

A likelihood-based density modification approach is developed that can incorporate expected electron-density information from a wide variety of sources.
Keywords: density modification, likelihood functions
Abstract
A likelihood-based approach to density modification is developed that can be applied to a wide variety of cases where some information about the electron density at various points in the unit cell is available. The key to the approach consists of developing likelihood functions that represent the probability that a particular value of electron density is consistent with prior expectations for the electron density at that point in the unit cell. These likelihood functions are then combined with likelihood functions based on experimental observations and with others containing any prior knowledge about structure factors to form a combined likelihood function for each structure factor. A simple and general approach to maximizing the combined likelihood function is developed. It is found that this likelihood-based approach yields greater phase improvement in model and real test cases than either conventional solvent flattening and histogram matching or a recent reciprocal-space solvent-flattening procedure [Terwilliger (1999 ▶), Acta Cryst. D55, 1863–1871].
1. Introduction
The phase information obtained from experimental measurements on macromolecules using either multiple isomorphous replacement or multiwavelength anomalous diffraction is often insufficient by itself for constructing a electron-density map useful for model building and interpretation. Many density-modification methods have been developed in recent years for improving the quality of electron-density maps by incorporation of prior knowledge about the features expected in these maps when they are obtained at high or moderate resolution (2–4 Å). Among the most powerful of these methods are solvent flattening, non-crystallographic symmetry averaging, histogram matching, phase extension, molecular replacement, entropy maximization and iterative model building (Abrahams, 1997 ▶; Bricogne, 1984 ▶, 1988 ▶; Cowtan & Main, 1993 ▶, 1996 ▶; Giacovazzo & Siliqi, 1997 ▶; Goldstein & Zhang, 1998 ▶; Gu et al., 1997 ▶; Lunin, 1993 ▶; Perrakis et al., 1997 ▶; Podjarny et al., 1987 ▶; Prince et al., 1988 ▶; Refaat et al., 1996 ▶; Roberts & Brünger, 1995 ▶; Rossmann & Arnold, 1993 ▶; Vellieux et al., 1995 ▶; Wilson & Agard, 1993 ▶; Xiang et al., 1993 ▶; Zhang & Main, 1990 ▶; Zhang, 1993 ▶; Zhang et al., 1997 ▶). The fundamental basis of density-modification methods is that there are many possible sets of structure-factor amplitudes and phases that are all reasonably probable based on the limited experimental data, and those structure factors that lead to maps that are most consistent with both the experimental data and the prior knowledge are the most likely overall. In these methods, the choice of prior information that is to be used and the procedure for combining prior information about electron density with experimentally derived phase information are crucial parts.
Until recently, density modification, the combination of knowledge about expected features of an electron-density map with experimental phase information, has generally been carried out in a two-step procedure that is iterated until convergence. In the first step, an electron-density map obtained experimentally is modified in real space in order to make it consistent with expectations. This can consist of flattening solvent regions, averaging non-crystallographic symmetry-related regions or histogram matching, for example. In the second step, phases are calculated from the modified map and are combined with the experimental phases to form a new phase set.
The disadvantage of this real-space modification approach is that it is not at all clear how to weight the observed phases with those obtained from the modified map. This is a consequence of the fact that the modified map contains some of the same information as the original map and some new information. This difficulty has been recognized for a long time and a number of approaches have been designed to improve the relative weighting from these two sources, recently including the use of maximum-entropy methods and the use of weighting optimized using cross-validation (Xiang et al., 1993 ▶; Roberts & Brünger, 1995 ▶; Cowtan & Main, 1996 ▶) and ‘solvent flipping’ (Abrahams, 1997 ▶).
