied biological actions than the parent NSAID. However, the activity of NO-flurbiprofen has infrequently been compared to combinations of flurbiprofen and organic nitrate; in this work, study of the clinical nitrate ISMN is included to provide comparison.
It is useful briefly to review the relevant reports on HCT-1026. Several studies have reported on the anti-inflammatory activity of flurbiprofen and HCT-1026 in vitro (Ajmone-Cat et al. 2001) and in vivo (Furlan et al. 2004). Despite the number of in vitro studies on anti-inflammatory actions of HCT-1026, the mechanisms that differentiate this drug from flurbiprofen remain a matter of debate; most recently proposed are NO-independent and NSAID-independent actions on NFκB and MAPK/ERK signaling pathways (Idris et al. 2009). There are a number of reports on the anti-inflammatory activity of HCT-1026 in LPS-induced chronic neuroinflammation (Hauss-Wegrzyniak et al. 1999b, Wenk et al. 2002); in one HCT-1026 was active, but flurbiprofen was not (Rosi et al. 2003). LPS infusion increased activated microglia in rats which was attenuated by HCT-1026 in young and adult, but not in old rats; whereas in the same study, HCT-1026 improved the resultant impaired spatial memory in young rats, but had no effect on adult or old rats (Hauss-Wegrzyniak et al. 1999a).
NCX-2216 is an NO-flurbiprofen containing the antioxidant ferulic acid that is released by esterase bioactivation in addition to flurbiprofen and 4-hydroxybutyl nitrate. NCX-2216 was reported transiently to inhibit TNFα and iNOS elevation in LPS-induced microglia and to activate peroxisome proliferator-activated receptor-γ (PPARγ) (Bernardo et al. 2006). A hybrid nitrate containing the ferulic acid and 4-hydroxybutyl nitrate moieties of NCX-2216 was reported to inhibit iNOS expression in LPS/IFNγ-induced RAW 264.7 cells (Ronchetti et al. 2006), suggesting that ferulic acid contributed to the activity of this NO-flurbiprofen.
NO-flurbiprofen and Aβ
NCX-2216 was reported to give a 40–45% reduction in immunoreactive amyloid deposits in the cerebral cortex and hippocampus of amyloid transgenic mice, twice the reduction observed after administration of ibuprofen (Jantzen et al. 2002). Increased CNS bioavailability, enhancing the inherent amyloid “production-modifying effects proposed for flurbiprofen”, was suggested as an explanation (Wilcock et al. 2007). In a separate study, HCT-1026 was reported to reduce Aβ load in a different amyloid transgenic mouse model (van Groen & Kadish 2005). Upon activation, monocytes release neurotoxic inflammatory mediators, the levels of which are elevated in the brains of AD patients (Akiyama et al. 2000). Activation of microglia and astrocytes is closely associated with the neuroinflammatory response that contributes to neurodegeneration. In an amyloid transgenic mouse, NCX-2216 was observed to activate microglia in a manner leading to clearance of amyloid, but not to neurotoxic inflammation; and in a further study to inhibit COX-2 expression and prostaglandin synthesis in the rat brain for a prolonged period compared to flurbiprofen, although NCX-2216 was not detected in brain tissue (Wallace et al. 2004). Administration of HCT-1026 to amyloid transgenic mice was reported to inhibit microglial activation surrounding plaques (van Groen & Kadish 2005), and a direct comparison of flurbiprofen, HCT-1026 and NCX-2216 in adult rats injected in the nucleus basalis with Aβ1–42, showed a reduction in inflammatory markers including iNOS (Prosperi et al. 2004). Thus, both NO-flurbiprofens have been shown to lower Aβ load in mice, but to modulate inflammatory markers differently from each other and from flurbiprofen (Gasparini et al. 2005, Gasparini et al. 2004).
