

Abstract
Cisplatin, a widely used chemotherapy drug, induces acute kidney injury, which limits its use and efficacy in cancer treatment. However, the molecular mechanism of cisplatin-induced nephrotoxicity is currently unclear. Using pharmacological and gene knockout models, we now demonstrate a pathological role for p53 in cisplatin nephrotoxicity. In C57BL/6 mice, cisplatin treatment induced p53 phosphorylation and protein accumulation, which was accompanied by the development of acute kidney injury. p53 was induced in both proximal and distal tubular cells and partially colocalized with apoptosis. Pifithrin-α, a pharmacological inhibitor of p53, suppressed p53 activation and ameliorated kidney injury during cisplatin treatment. Moreover, cisplatin-induced nephrotoxicity was abrogated in p53-deficient mice. Compared with wild-type animals, p53-deficient mice showed a better renal function, less tissue damage, and fewer apoptotic cells. In addition, cisplatin induced less apoptosis in proximal tubular cells isolated from p53-deficient mice than the cells from wild-type animals. Together these results suggest the involvement of p53 in cisplatin-induced renal cell apoptosis and nephrotoxicity.
Keywords: cisplatin, nephrotoxicity, p53, apoptosis, acute kidney injury
Cisplatin is one of the most widely used and most potent chemotherapeutic agents (5, 33, 39). It is being used for the treatment of cancers of the testis, ovary, head, neck, lung, and many other origins. It is particularly effective in treating testicular and ovarian cancer, with an impressive cure rate (5, 33, 39). However, the use of cisplatin is limited by its side effects in normal tissues, particularly injury to the kidneys (2, 7, 35). Following cisplatin treatment, over 30% of patients develop renal problems. The nephrotoxicity of cisplatin is indicated by tubular cell death, tissue damage, and the loss of renal function or acute renal failure (1, 3, 9, 16, 19–21, 23, 24, 26, 28, 31, 32, 37, 41, 44, 45).
Although tubular cell death is recognized as a major determinant of cisplatin nephrotoxicity, how these cells are terminally injured under the pathological condition is unclear. In vivo in animals, cisplatin induces both necrosis and apoptosis in renal tubular cells (9, 19, 21, 23, 27, 37, 41). In vitro in cultured tubular cells, whether apoptosis or/and necrosis is induced depends on the concentrations of cisplatin (20). Using these experimental models, recent studies have demonstrated the activation of multiple signaling pathways during cisplatin-induced tubular cell injury and nephrotoxicity (1, 3, 16, 19–21, 23, 24, 26, 28, 31, 32, 37, 41, 44, 45).
One of the cell killing pathways that are activated by cisplatin may involve p53 (5, 33, 39). p53, as a tumor suppressor protein, can induce apoptosis by transcription of proapoptotic genes or by direct interaction and activation of existing proapoptotic molecules (22, 25, 38). In cultured renal tubular cells, p53 is activated by cisplatin (6, 11–14, 30). Importantly, cisplatin-induced apoptosis is inhibited by pifithrin-α (a pharmacological inhibitor of p53) and also by a dominant negative mutant of p53 (6, 14). Mechanistically, p53 may upregulate the proapoptotic genes such as p53-upregulated modulator of apoptosis-α, leading to mitochondrial outer membrane permeabilization and the release of apoptogenic factors, including cytochrome c and apoptosis-inducing factor (11, 13, 30). Together, these studies have suggested the involvement of p53 and related signaling pathway in cisplatin-induced tubular cell apoptosis. Despite these findings, it is unclear whether and to what extents p53 contributes to cisplatin nephrotoxicity in vivo. Therefore, the current study was designed to: 1) analyze p53 activation during cisplatin nephrotoxicity in C57BL/6 mice; 2) determine the effects of pifithrin-α in these animals; and 3) compare cisplatin-induced nephrotoxicity in wild-type and p53-deficient mice.
Materials and Methods
Animals
p53-deficient (C57BL/6J-Trp53tm1Tyj) and wild-type C57BL/6 mice were originally from the Jackson Laboratory (Bar Harbor, ME). The animals were maintained under a 12:12-h light-dark cycle with free access to food and water in the animal facility of the Veterans Affairs Medical Center at Augusta, GA. All animal protocols were approved by The Institutional Animal Care and Use Committees of The Medical College of Georgia and The VA Medical Center at Augusta. For the p53-deficient mouse colonies, genomic DNA samples were extracted from the tail for PCR-based genotyping. Male mice of 8–10 wk were used in this study. To compare p53-deficient and p53-proficient mice, both littermates and animals from separate colonies were used, and the results were similar and thus calculated together.
Cisplatin treatment of animals
In the majority of experiments, cisplatin was freshly prepared in saline at 1 mg/ml and injected intraperitoneally in mice at a dose of 30 mg/kg as previously described (8, 41, 42); control animals were injected with a comparable volume of saline. In the experiment to test the effects of pifithrin-α, pifithrin-α was dissolved in dimethyl sulfoxide (DMSO) and then mixed with cisplatin solution for intraperitoneal injection; control animals were injected with saline containing DMSO, and the cisplatin-only group was injected saline containing DMSO and cisplatin. The dose of pifithrin-α (2.2 mg/kg) was chosen accordingly to a previous study (18).
Renal function and histology
Serum creatinine and blood urine nitrogen (BUN) were determined to monitor renal function as previously described (41–43). For histology, kidneys were fixed with 4% paraformaldehyde and stained with hematoxylin and eosin. Tissues damage was indicated by tubular lysis, dilation, disruption, and cast formation. The degree of tissue damage was scored based on the percentage of damaged tubules as previously described (41–43): 0: no damage; 1: <25%; 2: 25–50%; 3: 50–75%; 4: >75%.
TdT-UTP nick end labeling assay
Apoptosis in renal tissues was determined by TdT-UTP nick end labeling (TUNEL) assay using the in situ Cell Death Detection kit (Roche Applied Science, Indianapolis, IN) as previously described (41–43). Briefly, renal tissues were fixed with 4% paraformaldehyde and paraffin embedded. Tissue sections of 4 μm were exposed to a TUNEL reaction mixture containing terminal deoxynucleotidyl transferase and nucleotides, including fluorescein isothiocyanate (FITC)-labeled dUTP. The slides were examined by fluorescence microscopy.
