

Abstract
Transforming growth factor (TGF) β1) is an immunoregulatory cytokine involved in self-tolerance and lymphocyte homeostasis. Tgfb1 knock-out (KO) mice develop severe multi-focal autoimmune inflammatory lesions due to [Ca2+]i deregulation in T cells, and die within 3 weeks after birth. Because the calcineurin inhibitor FK506 inhibits the hyperresponsiveness of Tgfb1−/− thymocytes, and because calcineurin Aβ (CNAβ)-deficient mice do not reject allogenic tumours, we have generated Tgfb1−/−Cnab−/− mice to address whether CNAβ deficiency prevents T cell activation and inflammation in Tgfb1−/− mice. Here we show that in Tgfb1−/−Cnab−/− mice inflammation is reduced significantly relative to that in Tgfb1−/− mice. However, both CD4+ and CD8+ T cells in double knock-out (DKO) mice are activated, as revealed by up-regulation of CD11a lymphocyte function-associated antigen-1 (LFA-1), CD44 and CD69 and down-regulation of CD62L. These data suggest that deficiency of CNAβ decreases inflammatory lesions but does not prevent activation of autoreactive T cells. Also Tgfb1−/− T cells can undergo activation in the absence of CNAβ, probably by using the other isoform of calcineurin (CNAα) in a compensatory manner. CNAβ-deficient T cells undergo spontaneous activation in vivo and are activated upon anti-T cell receptor stimulation in vitro. Understanding the role of calcineurin in T cell regulation should open up new therapeutic opportunities for inflammation and cancer.
Keywords: autoimmunity, CNAβ, inflammation, knockout, TGFβ1, T cells
Introduction
Calcineurin (CN), a protein phosphatase activated by Ca2+-calmodulin, is involved in activation of nuclear factor of activated T cells (NFAT). CN is composed of two subunits, a catalytic subunit (A) and a regulatory subunit (B). Three isoforms of catalytic subunit (Aα, Aβ and Aγ) are expressed in vertebrate species. While Aγ is expressed only in testes and brain, Aα and Aβ are expressed ubiquitously [1,2]. CN inhibitors such as cyclosporin A (CsA) and FK506 are potent inhibitors of T cell responses and are used commonly to prevent graft rejection in transplant patients [3]. Upon activation, CN activates NFAT by dephosphorylation. Activated NFAT translocates to the nucleus, where it induces several cytokine genes involved in T cell responses and anergy [4]. Under optimal stimulatory conditions which activate AP-1, NFAT induces Il2, Il4, and Ifng, resulting in productive immune responses [5]. Under tolerogenic conditions, it forms a complex with forkhead box P3 (FOXP3) and together induces regulatory T cell (Treg) generation by induction of Il2ra (CD25) and Ctla4[6]. Because CsA inhibits not only T cell activation but also Treg generation in mice [7] and humans [8], it is important to determine the role of CN in autoimmune T cell responses.
We have found that TGFβ1 prevents autoimmune disease by elevating the threshold level of activation through the Ca2+-CN signalling pathway [9]. Tgfb1−/− thymocytes from young mice with no inflammation have elevated [Ca2+]i levels and exhibit an activated phenotype after suboptimal stimulation. Consistently, Tgfb1−/− thymocytes are more resistant to FK506-mediated inhibition of activation which is dependent upon Ca2+-CN signalling. In the periphery, Tgfb1−/− T cells exhibit an anergic response to receptor-mediated T cell activation, increased activation-induced cell death (AICD) and down-modulation of T cell receptor (TCR), suggesting a previously activated state [10,11]. Increased AICD of Tgfb1−/− T cells is due to a T cell-intrinsic deficiency of TGFβ1, as the addition of exogenous TGFβ1 does not prevent AICD, and induction of the survival factor Bcl-xl is greatly reduced upon activation of Tgfb1−/− T cells [12]. This protective effect is SMAD3-independent as SMAD3-deficient T cells also respond to TGFβ1 [13]. Addition of TGFβ1 to CD4+ T cells during activation with αCD3 +αCD28 prevents Ca2+ influx, NFATc activation and nuclear translocation, which are important for cytokine production and naive T cell proliferation [14]. Addition of TGFβ1 to Tgfb1−/− T cells during in vitro stimulation also prevents their hyperresponsiveness [15]. These studies together suggest that TGFβ1 functions in both cell intrinsic and extrinsic manners.
CNAβ deficiency causes a severe reduction of mature T cells both in the thymus and periphery [2]. The fact that the CN inhibitors CsA and FK506 inhibit allograft rejection suggests that CN signalling is important for proper immune responses [16]. Consistently, Cnab−/− mice do not reject allogenic tumours, suggesting that CNAβ-deficient T cells do not mount a strong immune response against allografts [2]. To test the hypothesis that blocking CN activity in T cells prevents T cell activation and autoimmunity, we combined deficiencies genetically in CNAβ and TGFβ1. As expected, we see a drastic reduction in severity of inflammation in the DKO mice, but surprisingly, both CD4+ and CD8+ splenic T cells are activated in the DKO mice, suggesting that either CNAβ is not required for activation of T cells or CNAα may compensate for the loss of CNAβ. Further analysis of T cells from CNAβ KO mice revealed that mature T cells undergo activation upon stimulation in vitro, confirming that T cell activation does not depend on CNAβ.