NO-flurbiprofen is neuroprotective in neuronal cells
The RAW 264.7 cell line is routinely used to examine the ability of agents to block cellular inflammatory response, modeling the activity of anti-inflammatory drugs toward activated macrophages. LPS treatment induces various cytokines and expression of iNOS. In RAW 264.7 cell cultures, furbiprofen was observed to be more potent than HCT-1026 in inhibition of prostaglandin synthesis, but less potent than HCT-1026 in inhibition of iNOS induction. The mononitrate, ISMN, had no effect alone or in combination with flurbiprofen, showing that this was a property of the intact NO-flurbiprofen molecule. Observations were extended to a more relevant system, LPS/IFN-induced primary astroglial cultures, in which flurbiprofen and HCT-1026 showed comparable anti-inflammatory potency towards inhibition of cytokine and iNOS elevation, providing similar observations to those in microglial cultures (Ajmone-Cat et al. 2001).
Inflammatory processes, such as exposure to LPS, which activate glia can lead to the release of inflammatory mediators including cytokines, NO, and prostaglandins, and initiate ROS generation, the combination of which can lead to cell death (Klegeris & McGeer 2002). Although RAW 264.7 cell culture appears a rudimentary system, the co-culture of these cells with the N2a mouse neuroblastoma cell line allows assay of neuroprotection. The objective was to examine if the neuroprotective properties of HCT-1026 observed in one in vivo study (Prosperi et al. 2004), could be recapitulated in an in vitro model of potential use in drug screening. That the anti-inflammatory activity of HCT-1026 could translate into neuroprotection was demonstrated in a simple co-culture experiment of LPS-induced RAW cells with a neuroblastoma cell culture: both flurbiprofen and HCT-1026 were observed to be highly efficacious neuroprotectants.
NO-flurbiprofen lowers Aβ in neuronal cells
In contrast to anti-inflammatory studies, in vitro studies on anti-amyloid activity of HCT-1026 and NO-NSAIDs have not been reported. The stably transfected N2a/APPsw neuroblastoma cell line secretes human amyloid fragments that are readily identified and quantified by ELISA and IP-MALDI-TOF mass spectroscopy (Abdul-Hay, 2009). The IP-MALDI-TOF assay in this cell system replicated the reported SALA activity of flurbiprofen and sulindac, and the SARA activity of fenofibrate was also replicated, although the modulation of Aβ1–42 levels appeared to depend on amyloid clearance rather than production (Abdul-Hay, 2009). HCT-1026 and fenofibrate, both carboxylate esters of SALAs, elevated Aβ1–42 at 100 μM, both in N2a/APPsw cell cultures and when exogenous Aβ1–40/Aβ1–42 was added to cells (Abdul-Hay, 2009). The mononitrate, ISMN, had no effect on Aβ1–42 secretion. Most interestingly as drug concentration was lowered, HCT-1026 switched from raising levels of Aβ1–42 to manifesting SALA activity equi-efficacious with flurbiprofen.
NO-flurbiprofen reverses cholinergic cognition deficits
In many regions of the CNS, activation of glutamatergic and cholinergic muscarinic receptors, which are disrupted in AD, will lead to the activation of nitric oxide synthase (NOS) and an elevation in the level of cGMP (Bredt & Snyder 1989, Garthwaite et al. 1989, Tonnaer et al. 1991). NO-stimulated elevation of cGMP is decreased in the cerebral cortex of patients with AD (Bonkale et al. 1995), and in hippocampal slices, disruption of LTP by Aβ peptides was shown to result from attenuated NO/cGMP signaling (Puzzo et al. 2005). Impaired memory formation in rats has been reported for NOS inhibitors, which was ameliorated by administration of cGMP-phosphodiesterase (PDE5) inhibitors (Ingram et al. 1998, Devan et al. 2006). Inhibition of PDE5 also reversed a cognitive impairment induced by blockade of muscarinic cholinergic receptors with scopolamine (Devan et al. 2004). HCT-1026 was observed to reverse aversive memory deficits in the STPA task and spatial working and reference memory in the MWM task, both induced by scopolamine. In STPA, an equimolar dose of flurbiprofen was without effect, whereas the nitrate drug ISMN at an equimolar dosage reversed the cognitive deficit. Disruption of cholinergic signaling is a feature of AD and scopolamine amnesia in human and non-human subjects remains an important tool in drug discovery (Terry & Buccafusco 2003, Thomas et al. 2008).