Immunofluorescence staining
The immunofluorescence staining of kidney tissue was conducted as described in our recent work (41). Briefly, freshly frozen renal tissues were fixed with 4% paraformaldehyde, permeabilized in 1% Triton X-100, and then incubated with a blocking buffer. Subsequently, the samples were incubated with the primary antibodies [anti-p53 or anti-phospho-p53 (Cell Signaling, Danvers, MA); anti-active caspase-3 (Idun Pharmaceuticals, La Jolla, CA)]. Finally, the slides were exposed to Cy3 or FITC-labeled secondary antibody (Chemicon, Temecula, CA). For double staining, the tissue sections were first stained for p53 immunofluorescence and then incubated with the TUNEL reaction mixture or FITC-labeled phaseolus vulgaris agglutinin (PHA) or peanut agglutinin (PNA) (Vector Laboratories, Burlingame, CA) as described recently (41). The staining was examined by fluorescence and confocal microscopy.
Primary proximal tubular cell culture
Proximal tubular cells were isolated from mice for primary culture as described recently (41). Briefly, renal cortical tissues were minced thoroughly and digested with 0.75 mg/ml collagenase 4 (Worthington, Lakewood, NJ). Proximal tubular cells were then purified by centrifugation at 2,000 g for 10 min in DMEM/F-12 medium with 32% Percoll (Amersham, Piscataway, NJ). After washes, the cells were plated in collagen-coated dishes and cultured in DMEM/F-12 medium supplemented with 5 μg/ml transferrin, 5 μg/ml insulin, 0.05 μM hydrocortisone, 50 μM vitamin c.
Cisplatin treatment of primary proximal tubular cells and morphological analysis
After 5–7 days of growth, the isolated proximal tubular cells were replated at 0.3 × 106/35-mm dish. For experimental treatment, the cells were incubated with 30 μM cisplatin in fresh culture medium. To examine apoptosis, the cells were stained with Hoechst 33342 to monitor apoptotic morphology by phase-contrast and fluorescent microscopy. The typical apoptotic morphology examined included cellular shrinkage, formation of apoptotic bodies, nuclear condensation, and fragmentation. To examine long-term survival, cells after 48 h of cisplatin treatment were returned to fresh medium without cisplatin and cultured for another 5 days.
Statistics
Data obtained from indicated numbers of animals or separate cell culture experiments were expressed as means ± SD. Statistical differences between two groups were determined by Student's t-test with Microsoft Excel 2000. P < 0.05 was considered significantly different.
Results
Phosphorylation and nuclear accumulation of p53 during cisplatin nephrotoxicity. Nephrotoxicity was induced by a single injection of cisplatin in C57BL/6 mice. As shown in Fig. 1A, three days after cisplatin administration, BUN increased from 60 to 192 mg/dl, indicative of a severe loss of renal function. Immunoblot analysis showed an increase of total p53 protein in kidney tissue lysates after cisplatin treatment (Fig. 1B). The increase started from day 2 and intensified at day 3. In parallel to p53 accumulation, there was p53 phosphorylation (Fig. 1C). In immunofluorescence analysis, p53 accumulation and phosphorylation were shown mainly in renal cortical cell nucleus (Fig. 1C). The results suggest that p53 is phosphorylated, stabilized, and accumulated to nucleus during cisplatin nephrotoxicity.
Fig. 1.
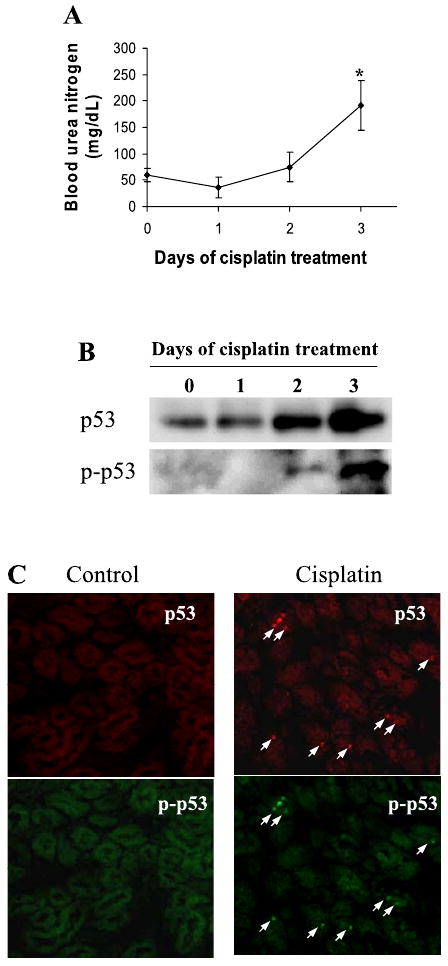
p53 phosphorylation and accumulation during cisplatin nephrotoxicity. Male C57BL/6 mice were injected with a single dose (30 mg/kg ip) of cisplatin. A: blood urea nitrogen. Blood samples were collected at indicated time points to determine the levels of blood urea nitrogen. Data are expressed as means ± SD; n ≥ 8 mice. *Significantly different from day 0. B: total p53 and phosphorylated p53. Kidney tissues were collected at indicated time points for immunoprecipitation of total p53. The resultant immunoprecipitates were analyzed for total p53 and phosphorylated p53 by immunoblots using specific antibodies. C: immunofluorescence of p53 and phospho-p53 (Ser15). Renal cortical tissues were collected from control and 3-day cisplatin-treated animals for immunofluorescence using specific antibodies. Arrows: positive staining of total and phosphorylated p53.
Localization of p53 activation in renal tubular cells during cisplatin nephrotoxicity
Immunofluorescence staining showed p53 induction in the cells of renal cortex and outer medulla, but not in the inner medulla (data not shown). In addition, p53 was not induced in glomeruli (data not shown). To further identify the cell types that induced p53, we determined whether p53 was induced in proximal tubules or distal tubules in renal cortex. To this end, we costained p53 with either FITC-labeled PHA or PNA, lectins that specifically bind proximal and distal tubular cells, respectively (4, 34, 41). As shown in Fig. 2A, the majority of p53-positive cells were localized in PHA-stained tubules, although occasionally there were cells showing p53 staining in PNA-stained tubules. Cell counting indicated that >80% of p53-positive cells were in PHA-stained tubules and about 20% in PNA-stained tubules (Fig. 2B). It is suggested that, although p53 is induced in both proximal and distal tubular cells during cisplatin nephrotoxicity, proximal tubules are the main site for p53 induction.