2. Density modification by reciprocal-space-based likelihood optimization
We have recently developed a very different approach to combinining experimental phase information with prior knowledge about expected electron-density distributions in maps. Our approach is based on maximization of a combined likelihood function (Terwilliger, 1999 ▶). The fundamental idea is to express our knowledge about the probability of a set of structure factors {F
h} in terms of two quantities: (i) the likelihood of having measured the observed set of structure factors 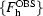 if this structure-factor set were correct and (ii) the likelihood that the map resulting from this structure-factor set {F
h} is consistent with our prior knowledge about this and other macromolecular structures.
if this structure-factor set were correct and (ii) the likelihood that the map resulting from this structure-factor set {F
h} is consistent with our prior knowledge about this and other macromolecular structures.
When set up in this way, the overlap of information that occurred in the real-space modification methods is not present because the experimental and prior information are kept separate. Consequently, proper weighting of experimental and prior information only requires estimates of probability functions for each source of information.
The likelihood-based density-modification approach has a second very important advantage. This is that the derivatives of the likelihood functions with respect to individual structure factors can be readily calculated in reciprocal space by FFT-based methods. As a consequence, density modification simply becomes an optimization of a combined likelihood function by adjustment of structure factors. This makes density modification a remarkably simple but powerful approach, only requiring that suitable likelihood functions be constructed for each aspect of prior knowledge that is to be incorporated. We previously showed that such an approach could be applied to solvent flattening and that the resulting algorithm was greatly improved over methods depending on real-space modification and phase recombination (Terwilliger, 1999 ▶).
Here, we extend the idea of likelihood-based density modification to include prior information on the electron-density distribution from a wide variety of potential sources and demonstrate it on both the electron density in the solvent region and the region occupied by a macromolecule. First, we describe the mathematics of likelihood-based density modification in a practical formulation that is modified somewhat from the one we used for reciprocal-space solvent flattening (Terwilliger, 1999 ▶). We then show how a likelihood function for a map that includes information on both the solvent- and macromolecule-containing regions can be constructed and used.
3. Likelihood-based density modification
The basic idea of our likelihood-based density-modification procedure is that there are two key kinds of information about the structure factors for a crystal of a macromolecule. The first is the experimental phase and amplitude information. This can be thought of in terms of a likelihood (or log-likelihood) function LLOBS(F h) for each structure factor F h, where the probability distribution for the structure factor p OBS(F h) is given by
For reflections with accurately measured amplitudes, the chief uncertainty in F h will be in the phase, while for unmeasured or poorly measured reflections it will be in both phase and amplitude.
The second kind of information about structure factors in this formulation is the likelihood of the map resulting from them. For example, for most macromolecular crystals a set of structure factors {F h} that leads to a map with a flat region corresponding to solvent is more likely to be correct than one that leads to a map with uniform variation everywhere. This map-likelihood function describes the probability that the map obtained from a set of structure factors is compatible with our expectations,
We then combine our two principal sources of information along with any prior knowledge of the structure factors to yield the likelihood of a particular set of structure factors,
where LLo({F h}) includes any structure-factor information that is known in advance, such as the distribution of intensities of structure factors (Wilson, 1949 ▶).
3.1. Approximating the likelihood function to simplify the procedure
In order to maximize the overall likelihood function in (3) we are going to need to know how the map-likelihood function changes in response to changes in structure factors. In the case of the map-likelihood function LLMAP({F h}) this can be thought of as two separate relationships, the response of the likelihood function to changes in electron density and the changes in electron density as a function of changes in structure factors. In principle, the likelihood of a particular map is a complicated function of the electron density over the entire map. Furthermore, the value of any structure factor affects the electron density everywhere in the map. To simplify the mathematics, we explicitly use a low-order approximation to the likelihood function for a map instead of attempting to evaluate the function precisely. As Fourier transformation is a linear process, each reflection contributes independently to the electron density at a given point in the cell. Although the log-likelihood of the electron density might have any form, we expect that for sufficiently small changes in structure factors, a first-order approximation to the log-likelihood function would apply and each reflection would also contribute relatively independently to changes in the log-likelihood function.
Consequently, we construct a local approximation to the map-likelihood function, neglecting correlations among different points in the map and between reflections, expecting that it might describe reasonably accurately how the likelihood function would vary in response to small changes in structure factors.