The NO/sGC/cGMP signal transduction system is considered to be important for modulating synaptic transmission and plasticity in brain regions such as the hippocampus, cerebral cortex, and cerebellum (O’Dell et al. 1991). We have previously reported on the ability of two organic nitrate NO chimeras to activate the NO/cGMP pathway in the brain and to reverse memory impairments, including scopolamine amnesia, in a variety of behavioral models, including STPA and water maze tasks (Thatcher et al. 2004, Thatcher et al. 2005, Thatcher et al. 2006, Bennett et al. 2007, Smith et al. 2000). One of these NO chimeras showed excellent brain bioavailability, whereas the plasma and brain levels of HCT-1026 were reported to be below detection limits (Govoni et al. 2006). It is possible that the HCT-1026 metabolite, 4-hydroxybutyl nitrate, has brain bioavailaibility since animal studies have shown that the level of inorganic nitrite in the brain increases after oral administration of HCT-1026 (Prosperi et al. 2004, Prosperi et al. 2001). Plasma nitrite itself has been shown to provide a source of NO under certain conditions (Lundberg et al. 2008).
Conclusions
Flurbiprofen is an NSAID and a SALA; the combination of these properties support the therapeutic use in AD of appropriate formulations or structural derivatives with improved safety profiles. The hybrid nitrate, NO-flurbiprofen, HCT-1026, is one such structural derivative that has been previously reported to lower amyloid levels in a transgenic mouse model and modulate neuroinflammation. In this study, HCT-1026 was observed to retain the anti-inflammatory properties of flurbiprofen and these translated to neuroprotective activity in neuronal cell culture. HCT-1026 was shown for the first time to act as a SALA in cell culture, but with higher potency and a concentration-response very different from flurbiprofen. Finally, in two animal behavioral models, HCT-1026 delivered cognition enhancing activity that reversed a cholinergic blockade, whereas flurbiprofen had no effect. Only the cognition enhancing effect of HCT-1026 was replicated by a simple organic nitrate. These data add positively to the preclinical efficacy profile of HCT-1026. The good safety profile reported for HCT-1026 is ascribed to the nitrate group, as are the cognition enhancing properties. Assessment of the contribution of the nitrate group to SALA activity awaits further structure-activity studies.
Acknowledgments
Portions of this work were funded by NIH grant AG027425 and an ISOA/ELAN award from the Institute for the Study of Aging. Dr Mark Z. Wang is thanked for preparation of HCT-1026 and Tewolde S. Tewolde is acknowledged for technical assistance with cell culture. No conflicts exist.