Fig. 2.
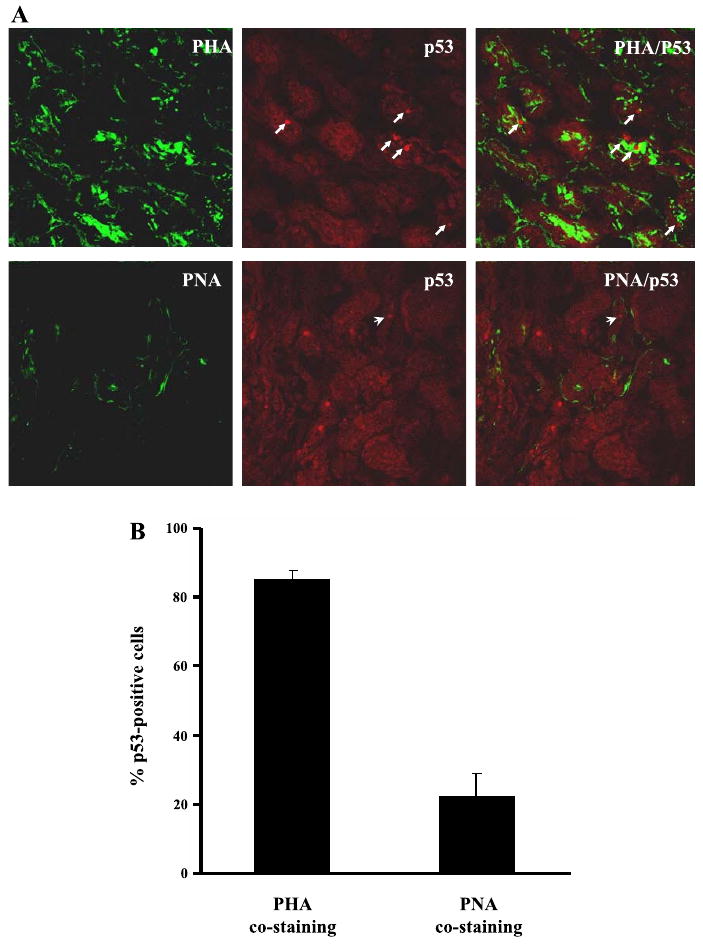
Localization of p53 in renal tubules during cisplatin nephrotoxicity. Male renal cortical tissues were collected 3 days after cisplatin injection. A: tissues were fixed for immunofluorescence staining of p53, followed by costaining with fluorescein isothiocyanate (FITC)-labeled phaseolus vulgaris agglutinin (PHA; proximal tubule marker) or peanut agglutinin (PNA; distal tubule marker). Arrows: p53 staining in PHA-positive tubules. Arrowhead: p53 staining in PNA-positive tubules. B: percentages of p53 positive cells in PHA-stained and PNA-stained tubules were determined by cell counting. Data are expressed as mean ± SD (n = 4).
Colocalization of p53 induction with tubular cell apoptosis
Using in vitro models of cultured cells, previous studies have suggested the involvement of p53 in tubular cell apoptosis during cisplatin treatment (6, 11–14, 30). To determine the relationship between p53 induction and tubular cell apoptosis in vivo, we examined p53 and apoptosis in the same tissues by immunofluorescence and TUNEL assay (Fig. 3A). After cisplatin injection (3 days), significant amounts of apoptotic cells were detected in renal cortical and outer medullary tissues (Fig. 3A). Although many of the apoptotic cells showed p53 induction, others did not. Quantitative analysis by cell counting indicated that about 50% apoptotic cells (TUNEL positive) had p53 induction (Fig. 3B).
Fig. 3.
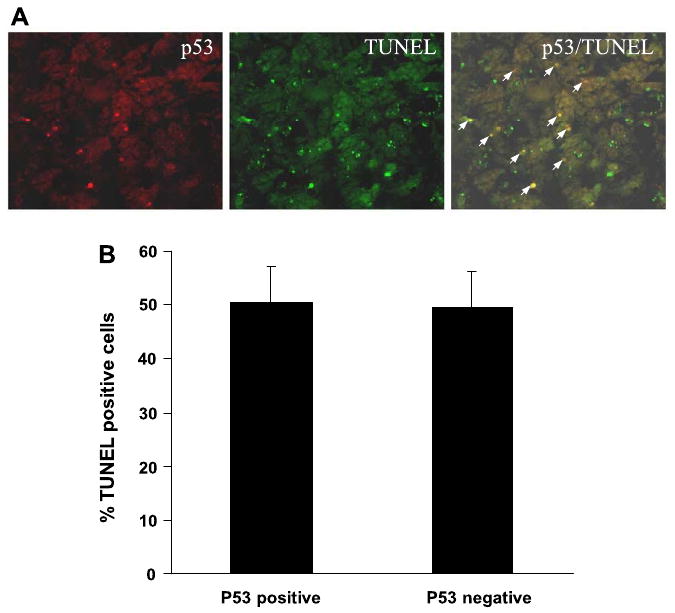
Colocalization of p53 with apoptosis in cisplatin-treated renal cortical tissues. Renal cortical tissues were collected 3 days after cisplatin injection and subjected to p53 immunofluorescence and TdT-UTP nick end labeling (TUNEL) staining. A: representative image of p53 and TUNEL staining. Arrows: colocalization of p53 and TUNEL staining. B: percentages of TUNEL positive cells with or without p53 accumulation were determined by cell counting. Data are expressed as means ± SD (n = 4).