By neglecting correlations among different points in the map, we can write the log-likelihood for the whole electron-density map as the sum of the log-likelihoods of the densities at each point in the map, normalized to the volume of the unit cell and the number of reflections used to construct it (Terwilliger, 1999 ▶),
Additionally, by treating each reflection as independently contributing to the likelihood function, we can write a local approximation to the log-likelihood of the density at each point. This approximation is given by the sum over all reflections of first few terms of a Taylor’s series expansion around the value obtained with the starting structure factors  used in a cycle of density modification,
used in a cycle of density modification, 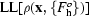 ,
,
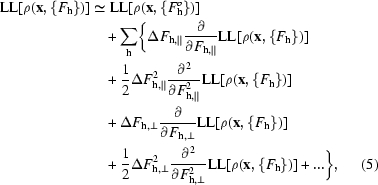 |
where 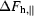 and
and  are the differences between F
h and
are the differences between F
h and 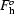 along the directions of
along the directions of  and
and 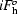 , respectively.
, respectively.
Combining (4) and (5), we can write an expression for the map log-likelihood function,
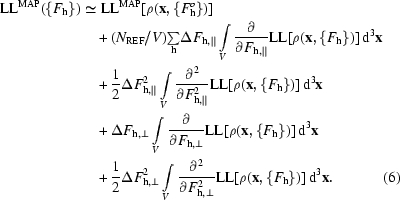 |
3.2. FFT-based calculation of the reciprocal-space derivatives of log-likelihood of electron density LL[ρ(x, {F h})]
The integrals in (6) can be rewritten in a form that is suitable for evaluation by an FFT-based approach. Considering the first integral in (6), we use the chain rule to write that
and note that the derivative of ρ(x) with respect to  for a particular index h is given by
for a particular index h is given by
Now we can rearrange and rewrite the first integral in (6) in the form
where the complex number a
h is a term in the Fourier transform of 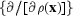 LL[ρ(x, {F
h})],
LL[ρ(x, {F
h})],
In space groups other than P1, only a unique set of structure factors need to be specified to calculate an electron-density map. Taking space-group symmetry into account, (9) can be generalized (Terwilliger, 1999 ▶) to read
where the indices h′ are all indices equivalent to h owing to space-group symmetry.
A similar procedure can be used to rewrite the second integral in (6), yielding the expression
 |
where the indices h′ and k′ are each all indices equivalent to h owing to space-group symmetry and where the coefficients b h are again terms in a Fourier transform, this time of the second derivative of the log-likelihood of the electron density,
The third and fourth integrals in (6) can be rewritten in a similar way, yielding the expressions
and
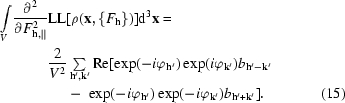 |
The significance of (4) through (15) is that we now have a simple expression (6) describing how the map-likelihood function LLMAP({F h}) varies when small changes are made in the structure factors. Evaluating this expression only requires that we be able to calculate the first and second derivatives of log-likelihood of the electron density with respect to electron density at each point in the map and carry out an FFT. Furthermore, maximization of the (local) overall likelihood function (3) becomes straightforward, as every reflection is treated independently. It consists simply of adjusting each structure factor to maximize its contribution to the approximation to the likelihood function through (3) to (15).
In practice, instead of directly maximizing the overall likelihood function, we use it here to estimate the probability distribution for each structure factor (Terwilliger, 1999 ▶) and then integrate this probability distribution over the phase (or phase and amplitude) of the reflection to obtain a weighted mean estimate of the structure factor. Using (3) to (15), the probability distribution for an individual structure factor can be written as
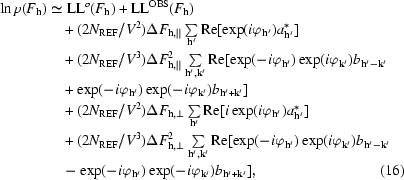 |
where, as above, the indices h′ and k′ are each all indices equivalent to h owing to space-group symmetry and the coefficients a
h and b
h are given in (10) and (13). Also as before, and are the differences between F
h and along the directions of and  , respectively. All the quantities in (16) can be readily calculated once a likelihood function for the electron density and its derivatives are obtained.