Abbreviations
- AD
Alzheimer’s disease
- Aβ
Amyloid beta
- NSAIDs
non-steroidal anti-inflammatory drugs (NSAIDs)
- SALA
selective amyloid lowering agent
- SARA
selective amyloid raising agent
- APP
amyloid precursor protein
- IP
immunoprecipitation
- PBS
phosphate buffered saline
- DMEM
Dulbecco’s modified Eagle’s medium
- COX
cyclooxygenase
- NO
nitric oxide
- cGMP
Cyclic guanosine monophosphate
- ELISA
enzyme-linked immunosorbent assay
- APPsw
amyloid precursor protein with swedish mutation
- AEBSF
4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride
- EDTA
ethylenediaminetetraacetic acid
- MALDI
matrix-assisted laser desorption/ionization
- TOF
time of flight
- LPS
Lipopolysaccharide
- IFN
Interferon
- HPLC
High-performance liquid chromatography
- LC
liquid chromatography
- MS
Mass spectrometry
- STPA
step-through passive avoidance
- RWM
radial water maze
- NOS
nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- IL
interleukin
- sGC
soluble guanylyl cyclase
- MAPK
Mitogen-activated protein kinases
- ERK
extracellular signal-regulated kinases
- PDE
phosphodiesterase
- ANOVA
analysis of variance
References
- Abdul-Hay SO, Edirisinghe P, Thatcher GRJ. Selective modulation of amyloid β peptide degradation by flurbiprofen, fenofibrate, and related compounds regulates Aβ levels. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.06355.x. [DOI] [PubMed] [Google Scholar]
- Ajmone-Cat MA, Nicolini A, Minghetti L. Differential effects of the nonsteroidal antiinflammatory drug flurbiprofen and its nitric oxide-releasing derivative, nitroflurbiprofen, on prostaglandin E(2), interleukin-1beta, and nitric oxide synthesis by activated microglia. J Neurosci Res. 2001;66:715–722. doi: 10.1002/jnr.10038. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- Bennett BM, Reynolds JN, Prusky GT, Douglas RM, Sutherland RJ, Thatcher GR. Cognitive deficits in rats after forebrain cholinergic depletion are reversed by a novel NO mimetic nitrate ester. Neuropsychopharmacology. 2007;32:505–513. doi: 10.1038/sj.npp.1301054. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Gasparini L, Ongini E, Minghetti L. Dynamic regulation of microglial functions by the non-steroidal anti-inflammatory drug NCX 2216: implications for chronic treatments of neurodegenerative diseases. Neurobiol Dis. 2006;22:25–32. doi: 10.1016/j.nbd.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Feinstein DL, Shen Q, Bhat AN. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells. Roles of nuclear factors, nuclear factor kappa B, cAMP response element-binding protein, CCAAT/enhancer-binding protein-beta, and activating transcription factor-2. J Biol Chem. 2002;277:29584–29592. doi: 10.1074/jbc.M204994200. [DOI] [PubMed] [Google Scholar]
- Bolla M, Almirante N, Benedini F. Therapeutic potential of nitrate esters of commonly used drugs. Curr Top Med Chem. 2005;5:707–720. doi: 10.2174/1568026054679335. [DOI] [PubMed] [Google Scholar]
- Bonkale WL, Winblad B, Ravid R, Cowburn RF. Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer’s disease. Neurosci Lett. 1995;187:5–8. doi: 10.1016/0304-3940(95)11323-o. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci U S A. 1989;86:9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009;72:1899–1905. doi: 10.1212/WNL.0b013e3181a18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Griffin S, Munch G, Pasinetti GM. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer’s disease brain exists. J Alzheimers Dis. 2002;4:193–201. doi: 10.3233/jad-2002-4309. [DOI] [PubMed] [Google Scholar]