Suppression of cisplatin nephrotoxicity by pifithrin-α
Pifithrin-α is a pharmacological inhibitor of p53 (18). To determine the role of p53 in cisplatin nephrotoxicity in vivo, we initially examined the effects of pifithrin-α in C57BL/6 mice. As shown in Fig. 4A, cisplatin-induced p53 accumulation and phosphorylation in kidney tissues were suppressed by coadministration of pifithrin-α. Importantly, the development of acute renal failure during cisplatin treatment was ameliorated by pifithrin-α. BUN was decreased from 135 mg/dl in cisplatin-treated animals to 70 mg/dl in the cisplatin+pifithrin-α treated group (Fig. 5A). Consistently, serum creatinine was reduced from 1.7 to 0.9 mg/dl by pifithrin-α during cisplatin treatment (Fig. 5B). Of note, compared with the results shown in Fig. 1, BUN and serum creatinine increases in cisplatin-treated animals were lower in this experiment. This was likely because of the inclusion during cisplatin injection of DMSO, the control vehicle solution for pifithrin-α in this experiment. Renal protective effects of DMSO were shown previously in a nephrotoxic model of mercuric chloride (15).
Fig. 4.
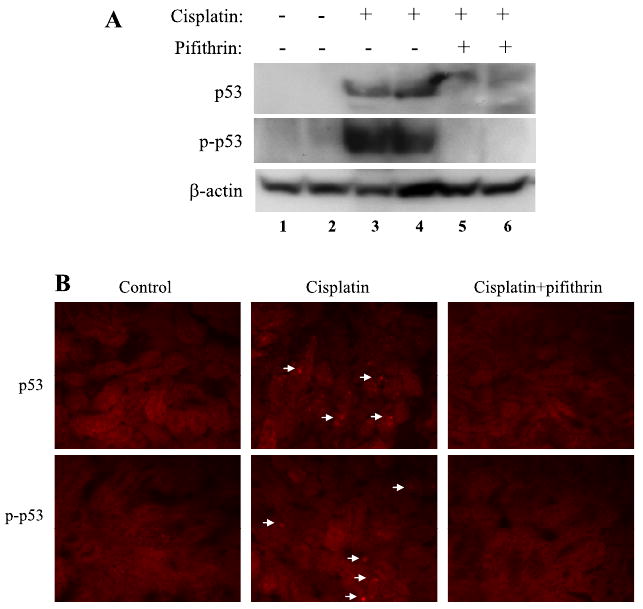
Suppression of p53 activation during cisplatin treatment by pifithrin-α. C57BL/6 mice were treated for 3 days with cisplatin only or cisplatin together with 2.2 mg/kg pifithrin-α to collect renal tissues. A: immunoblot analysis of p53 and phospho-p53 (Ser15) in renal tissue lysates. B: immunofluorescence of p53 and phospho-p53 (Ser15) in renal cortex. Arrows: cells with p53 and phospho-p53 induction. The results are representatives of 4 separate experiments.
Fig. 5.

Effects of pifithrin-α on cisplatin-induced acute renal failure. C57BL/6 mice were injected with saline as control, cisplatin, or cisplatin and 2.2 mg/kg pifithrin-α. Blood samples were collected 3 days later for measurements of blood urea nitrogen (A) and serum creatinine (B). Data are expressed as means ± SD; n = 7 for A and n ≥ 5 for B. *Statistically significant difference from the control group; #statistically significant difference from the cisplatin only group.
At the histological level, cisplatin induced obvious tissue damage in the kidneys, particularly in renal cortex and outer medulla (Fig. 6). Coadministration of pifithrin-α afforded significant protective effects against the tissue damage. Pifithrin-α also attenuated tubular cell apoptosis during cisplatin treatment (Fig. 6A). The protective effects of pifithrin-α were further indicated by semiquantitative evaluation of tissue pathology (Fig. 6B) and counting of apoptotic cells (Fig. 6C). Together these pharmacological results support a role of p53 in cisplatin nephrotoxicity in vivo.
Fig. 6.
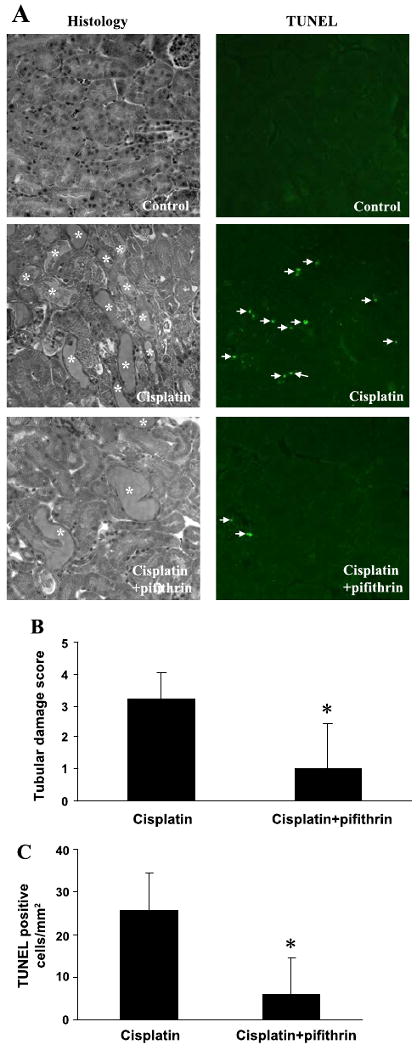
Effects of pifithrin-α on cisplatin-induced renal tissue damage and apoptosis. C57BL/6 mice were injected with saline (control), cisplatin, or cisplatin and 2.2 mg/kg pifithrin-α. Renal cortical tissues were collected 3 days later for analysis of histology by hematoxylin and eosin (H & E) staining and apoptosis by TUNEL assay. A: representative images of H & E staining and TUNEL assay. *Severely damaged tubules; arrows: TUNEL positive cells. B: pathological score of tissue damage. C: nos. of TUNEL positive cells/ mm2 cortical tissue. Data are expressed as means ± SD; n = 4. *Statistically significant difference from the cisplatin-only group.