, respectively. All the quantities in (16) can be readily calculated once a likelihood function for the electron density and its derivatives are obtained.
4. Likelihood function for an electron-density map with errors
A key step in likelihood-based density modification is the decision as to the likelihood function for values of the electron density at a particular location in the map. For the present purpose, an expression for the log-likelihood of the electron density LL[ρ(x, {F h})] at a particular location x in a map is needed that depends on whether the point x is within the solvent region or the protein region. In general, this function might depend on whether the point satisfies any of a wide variety of conditions, such as being at a certain location in a known fragment of structure or being at a certain distance from some other feature of the map. We discussed previously (Terwilliger, 1999 ▶) how one might incorporate information on the environment of x by writing the log-likelihood function as the log of the sum of conditional probabilities dependent on the environment of x,
where p PROT(x) is the probability that x is in the protein region and p[ρ(x)|PROT] is the conditional probability for ρ(x) given that x is in the protein region, and p SOLV(x) and p[ρ(x)|SOLV] are the corresponding quantities for the solvent region. The probability that x is in the protein or solvent regions is estimated by a modification of the methods of Wang (1985 ▶) and Leslie (1987 ▶) as described previously (Terwilliger, 1999 ▶). If there were more than just solvent and protein regions that identified the environment of each point, then (17) could be modified to include those as well.
In developing (13) to (15), the derivatives of the likelihood function for electron density were intended to represent how the likelihood function changed when small changes in one structure factor were made. Surprisingly, the likelihood function that is most appropriate for our present purposes in this case is not a globally correct one. Instead, it is a likelihood function that represents how the overall likelihood function varies in response to small changes in one structure factor, keeping all others constant. To see the difference, consider the electron density in the solvent region of a macromolecular crystal. In an idealized situation with all possible reflections included, the electron density might be exactly equal to a constant in this region. The goal in using (16) is to obtain the relative probabilites for each possible value of a particular unknown structure factor F h. If all other structure factors were exact, then the globally correct likelihood function for the electron density (zero unless the solvent region is perfectly flat) would correctly identify the correct value of the unknown structure factor. Now suppose we had imperfect phase information. The solvent region would have a significant amount of noise and its value would no longer be a constant. If we use the globally correct likelihood function for the electron density, we would assign a zero probability to any value of the structure factor that did not lead to an absolutely flat solvent region. This is clearly unreasonable, because all the other (incorrect) structure factors are contributing noise that exists regardless of the value of this structure factor.
This situation is very similar to the one encountered in structure refinement of macromolecular structures where there is a substantial deficiency in the model. The errors in all the other structure factors in the present discussion correspond to the deficiency in the macromolecular model in the refinement case. The appropriate variance to use as a weighting factor in refinement includes the estimated model error as well as the error in measurement (e.g. Terwilliger & Berendzen, 1996 ▶; Pannu & Read, 1996 ▶). Similarly, the appropriate likelihood function for electron density for use in the present method is one in which the overall uncertainty in the electron density arising from all reflections other than the one being considered is included in the variance.
A likelihood function of this kind for the electron density can be developed using a model in which the electron density arising from all reflections but one is treated as a random variable (Terwilliger & Berendzen, 1996 ▶; Pannu & Read, 1996 ▶). Suppose that the true value of the electron density at x was known and was given by ρT. Then consider that we have estimates of all the structure factors, but that substantial errors exist in each one. The expected value of the estimate of this electron density obtained from current estimates of all the structure factors (ρOBS) will be given by 〈ρOBS〉 = βρT and the expected value of the variance by 〈(ρOBS − βρT)2〉 =  . The factor β represents the expectation that the calculated value of ρ will be smaller than the true value. This is for two reasons. One is that such a estimate may be calculated using figure-of-merit weighted estimates of structure factors, which will be smaller than the correct ones. The other is that phase error in the structure factors systematically leads to a bias towards a smaller component of the structure factor along the direction of the true structure factor. This is the same effect that leads to the D correction factor in maximum-likelihood refinement (Pannu & Read, 1996 ▶).