- Chalmers IM, Cathcart BJ, Kumar EB, Dick WC, Buchanan WW. Clinico-pharmacological studies and clinical evaluation of flurbiprofen. A new non-steroidal antirheumatic agent. Ann Rheum Dis. 1972;31:319–324. doi: 10.1136/ard.31.4.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- Devan BD, Bowker JL, Duffy KB, Bharati IS, Jimenez M, Sierra-Mercado D, Jr, Nelson CM, Spangler EL, Ingram DK. Phosphodiesterase inhibition by sildenafil citrate attenuates a maze learning impairment in rats induced by nitric oxide synthase inhibition. Psychopharmacology (Berl) 2006;183:439–445. doi: 10.1007/s00213-005-0232-z. [DOI] [PubMed] [Google Scholar]
- Devan BD, Sierra-Mercado D, Jr, Jimenez M, Bowker JL, Duffy KB, Spangler EL, Ingram DK. Phosphodiesterase inhibition by sildenafil citrate attenuates the learning impairment induced by blockade of cholinergic muscarinic receptors in rats. Pharmacol Biochem Behav. 2004;79:691–699. doi: 10.1016/j.pbb.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan R, Kurne A, Bergami A, et al. A nitric oxide releasing derivative of flurbiprofen inhibits experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;150:10–19. doi: 10.1016/j.jneuroim.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Galasko DR, Graff-Radford N, May S, et al. Safety, tolerability, pharmacokinetics, and Abeta levels after short-term administration of R-flurbiprofen in healthy elderly individuals. Alzheimer Dis Assoc Disord. 2007;21:292–299. doi: 10.1097/WAD.0b013e31815d1048. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Garthwaite G, Palmer RM, Moncada S. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur J Pharmacol. 1989;172:413–416. doi: 10.1016/0922-4106(89)90023-0. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Ongini E, Wenk G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J Neurochem. 2004;91:521–536. doi: 10.1111/j.1471-4159.2004.02743.x. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Ongini E, Wilcock D, Morgan D. Activity of flurbiprofen and chemically related anti-inflammatory drugs in models of Alzheimer’s disease. Brain Res Brain Res Rev. 2005;48:400–408. doi: 10.1016/j.brainresrev.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Govoni M, Casagrande S, Maucci R, Chiroli V, Tocchetti P. In vitro metabolism of (nitrooxy)butyl ester nitric oxide-releasing compounds: comparison with glyceryl trinitrate. J Pharmacol Exp Ther. 2006;317:752–761. doi: 10.1124/jpet.105.097469. [DOI] [PubMed] [Google Scholar]
- Hagos GK, Abdul-Hay SO, Sohn J, Edirisinghe PD, Chandrasena RE, Wang Z, Li Q, Thatcher GR. Anti-inflammatory, antiproliferative, and cytoprotective activity of NO chimera nitrates of use in cancer chemoprevention. Mol Pharmacol. 2008;74:1381–1391. doi: 10.1124/mol.108.046664. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (USA) 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Vraniak P, Wenk GL. The effects of a novel NSAID on chronic neuroinflammation are age dependent. Neurobiol Aging. 1999a;20:305–313. doi: 10.1016/s0197-4580(99)00028-7. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Willard LB, Del Soldato P, Pepeu G, Wenk GL. Peripheral administration of novel anti-inflammatories can attenuate the effects of chronic inflammation within the CNS. Brain Res. 1999b;815:36–43. doi: 10.1016/s0006-8993(98)01081-6. [DOI] [PubMed] [Google Scholar]
- Hinz B, Brune K, Rau T, Pahl A. Flurbiprofen enantiomers inhibit inducible nitric oxide synthase expression in RAW 264.7 macrophages. Pharm Res. 2001;18:151–156. doi: 10.1023/a:1011020132140. [DOI] [PubMed] [Google Scholar]
- Idris AI, Ralston SH, van’t Hof RJ. The nitrosylated flurbiprofen derivative HCT1026 inhibits cytokine-induced signalling through a novel mechanism of action. Eur J Pharmacol. 2009;602:215–222. doi: 10.1016/j.ejphar.2008.11.023. [DOI] [PubMed] [Google Scholar]