Amelioration of cisplatin nephrotoxicity in p53-deficient mice
To further examine the role played by p53 in cisplatin nephrotoxicity, we tested a p53 knockout mouse model. p53 deficiency in these animals was confirmed by PCR-based genotyping (data not shown). In addition, cisplatin induced p53 in kidney tissues of wild-type but not the gene knockout animals (Fig. 7A). With this information, we first compared wild-type and p53-deficient mice for their renal function. Without treatment, renal function in these two genotypes of animals was similar, BUN around 50 mg/dl and serum creatinine around 0.4 mg/dl (Fig. 7, B and C). After cisplatin injection (3 days), wild-type animals developed severe renal failure, showing a BUN value of 190 mg/dl and serum creatinine level of 2.1 mg/dl (Fig. 7, B and C). In the same experiments, p53-deficient mice showed an average BUN value of 135 mg/dl and serum creatinine of 1.0 mg/dl (Fig. 7, B and C).
Fig. 7.
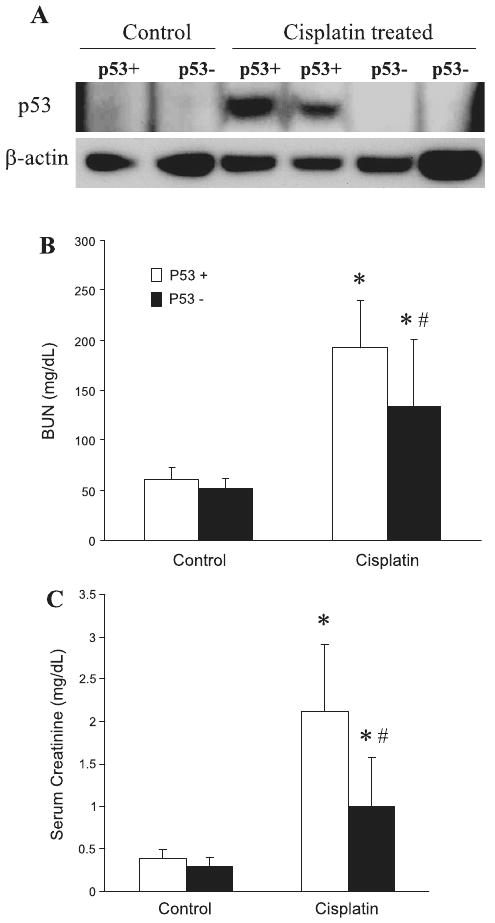
Renal function of wild-type and p53-deficient mice following cisplatin treatment. Wild-type (p53+) and p53-deficient (p53−) mice were injected with 30 mg/kg cisplatin. Renal tissues and blood samples were collected 3 days later for analysis. A: immunoblots of p53 and β-actin to show p53 induction by cisplatin in wild-type but not p53-deficient mice. B: blood urea nitrogen. C: serum creatinine. Data are expressed as means ± SD; n ≥ 29 for B and n ≥ 22 for C. *Statistically significant difference from control; #statistically significant difference from cisplatin-treated wild-type group.
We further examined the histology of renal tissues collected from representative animals. Under control conditions, both wild-type and p53-deficient kidneys showed a normal healthy histology (shown in Fig. 8 only for wild type). Cisplatin treatment induced severe tubular damage in wild-type mice (Fig. 8, top), which was partially suppressed in p53-deficient animals (Fig. 8, top). Further examination of the tissues by TUNEL assay indicated that cisplatin-induced apoptosis was markedly attenuated in p53-deficient animals (Fig. 8, middle). Consistently, caspase activation shown by active caspase-3 immunofluorescence was also lower in p53-deficient animals (Fig. 8, bottom).
Fig. 8.
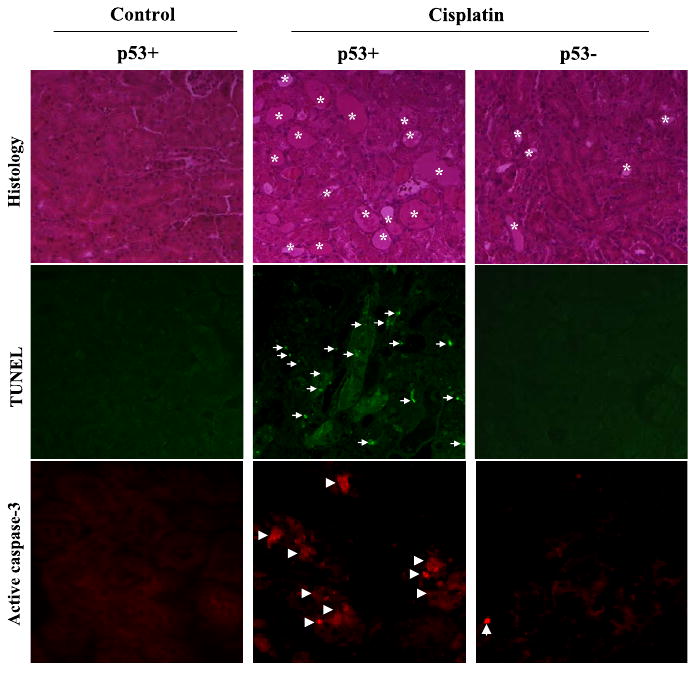
Renal tissue damage, apoptosis, and caspase activation in wild-type and p53-deficient mice following cisplatin treatment. Wild-type (p53+) and p53-deficient (p53−) mice were injected with 30 mg/kg cisplatin to collect renal cortical tissues 3 days later to analyze renal histology by H & E staining (top), apoptosis by TUNEL assay (middle), and caspase activation by immunofluorescence of active caspase-3 (bottom). *Severely damaged tubules; arrows: TUNEL-positive cells; arrowheads: active caspase-3 staining.