. The factor β represents the expectation that the calculated value of ρ will be smaller than the true value. This is for two reasons. One is that such a estimate may be calculated using figure-of-merit weighted estimates of structure factors, which will be smaller than the correct ones. The other is that phase error in the structure factors systematically leads to a bias towards a smaller component of the structure factor along the direction of the true structure factor. This is the same effect that leads to the D correction factor in maximum-likelihood refinement (Pannu & Read, 1996 ▶).
A probability function for the electron density at this point that is appropriate for assessing the probabilities of values of the structure factor for one reflection can now be written as
In a slightly more complicated case, where the value of ρT is not known exactly but rather has an uncertainty σT, (18) becomes
Finally, in the case where only a probability distribution p(ρT) for ρT is known, (18) becomes
4.1. Likelihood function for solvent- and macromolecule-containing regions of a map
Using (19) and (20), we are now in a position to use a histogram-based approach (Goldstein & Zhang, 1998 ▶; Lunin, 1993 ▶; Zhang & Main, 1990 ▶) to develop likelihood functions for the solvent region of a map and for the macromolecule-containing region of a map. The approach is simple. The probability distribution for true electron density in the solvent or macromolecule regions of a crystal structure is obtained from an analysis of model structures and represented as a sum of Gaussian functions of the form
If the values of β and σMAP were known for an experimental map with unknown errors but identified solvent and protein regions, then using (19) we could write the probability distribution for electron density in the each region of the map as
with the appropriate values of β and σ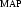 . In practice, the values of β and σMAP are estimated by a least-squares fitting of the probability distribution given in (22) to the one found in the experimental map. This procedure has the advantage that the scale of the experimental map does not have to be accurately determined. Then (22) is used with the refined values of β and σMAP as the probability function for electron density in the corresponding region (solvent or macromolecule) of the map.
. In practice, the values of β and σMAP are estimated by a least-squares fitting of the probability distribution given in (22) to the one found in the experimental map. This procedure has the advantage that the scale of the experimental map does not have to be accurately determined. Then (22) is used with the refined values of β and σMAP as the probability function for electron density in the corresponding region (solvent or macromolecule) of the map.
5. Evaluation of maximum-likelihood density modification with model and real data
To evaluate the utility of maximum-likelihood density modification as described here, we carried out tests using the same model and experimental data that we previously analyzed using reciprocal-space solvent flattening and by real-space solvent flattening (Terwilliger, 1999 ▶). The first test case consisted of a set of phases constructed from a model with 32–68% of the volume of the unit cell taken up by protein. The initial effective figure of merit of the phases overall [〈cos(Δϕ)〉] was about 0.40. In our previous tests, we showed that both real-space and reciprocal-space solvent flattening improved the quality of phasing considerably. In the current tests, the real-space density modification included both solvent flattening and histogram matching to be as comparable as possible to the maximum-likelihood density modification we have developed.
Table 1 ▶ shows the the quality of phases obtained after each method for density modification was applied to this model case. In all cases, maximum-likelihood density modification of this map resulted in phases with an effective figure of merit [〈cos(Δϕ)〉] higher than any of the other methods. When the fraction of solvent in the model unit cell was 50%, for example, maximum-likelihood density modification yielded an effective figure of merit of 0.83, while real-space solvent flattening and histogram matching resulted in an effective figure of merit of 0.62 and reciprocal-space solvent flattening gave an effective figure of merit of 0.67.
Table 1. Correlation of density-modified phases with true phases [〈cos(Δϕ)〉] for model data in a unit cell containing 32–68% solvent.