- in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Ingram DK, Spangler EL, Meyer RC, London ED. Learning in a 14-unit T-maze is impaired in rats following systemic treatment with N omega-nitro-L-arginine. Eur J Pharmacol. 1998;341:1–9. doi: 10.1016/s0014-2999(97)01426-x. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klegeris A, McGeer PL. Cyclooxygenase and 5-lipoxygenase inhibitors protect against mononuclear phagocyte neurotoxicity. Neurobiol Aging. 2002;23:787–794. doi: 10.1016/s0197-4580(02)00021-0. [DOI] [PubMed] [Google Scholar]
- Kowalska MA, Badellino K. beta-Amyloid protein induces platelet aggregation and supports platelet adhesion. Biochem Biophys Res Commun. 1994;205:1829–1835. doi: 10.1006/bbrc.1994.2883. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- MacNaughton WK, Cirino G, Wallace JL. Endothelium-derived relaxing factor (nitric oxide) has protective actions in the stomach. Life Sci. 1989;45:1869–1876. doi: 10.1016/0024-3205(89)90540-7. [DOI] [PubMed] [Google Scholar]
- Mariotto S, Menegazzi M, Carcereri de Prati A, Cuzzolin L, Adami A, Suzuki H, Benoni G. Protective effect of NO on gastric lesions and inhibition of expression of gastric inducible NOS by flurbiprofen and its nitro-derivative, nitroflurbiprofen. Br J Pharmacol. 1995;116:1713–1714. doi: 10.1111/j.1476-5381.1995.tb16650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E. Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol. 1995;10:509–519. [PubMed] [Google Scholar]
- Mattson MP, Guo ZH, Geiger JD. Secreted form of amyloid precursor protein enhances basal glucose and glutamate transport and protects against oxidative impairment of glucose and glutamate transport in synaptosomes by a cyclic GMP- mediated mechanism. J Neurochem. 1999;73:532–537. doi: 10.1046/j.1471-4159.1999.0730532.x. [DOI] [PubMed] [Google Scholar]
- Morihara T, Chu T, Ubeda O, Beech W, Cole GM. Selective inhibition of Abeta42 production by NSAID R-enantiomers. J Neurochem. 2002;83:1009–1012. doi: 10.1046/j.1471-4159.2002.01195.x. [DOI] [PubMed] [Google Scholar]
- O’Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci U S A. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosperi C, Scali C, Barba M, Bellucci A, Giovannini MG, Pepeu G, Casamenti F. Comparison between flurbiprofen and its nitric oxide-releasing derivatives HCT-1026 and NCX-2216 on Abeta(1–42)-induced brain inflammation and neuronal damage in the rat. Int J Immunopathol Pharmacol. 2004;17:317–330. doi: 10.1177/039463200401700312. [DOI] [PubMed] [Google Scholar]
- Prosperi C, Scali C, Pepeu G, Casamenti F. NO-flurbiprofen attenuates excitotoxin-induced brain inflammation, and releases nitric oxide in the brain. Jpn J Pharmacol. 2001;86:230–235. doi: 10.1254/jjp.86.230. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci. 2005;25:6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- Ronchetti D, Impagnatiello F, Guzzetta M, Gasparini L, Borgatti M, Gambari R, Ongini E. Modulation of iNOS expression by a nitric oxide-releasing derivative of the natural antioxidant ferulic acid in activated RAW 264.7 macrophages. Eur J Pharmacol. 2006;532:162–169. doi: 10.1016/j.ejphar.2005.12.034. [DOI] [PubMed] [Google Scholar]
- Rosi S, McGann K, Hauss-Wegrzyniak B, Wenk GL. The influence of brain inflammation upon neuronal adenosine A2B receptors. J Neurochem. 2003;86:220–227. doi: 10.1046/j.1471-4159.2003.01825.x. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-b attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature (London) 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Smith S, Dringenberg HC, Bennett BM, Thatcher GRJ, Reynolds JN. A novel nitrate ester reverses the cognitive impairment caused by scopolamine in the Morris water maze. Neuroreport. 2000;11:3883–3886. doi: 10.1097/00001756-200011270-00055. [DOI] [PubMed] [Google Scholar]