Proximal tubular cells isolated from p53-deficient mice are resistant to cisplatin-induced apoptosis. We hypothesized that the lower sensitivity of p53-deficient mice to cisplatin nephrotoxicity was largely because of p53 deficiency in proximal tubular cells. To test this possibility, we isolated proximal tubular cells from wild-type (p53+) and p53-deficient (p53−) mice. Primary cultures of the isolated cells were then treated with 30 μM cisplatin. Representative cell morphology was shown in Fig. 9A. Without treatment, there was a low basal level of apoptosis in both p53+ and p53− cells. After 24 h of cisplatin treatment, significant apoptosis was detected in p53+ proximal tubular cells, which showed a typical apoptotic morphology characterized by cellular shrinkage, apoptotic body formation, and nuclear condensation and fragmentation; these cells also had positive TUNEL staining (Fig. 9A). Apoptosis was obviously lower in p53− cells (Fig. 9A). Under these experimental conditions, cisplatin did not induce significant necrosis as shown by the lack of propidium iodide staining (data not shown). We further quantified apoptosis by cell counting. As shown in Fig. 9B, cisplatin treatment for 24 h induced about 30% apoptosis in p53+ tubular cells, but <10% in p53− cells. After 40–48 h of cisplatin treatment, >80% apoptosis was shown in p53+ cells, whereas about 50% was shown in p53+ cells (Fig. 9B). We further determined whether the resistance of p53− cells to cisplatin-induced apoptosis was associated with long-term survival. To this end, the cells after 48 h of treatment were returned to cisplatin-free culture medium for recovery. In 5 days, some p53− cells recovered and proliferated to reach confluence in the center area of the dish, whereas p53+ cells did not recover or only recovered to show a few cell islands that appeared to be quite stressed and less proliferative (cell morphology not shown). For a quantitative purpose, the recovered cells were harvested to measure total protein. As shown in Fig. 9C, 43 μg protein were recovered in p53− cells, whereas only 8 μg were recovered from the p53+ dish. Control dishes without cisplatin exposure had ∼110 μg protein.
Fig. 9.

Cisplatin-induced apoptosis in primary cultures of proximal tubular cells isolated from wild-type and p53-deficient mice. Proximal tubular cells were isolated from wild-type (p53+) and p53-deficient (p53-) mice for primary culture and then treated with 30 μM cisplatin. A: representative morphology of cells after 24 h of cisplatin treatment. Cells were fixed for Hoechst 33342 staining and TUNEL assay. Cell morphology, Hoechst staining, and TUNEL staining of the same fields of cells were recorded by phase-contrast and fluorescence microscopy. B: percentages of apoptosis were determined by counting the cells with typical apoptotic morphology. C: cell survival. After 48 h of cisplatin treatment, the cells were returned to normal culture medium without cisplatin for 5 days, and then whole cell lysates were collected for measurement of total protein. Data are expressed as means ± SD; n = 3. *Statistically significant difference from cisplatin-treated wild-type group.
Discussion
Using pharmacological and gene knockout models, this study has provided the first in vivo evidence for a role of p53 and related signaling pathway in cisplatin nephrotoxicity. The results have characterized p53 activation during cisplatin treatment of C57BL/6 mice, demonstrated the protective effects of pifithrin-α in this in vivo model, and further shown the resistance of p53-deficient mice and isolated proximal tubular cells to cisplatin-induced acute kidney injury.
In this study, p53 activation started at day 2 of cisplatin treatment and was intensified at day 3, showing a temporal correlation with the development of renal injury and renal failure. In addition, p53 was shown to be activated in renal tubular cells, the main cell types that were injured by cisplatin. Costaining with FITC-labeled lectins specific for proximal and distal tubules indicated that p53 was induced in both proximal and distal tubular cells under the pathological condition (Fig. 2). Although earlier work suggested that distal tubules were the primary site of apoptosis during cisplatin nephrotoxicity (23), more recent studies also showed apoptosis in proximal tubules (19, 37). Cisplatin-induced apoptosis in proximal tubules was further detected in our latest work by colocalizing apoptotic cells with PHA lectin staining. Thus the spatial analysis of p53 activation is consistent with the involvement of p53 in cisplatin-induced renal tubular cell injury. This inference is further supported by our results showing that the cells that had p53 activation underwent apoptosis as indicated by positive TUNEL staining (Fig. 3). Of note, although TUNEL staining may not be very specific for apoptosis, it has been demonstrated to be relatively specific for apoptosis analysis in acute kidney injury induced by renal ischemia and cisplatin (17).
With clear evidence for p53 activation as indicated by its phosphorylation and protein accumulation, this study has not investigated the mechanism of p53 activation during cisplatin nephrotoxicity. For a mechanistic study, it would be preferable to be initially conducted in in vitro models. Nevertheless, it can be speculated that cisplatin may induce DNA damage by its intra- and interstrand cross-linking property (5, 33, 39). This may then activate the signaling pathways of DNA damage response, including ataxia telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR), leading to p53 phosphorylation and activation. Our recent work has shown the cleavage and inactivation of ATM, but not ATR, during cisplatin treatment of renal tubular cells (40). However, the role of ATM and ATR in p53 activation under these experimental conditions remains to be clarified. It is also noteworthy that, in addition of DNA damage, cisplatin may activate p53 via other injury or stress-related signaling pathways (38). In this regard, our recent work demonstrated the inhibitory effects of free radical scavenging chemicals on p53 activation and cisplatin-induced nephrotoxicity, suggesting that oxidative stress may contributes to p53 activation (12).
Our results showed significant protective effects of pifithrin-α on cisplatin nephrotoxicity (Figs. 4–6). Moreover, p53-deficeint mice were shown to be resistant to cisplatin injury (Figs. 7–8). The effects of pifithrin-α and p53 deficiency were shown at the levels of renal function, histology, and tubular cell apoptosis. Nevertheless, it is important to recognize that the protective effects were only partial. This is not surprising, since the pathogenesis of cisplatin nephrotoxicity is generally considered to be multifactorial. Indeed, our results showed that, while many apoptotic cells in cisplatin-treated tissues had p53 induction, others did not induce p53 (Fig. 3), suggesting that apoptosis of the latter cell population is independent of p53. Consistently, in primary cultures of proximal tubular cells, p53 deficiency did not completely block cisplatin-induced apoptosis (Fig. 9). It is important to recognize that tubular cell apoptosis during acute kidney injury is dynamic and progressive. Thus analysis of apoptosis at a fixed time point (as in our study) only shows a snapshot image (17), which likely underestimates the amount of apoptotic cells developing during the whole study period of 3 days. As a result, partial protection of apoptosis may have significant effects on renal histology and function.