Data and analysis using reciprocal-space solvent flattening are from Terwilliger (1999 ▶). Phases with simulated errors for 6906 data from ∞ to 3.0 Å were constructed using a model consisting of coordinates from a dehalogenase enzyme from Rhodococcus species ATCC 55388 (American Type Culture Collection, 1992 ▶) determined recently in our laboratory (Newman et al., 1999 ▶; PDB entry 1bn7), except that some of the atoms were not included in order to vary the fraction of solvent in the unit cell. The cell was in space group P21212, with unit-cell parameters a = 94, b = 80, c = 43 Å and one molecule in the asymmetric unit. Phases with simulated errors were generated by adding phase errors as described in Terwilliger (1999 ▶) to yield an average value of the cosine of the phase error (i.e. the true figure of merit of the phasing) of 〈cos(Δϕ)〉 = 0.42 for acentric and 0.39 for centric reflections. The model data with simulated errors was then density modified by the maximum-likelihood method described here, by reciprocal-space solvent flattening (Terwilliger, 1999 ▶) and by a real-space method as implemented in the program dm (Cowtan & Main, 1996 ▶), version 1.8, using solvent flattening and histogram matching.
| Fraction protein (%) | Starting | Real-space solvent flattening | Reciprocal-space solvent flattening | Maximum-likelihood solvent flattening |
|---|---|---|---|---|
| 32 | 0.41 | 0.64 | 0.85 | 0.87 |
| 42 | 0.40 | 0.62 | 0.67 | 0.83 |
| 50 | 0.41 | 0.54 | 0.56 | 0.77 |
| 68 | 0.42 | 0.48 | 0.41 | 0.53 |
The utility of maximum-likelihood density modification was also compared with real-space density modification and with reciprocal-space solvent flattening using experimental multiwavelength (MAD) data on initiation factor 5A (IF-5A) recently determined in our laboratory (Peat et al., 1998 ▶). IF-5A crystallizes in space group I4, with unit-cell parameters a = 114, b = 114, c = 33 Å, one molecule in the asymmetric unit and a solvent content of about 60%. The structure was solved using MAD phasing based on three Se atoms in the asymmetric unit at a resolution of 2.2 Å. For purposes of testing density-modification methods, only one of the three selenium sites was used in phasing here, resulting in a starting map with a correlation coefficient to the map calculated using the final refined structure of 0.37. The resulting electron-density map was improved by real-space density modification using solvent flattening and histogram matching with dm (Cowtan & Main, 1996 ▶), by real-space density modification using solvent flipping (Abrahams, 1997 ▶) and after maximum-likelihood density modification. The ‘experimental’ map, the dm-modified map and the maximum-likelihood map are shown in Fig. 1 ▶. As anticipated, the real-space modified map obtained with dm is improved over the starting map; it has a correlation coefficient of 0.65. Density modification including solvent flipping yielded a similar improvement, with a correlation coefficient of 0.61 to the model map. The maximimum-likelihood modified map was much more substantially improved, with a correlation coefficient to the map based on a refined model of 0.79.
Figure 1.

Sections of electron density obtained before and after density modification of phases obtained for IF-5A (Peat et al., 1998 ▶) phased using one Se atom in the asymmetric unit. Density modification was carried out by real-space solvent flattening and histogram matching or by maximum-likelihood solvent flattening. Values for real-space density modification were carried out using the program dm (Cowtan & Main, 1996 ▶), version 1.8, using solvent flattening with histogram matching. Starting phases were calculated with SOLVE (Terwilliger & Berendzen, 1999 ▶). The correlation coefficient between the map calculated based on the refined model of IF-5A and the starting map was 0.37, for the real-space modifed map it was 0.65 and for the maximum-likelihood map it was 0.79.
6. Discussion
We have shown here that a maximum-likelihood approach can be used to carry out density modification on macromolecular crystal structures and that this approach is much more powerful than either conventional density modification based on solvent flattening and histogram matching or our recent reciprocal-space solvent-flattening procedure (Terwilliger, 1999 ▶). The reason the approach works so well is that the relative weighting of experimental phase information and of expected electron-density distributions is taken care of automatically by keeping the two sources of information clearly delineated and by defining suitable probability distributions for each.