- Somasundaram S, Rafi S, Jacob M, et al. Intestinal tolerability of nitroxybutyl-flurbiprofen in rats. Gut. 1997;40:608–613. doi: 10.1136/gut.40.5.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Green RC, Breitner JC, et al. No advantage of A beta 42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology. 2008;70:2291–2298. doi: 10.1212/01.wnl.0000313933.17796.f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry AV, Jr, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. 2003;306:821–827. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- Thatcher GRJ, Bennett BM, Dringenberg HC, Reynolds JN. Novel nitrates as NO mimetics directed at Alzheimer’s disease. J Alzheimers Dis. 2004;6:S75–84. doi: 10.3233/jad-2004-6s614. [DOI] [PubMed] [Google Scholar]
- Thatcher GRJ, Bennett BM, Reynolds JN. Nitric oxide mimetic molecules as therapeutic agents in Alzheimer’s disease. Curr Alzheimer Res. 2005;2:171–182. doi: 10.2174/1567205053585945. [DOI] [PubMed] [Google Scholar]
- Thatcher GRJ, Bennett BM, Reynolds JN. NO chimeras as therapeutic agents in Alzheimer’s disease. Curr Alzheimer Res. 2006;3:237–245. doi: 10.2174/156720506777632925. [DOI] [PubMed] [Google Scholar]
- Thomas E, Snyder PJ, Pietrzak RH, Jackson CE, Bednar M, Maruff P. Specific impairments in visuospatial working and short-term memory following low-dose scopolamine challenge in healthy older adults. Neuropsychologia. 2008;46:2476–2484. doi: 10.1016/j.neuropsychologia.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Ting KK, Brew B, Guillemin G. The involvement of astrocytes and kynurenine pathway in Alzheimer’s disease. Neurotox Res. 2007;12:247–262. doi: 10.1007/BF03033908. [DOI] [PubMed] [Google Scholar]
- Tonnaer JA, Cheung CL, De Boer T. cGMP formation and phosphoinositide turnover in rat brain slices are mediated by pharmacologically distinct muscarinic acetylcholine receptors. Eur J Pharmacol. 1991;207:183–188. doi: 10.1016/0922-4106(91)90029-h. [DOI] [PubMed] [Google Scholar]
- van Groen T, Kadish I. Transgenic AD model mice, effects of potential anti-AD treatments on inflammation and pathology. Brain Res Brain Res Rev. 2005;48:370–378. doi: 10.1016/j.brainresrev.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- Venault P, Chapouthier G, de Carvalho LP, Simiand J, Morre M, Dodd RH, Rossier J. Benzodiazepine impairs and beta-carboline enhances performance in learning and memory tasks. Nature. 1986;321:864–866. doi: 10.1038/321864a0. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Muscara MN, de Nucci G, Zamuner S, Cirino G, del Soldato P, Ongini E. Gastric tolerability and prolonged prostaglandin inhibition in the brain with a nitric oxide-releasing flurbiprofen derivative, NCX-2216 [3-[4-(2-fluoro-alpha-methyl-[1,1′-biphenyl]-4-acetyloxy)-3-methoxyphenyl]-2-propenoic acid 4-nitrooxy butyl ester] J Pharmacol Exp Ther. 2004;309:626–633. doi: 10.1124/jpet.103.063453. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Reuter B, Cicala C, McKnight W, Grisham MB, Cirino G. Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology. 1994;107:173–179. doi: 10.1016/0016-5085(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Rosi S, McGann K, Hauss-Wegrzyniak B. A nitric oxide-donating flurbiprofen derivative reduces neuroinflammation without interacting with galantamine in the rat. Eur J Pharmacol. 2002;453:319–324. doi: 10.1016/s0014-2999(02)02387-7. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–960. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: a randomised phase II trial. Lancet Neurol. 2008;7:483–493. doi: 10.1016/S1474-4422(08)70090-5. [DOI] [PubMed] [Google Scholar]
- Yang P, Felix E, Madden T, Fischer SM, Newman RA. Quantitative high-performance liquid chromatography/electrospray ionization tandem mass spectrometric analysis of 2- and 3-series prostaglandins in cultured tumor cells. Anal Biochem. 2002;308:168–177. doi: 10.1016/s0003-2697(02)00218-x. [DOI] [PubMed] [Google Scholar]