In addition, we noticed that the p53-deficient mice used in our experiments can be roughly divided into two groups, depending on their sensitivity to cisplatin injury. One group (about two-third of the tested animals) was clearly resistant to cisplatin injury and only showed small to moderate increases of BUN and serum creatinine at day 3 of cisplatin treatment. In contrast, the other group (about one-third of the tested animals) showed medium to severe renal injury and renal failure during cisplatin treatment, which was comparable to wild-type animals. Frequently, both resistant and sensitive animals were found in the same litter, suggesting that the variation was not because of the abnormality of specific litters. It was reported that the developing kidneys of p53-deficient mice had structural abnormalities, including hyperplastic cyst formation in the differentiated zone (29). Our study used young adult mice that did not show obvious structural or functional abnormalities in the kidneys. Occasionally, there were renal cysts, but the majority of the animals was normal. Although the exact cause of the varied cisplatin sensitivity in p53-deficient mice is unclear, knockout of p53 is expected to promote cancer development. According to the information from Jackson Laboratory, the p53-deficient mice develop tumors (principally lymphomas and osteosarcoma) at around 3–6 mo of age. In our study, the animals were used at 8–10 wk (2–2.5 mo). Thus it is possible that some of the animals had already undergone homeostatic changes of early tumor development by the time of experiment, which may affect their sensitivity to cisplatin. It would be advantageous to use conditional p53 knockout models that specifically downregulate p53 in renal tubular cells to further investigate this possibility and determine the role of p53 in cisplatin nephrotoxicity.
The involvement of p53 in cisplatin nephrotoxicity suggests that it is possible to protect kidneys during cisplatin chemotherapy by blocking p53. However, we have to recognize that inhibition of p53 may also limit apoptosis of cancer cells and thus reduce the therapeutic effects in tumors. In this regard, over 50% of cancers have p53 mutations, yet cisplatin is frequently effective in treating them, indicating that cisplatin therapy in these cancer types may not be stringently p53 dependent (10, 33). During cisplatin therapy of these cancers, targeting p53 should be a useful strategy for renoprotection. On the other hand, in cancers where the therapeutic effects of cisplatin depend on p53, restrictive renal delivery of p53 inhibitory chemicals, antisense oligonucleotides, and small-interfering RNAs (siRNAs) may be considered. Renal tissues have an enormous capacity of reabsorption and are thus particularly accessible to drugs, including antisense oligonucleotides and siRNA (36). Whether these p53-targeting approaches can protect kidneys against cisplatin nephrotoxicity without limiting the therapeutic effects in cancers warrants further investigation.
Acknowledgments
We thank Drs. Nahid Mivechi and Dimitrios Moskophidis at the Center for Molecular Chaperone/Radiobiology & Cancer Virology of the Medical College of Georgia for breeding pairs of p53-deficeinct mice.
GRANTS: This study was supported in part by grants from the National Institutes of Health and the Department of Veterans Affairs.
References
- 1.Arany I, Megyesi JK, Kaneto H, Price PM, Safirstein RL. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am J Physiol Renal Physiol. 2004;287:F543–F549. doi: 10.1152/ajprenal.00112.2004. [DOI] [PubMed] [Google Scholar]
- 2.Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol. 2003;23:460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 3.Baliga R, Zhang Z, Baliga M, Ueda N, Shah SV. In vitro and in vivo evidence suggesting a role for iron in cisplatin-induced nephrotoxicity. Kidney Int. 1998;53:394–401. doi: 10.1046/j.1523-1755.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- 4.Balster DA, O'Dorisio MS, Summers MA, Turman MA. Segmental expression of somatostatin receptor subtypes sst(1) and sst(2) in tubules and glomeruli of human kidney. Am J Physiol Renal Physiol. 2001;280:F457–F465. doi: 10.1152/ajprenal.2001.280.3.F457. [DOI] [PubMed] [Google Scholar]
- 5.Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Perez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7:3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 6.Cummings BS, Schnellmann RG. Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther. 2002;302:8–17. doi: 10.1124/jpet.302.1.8. [DOI] [PubMed] [Google Scholar]
- 7.Dong Z, Atherton SS. Tumor necrosis factor-alpha in cisplatin nephrotoxicity: a homebred foe? Kidney Int. 2007;72:5–7. doi: 10.1038/sj.ki.5002320. [DOI] [PubMed] [Google Scholar]
- 8.Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke T, Somerset H, Oh DJ, Lu L, Klein C, Dinarello CA, Edelstein CL. Cisplatin-induced ARF is associated with an increase in the cytokines IL-1β,IL-18, IL-6 and neutrophil infiltration in the kidney. J Pharmacol Exp Ther. 2007;322:8–15. doi: 10.1124/jpet.107.119792. [DOI] [PubMed] [Google Scholar]
- 9.Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA, Edelstein CL. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int. 2004;66:2202–2213. doi: 10.1111/j.1523-1755.2004.66010.x. [DOI] [PubMed] [Google Scholar]
- 10.Gudkov AV, Komarova EA. Prospective therapeutic applications of p53 inhibitors. Biochem Biophys Res Commun. 2005;331:726–736. doi: 10.1016/j.bbrc.2005.03.153. [DOI] [PubMed] [Google Scholar]
- 11.Jiang M, Pabla N, Murphy RF, Yang T, Yin XM, Degenhardt K, White E, Dong Z. Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J Biol Chem. 2007;282:2636–2645. doi: 10.1074/jbc.M606928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang M, Wei Q, Pabla N, Dong G, Wang CY, Yang T, Smith SB, Dong Z. Effects of hydroxyl radical scavenging on cisplatin-induced p53 activation, tubular cell apoptosis and nephrotoxicity. Biochem Pharmacol. 2007;73:1499–1510. doi: 10.1016/j.bcp.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang M, Wei Q, Wang J, Du C, Yu J, Zhang L, Dong Z. Regulation of PUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Oncogene. 2006;25:4056–4066. doi: 10.1038/sj.onc.1209440. [DOI] [PubMed] [Google Scholar]