The maximum-likelihood approach to improvement of crystallographic phases has been developed extensively by Bricogne and others (e.g. Bricogne, 1984 ▶, 1988 ▶; Lunin, 1993 ▶). The importance of the present work and of our recent work on reciprocal-space solvent flattening (Terwilliger, 1999 ▶) is that we have developed a simple, effective and general way to carry it out.
Although we have demonstrated here only two sources of expected electron-density distributions (probability distributions for solvent regions and for protein-containing regions), the methods developed here can be applied directly to a wide variety of sources of information. For example, any source of information about the expected electron density at a particular point in the unit cell that can be written in a form such as the one in (22) can be used in our procedure to describe the likelihood that a particular value of electron density is consistent with expectations.
Sources of expected electron-density information that are especially suitable for application to our method include non-crystallographic symmetry and the knowledge of the location of fragments of structure in the unit cell. In the case of non-crystallographic symmetry, the probability distribution for electron density at one point in the unit cell can be written using (22) with a value of ρT equal to the weighted mean at all non-crystallographically equivalent points in the cell. The value of σT could be calculated based on their variance and the value of σ. In the case of knowledge of locations of fragments in the unit cell, this knowledge can be used to calculate estimates of the electron-density distribution for each point in the neighborhood of the fragment. These electron-density distributions can then in turn be used just as described above to estimate ρT and σT in this region. An iterative process, in which fragment locations are identified by cross-correlation or related searches (density modification) is applied and additional searches are carried out to further generate a model for the electron density, could even be developed, in an extension of the iterative chain-tracing methods described by Wilson & Agard (1993 ▶). Such a process could potentially even be used to construct a complete probabilistic model of a macromolecular structure using structure-factor estimates obtained from molecular replacement with fragments of macromolecular structures as a starting point. In all these cases, the electron-density information could be included in much the same way as the probability distributions we used here for the solvent and protein regions of maps. In each case, the key is an estimate of the probability distribution for electron density at a point in the map that contains some information that restricts the likely values of electron density at that point. The procedure could be further extended by having probability distributions describing the likelihood that a particular point in the unit cell is within protein, within solvent, within a particular location in a fragment of protein structure, within a non-crystallographically related region and so on. These probability distributions could be overlapping or non-overlapping. Then for each category of points, the probability distribution for electron density within that category could be formulated as in (22) and our current methods applied.
The procedure described here differs from the reciprocal-space solvent-flattening procedure described previously (Terwilliger, 1999 ▶) in two important ways. One is that the expected electron-density distribution in the non-solvent region is included in the calculations and a formalism for incorporating information about the electron-density map from a wide variety of sources is developed. The second is that the probability distribution for the electron density is calculated using (22) for both solvent and non-solvent regions and values of the scaling parameter β and the map uncertainty σMAP are estimated by a fitting of model and observed electron-density distributions. This fitting process makes the whole procedure very robust with respect to scaling of the experimental data, which otherwise would have to be very accurate in order that the model electron-density distributions be applicable.
Software for carrying out maximum-likelihood density modification (‘Resolve’) and complete documentation is available on the WWW at http://resolve.lanl.gov.
Acknowledgments
The author would like to thank Joel Berendzen for helpful discussions and the NIH and the US Department of Energy for generous support.
References
- Abrahams, J. P. (1997). Acta Cryst. D53, 371–376. [DOI] [PubMed] [Google Scholar]
- American Type Culture Collection (1992). Catalogue of Bacteria and Bacteriophages, 18th ed., pp. 271–272.
- Bricogne, G. (1984). Acta Cryst. A40, 410–445. [Google Scholar]
- Bricogne, G. (1988). Acta Cryst. A44, 517–545. [Google Scholar]
- Cowtan, K. D. & Main, P. (1993). Acta Cryst. D49, 148–157 [DOI] [PubMed] [Google Scholar]
- Cowtan, K. D. & Main, P. (1996). Acta Cryst. D52, 43–48. [DOI] [PubMed] [Google Scholar]
- Giacovazzo, C. & Siliqi, D. (1997). Acta Cryst. A53, 789–798. [Google Scholar]
- Goldstein, A. & Zhang, K. Y. J. (1998). Acta Cryst. D54, 1230–1244. [DOI] [PubMed] [Google Scholar]
- Gu, Y., Zheng, Ch., Zhao, Y., Ke, H. & Fan, H. (1997). Acta Cryst. D53, 792–794. [DOI] [PubMed] [Google Scholar]
- Leslie, A. G. W. (1987). Proceedings of the CCP4 Study Weekend, pp. 25–31. Warrington: Daresbury Laboratory.
- Lunin, V. Y. (1993). Acta Cryst. D49, 90–99. [DOI] [PubMed] [Google Scholar]
- Newman, J., Peat, T. S., Richard, R., Kan, L., Swanson, P. W., Affholter, J. A., Holmes, I. H., Schindler, J. F., Unkefer, C. J. & Terwilliger, T. C. (1999). Biochemistry, 38, 16105–16114. [DOI] [PubMed] [Google Scholar]
- Pannu, N. S. & Read, R. J. (1996). Acta Cryst. A52, 659–668. [Google Scholar]
- Peat, T. S., Newman, J., Waldo, G. S., Berendzen, J. & Terwilliger, T. C. (1998). Structure, 15, 1207–1214. [DOI] [PubMed]
- Perrakis, A., Sixma, T. K., Wilson, K. S. & Lamzin, V. S. (1997). Acta Cryst. D53, 448–455. [DOI] [PubMed] [Google Scholar]
- Podjarny, A. D., Bhat, T. N. & Zwick, M. (1987). Annu. Rev. Biophys. Biophys. Chem.16, 351–373. [DOI] [PubMed] [Google Scholar]
- Prince, E., Sjolin, L. & Alenljung, R. (1988). Acta Cryst. A44, 216–222. [Google Scholar]
- Refaat, L. S., Tate, C. & Woolfson, M. M. (1996). Acta Cryst. D52, 252–256. [DOI] [PubMed] [Google Scholar]
- Roberts, A. L. U. & Brünger, A. T. (1995). Acta Cryst. D51, 990–1002. [DOI] [PubMed] [Google Scholar]
- Rossmann, M. G. & Arnold, E. (1993). International Tables for Crystallography. Vol. B, edited by U. Shmueli, pp. 230–258. Dordrecht/Boston/London: Kluwer Academic Publishers.
- Terwilliger, T. C. (1999). Acta Cryst. D55, 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. & Berendzen, J. (1996). Acta Cryst. D51, 609–618. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellieux, F. M. D. A. P., Hunt, J. F., Roy, S. & Read, R. J. (1995). J. Appl. Cryst.28, 347–351. [Google Scholar]
- Wang, B.-C. (1985). Methods Enzymol.115, 90–112. [DOI] [PubMed] [Google Scholar]
- Wilson, A. J. C. (1949). Acta Cryst.2, 318–321. [Google Scholar]
- Wilson, C. & Agard, D. A. (1993). Acta Cryst. A49, 97–104. [Google Scholar]
- Xiang, S., Carter, C. W. Jr, Bricogne, G. & Gilmore, C. J. (1993). Acta Cryst. D49, 193–212. [DOI] [PubMed] [Google Scholar]
- Zhang, K. Y. J. (1993). Acta Cryst. D49, 213–222. [DOI] [PubMed] [Google Scholar]
- Zhang, K. Y. J., Cowtan, K. D. & Main, P. (1997). Methods Enzymol.277, 53–64. [DOI] [PubMed]
- Zhang, K. Y. J. & Main, P. (1990). Acta Cryst. A46, 41–46. [Google Scholar]