- 14.Jiang M, Yi X, Hsu S, Wang CY, Dong Z. Role of p53 in cisplatin-induced tubular cell apoptosis: dependence on p53 transcriptional activity. Am J Physiol Renal Physiol. 2004;287:F1140–F1147. doi: 10.1152/ajprenal.00262.2004. [DOI] [PubMed] [Google Scholar]
- 15.Jo SK, Hu X, Yuen PS, Aslamkhan AG, Pritchard JB, Dear JW, Star RA. Delayed DMSO administration protects the kidney from mercuric chloride-induced injury. J Am Soc Nephrol. 2004;15:2648–2654. doi: 10.1097/01.ASN.0000139933.20109.CB. [DOI] [PubMed] [Google Scholar]
- 16.Kaushal GP, Kaushal V, Hong X, Shah SV. Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int. 2001;60:1726–1736. doi: 10.1046/j.1523-1755.2001.00026.x. [DOI] [PubMed] [Google Scholar]
- 17.Kelly KJ, Sandoval RM, Dunn KW, Molitoris BA, Dagher PC. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am J Physiol Cell Physiol. 2003;284:C1309–C1318. doi: 10.1152/ajpcell.00353.2002. [DOI] [PubMed] [Google Scholar]
- 18.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Basnakian A, Bhatt R, Megyesi J, Gokden N, Shah SV, Portilla D. PPAR-alpha ligand ameliorates acute renal failure by reducing cisplatin-induced increased expression of renal endonuclease G. Am J Physiol Renal Physiol. 2004;287:F990–F998. doi: 10.1152/ajprenal.00206.2004. [DOI] [PubMed] [Google Scholar]
- 20.Lieberthal W, Triaca V, Levine J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs necrosis. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;270:F700–F708. doi: 10.1152/ajprenal.1996.270.4.F700. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Baliga R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003;63:1687–1696. doi: 10.1046/j.1523-1755.2003.00908.x. [DOI] [PubMed] [Google Scholar]
- 22.Manfredi JJ. p53 and apoptosis: it's not just in the nucleus anymore. Mol Cell. 2003;11:552–554. doi: 10.1016/s1097-2765(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 23.Megyesi J, Safirstein RL, Price PM. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest. 1998;101:777–782. doi: 10.1172/JCI1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowak G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. 2002;277:43377–43388. doi: 10.1074/jbc.M206373200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 26.Park MS, De Leon M, Devarajan P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J Am Soc Nephrol. 2002;13:858–865. doi: 10.1681/ASN.V134858. [DOI] [PubMed] [Google Scholar]
- 27.Ramesh G, Reeves WB. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. Am J Physiol Renal Physiol. 2005;289:F166–F174. doi: 10.1152/ajprenal.00401.2004. [DOI] [PubMed] [Google Scholar]
- 28.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–842. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saifudeen Z, Dipp S, El-Dahr SS. A role for p53 in terminal epithelial cell differentiation. J Clin Invest. 2002;109:1021–1030. doi: 10.1172/JCI13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seth R, Yang C, Kaushal V, Shah SV, Kaushal GP. p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J Biol Chem. 2005;280:31230–31239. doi: 10.1074/jbc.M503305200. [DOI] [PubMed] [Google Scholar]
- 31.Sheikh-Hamad D, Cacini W, Buckley AR, Isaac J, Truong LD, Tsao CC, Kishore BK. Cellular and molecular studies on cisplatin-induced apoptotic cell death in rat kidney. Arch Toxicol. 2004;78:147–155. doi: 10.1007/s00204-003-0521-4. [DOI] [PubMed] [Google Scholar]
- 32.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol. 2000;278:F726–F736. doi: 10.1152/ajprenal.2000.278.5.F726. [DOI] [PubMed] [Google Scholar]
- 33.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 34.Silva FG, Nadasdy T, Laszik Z. Immunohistochemical and lectin dissection of the human nephron in health and disease. Arch Pathol Lab Med. 1993;117:1233–1239. [PubMed] [Google Scholar]
- 35.Taguchi T, Nazneen A, Abid MR, Razzaque MS. Cisplatin-associated nephrotoxicity and pathological events. Contrib Nephrol. 2005;148:107–121. doi: 10.1159/000086055. [DOI] [PubMed] [Google Scholar]
- 36.Tomita N, Azuma H, Kaneda Y, Ogihara T, Morishita R. Application of decoy oligodeoxynucleotides-based approach to renal diseases. Curr Drug Targets. 2004;5:717–733. doi: 10.2174/1389450043345146. [DOI] [PubMed] [Google Scholar]
- 37.Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, Fukuda K, Kanai H, Kishihara K, Hirakata H, Iida M. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003;63:72–82. doi: 10.1046/j.1523-1755.2003.00709.x. [DOI] [PubMed] [Google Scholar]
- 38.Vousden KH, Lane DL. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 39.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Pabla N, Wang CY, Wang W, Schoenlein PV, Dong Z. Caspase-mediated cleavage of ATM during cisplatin-induced tubular cell apoptosis: inactivation of its kinase activity toward p53. Am J Physiol Renal Physiol. 2006;291:F1300–F1307. doi: 10.1152/ajprenal.00509.2005. [DOI] [PubMed] [Google Scholar]
- 41.Wei Q, Dong G, Franklin J, Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007;72:53–62. doi: 10.1038/sj.ki.5002256. [DOI] [PubMed] [Google Scholar]
- 42.Wei Q, Wang MH, Dong Z. Differential gender differences in ischemic and nephrotoxic acute renal failure. Am J Nephrol. 2005;25:491–499. doi: 10.1159/000088171. [DOI] [PubMed] [Google Scholar]
- 43.Wei Q, Yin XM, Wang MH, Dong Z. Bid deficiency ameliorates ischemic renal failure and delays animal death in C57BL/6 mice. Am J Physiol Renal Physiol. 2006;290:F35–F42. doi: 10.1152/ajprenal.00184.2005. [DOI] [PubMed] [Google Scholar]
- 44.Yu F, Megyesi J, Safirstein RL, Price PM. Identification of the functional domain of p21WAF1/CIP1 that protects from cisplatin cytotoxicity. Am J Physiol Renal Physiol. 2005;289:F514–F520. doi: 10.1152/ajprenal.00101.2005. [DOI] [PubMed] [Google Scholar]
- 45.Zhang B, Ramesh G, Norbury CC, Reeves WB. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007;72:37–44. doi: 10.1038/sj.ki.5002242. [DOI] [PubMed] [Google Scholar]