Materials and methods
Mice
Tgfb1+/− (BALB/c, N7) (gift from James D. Gorham, Dartmouth Medical School) and Cnab−/− (129/C57BL/6) mice were combined genetically under a University of Cincinnati IACUC protocol.
Polymerase chain reaction (PCR) genotyping
The genotype of newborn pups from double heterozygous matings was determined by PCR amplification of tail DNA and size fractionation on agarose gels [17].
Flow cytometry analysis of splenocytes
Phenotype analysis of splenocytes was determined by four-colour flow cytometry, as described previously [18]. Fluorescein isothiocyanate (FITC-), phycoerythrin (PE-), peridinin chlorophyll (PerCP) or allophycocyanin (APC)-conjugated antibodies to cell surface molecules were purchased from either BD Biosciences (San Diego, CA, USA) or eBioscience (San Diego, CA, USA). Forkhead box P3 (FOXP3) staining kit (clone FKJ-16s) was purchased from eBioscience. R-PE-anti-mouse CD11a lymphocyte function-associated antigen-1 (LFA-1) was purchased from BioDesign (Saco, ME, USA). Splenocytes were surface-stained with fluorochrome conjugated antibodies after blocking with Fc blocking antibodies in fluorescence activated cell sorter (FACS) staining buffer at 4°C in the dark. Cells were washed once with FACS buffer, fixed in 2% paraformaldehyde and acquired using a BD-liquid silicone rubber (LSR) flow cytometer. Intracellular FOXP3 staining was performed according to the manufacturer's protocol, as described in our earlier studies [15]. Flow cytometry data were analysed by CellQuest software.
Cell culture and cytokine measurements
For cytokine measurements, splenocytes were isolated from Cnab−/− and control mice and stimulated with CD3/CD28 beads (Dynal Biotech, Oslo, Norway) in 48-well plates for 3 days. Culture supernatants were harvested and assayed for cytokines by enzyme-linked immunosorbent assay (ELISA) kits from BD Biosciences. Briefly, 96-well ELISA plates were coated with capture antibodies in coating buffer (NaHCO3/Na2CO3 buffer, pH 9·5) overnight at 4°C. Plates were then washed with buffer and blocked with assay diluent buffer with 1% bovine serum albumin (BSA). Plates were washed and incubated with diluted supernatants and standard cytokine, washed again, and incubated with biotinylated detection antibody/streptavidin-horseradish peroxidase (HRP). Plates were washed again and developed with substrate and then read in a microplate reader. Cytokine concentrations were calculated using serial dilutions of the standard run in the same plate.
Inflammation score
Animals were euthanized following institutional guidelines, and tissues were fixed in 10% neutral buffered formalin. Tissues were dehydrated through a gradient of alcohol and xylene, embedded in paraffin and 5-µm sections were cut and haematoxylin and eosin (H&E)-stained. An inflammation score was assigned to each tissue depending on the severity of the inflammatory cell infiltrate: 0 (no inflammation), 0·5 (very mild), 1·0 (mild), 2 (moderate), 3 (severe) and 4 (very severe) [10,19], as follows: very mild: the inflammatory cells are very infrequent and usually involve fewer than 10 cells per section; mild: inflammatory component is composed of fewer than 100 cells – the inflammation is confined to a few areas in the tissues; moderate: inflammation involves multiple areas in the tissue or is a large area composed of more than 100 inflammatory cells but fewer than 1000 cells – there may be associated tissue damage near the inflammatory component; severe: inflammatory cells comprise large multi-focal areas of the tissue and usually involve at least 20% of the tissue – there are greater than 1000 cells involved and there is clear alteration of the adjacent tissues either due to compression from the inflammatory component or necrosis of the adjacent tissue; very severe: similar to severe, only nearly all areas of the tissue are affected – there is alteration of the normal parenchyma appearance. Data for the most commonly affected organs are shown in the figures.
Results
Unaltered splenocyte and T cell numbers in DKO mice
CNAβ is important for normal T cell development, as Cnab−/− mice have a defect in T cell development with decreased numbers of mature T cells both in the thymus and periphery [2]. We postulated that a reduction in CN-mediated signalling in Tgfb1−/− T cells would prevent T cell activation and inflammation, not only because it would decrease the number of mature T cells, but also because it mediates hyperresponsiveness in the absence of TGFβ1 signalling [9].
Comparison of splenocyte counts from 3–4-week-old Cnab−/− and DKO mice reveals no differences (Fig. 1a). There is also no difference in percentages of CD4+ and CD8+ T cell subsets (Fig. 1b, dot-plots). This is in contrast to Tgfb1−/− mice, which exhibit a severe decrease in total splenocytes and T cells in their spleen [10,19]. However, there is a decrease in expression of CD8 on T cells in DKO mice. Similarly, there is a decrease in CD3 expression on DKO T cells (Fig. 1b, histograms). Consistent with the inflammation and consequent loss of T cells from the spleens of Tgfb1−/− mice [10], there is a decrease in CD4+ T cells in Tgfb1−/−Cnab+/− mice compared to control mice (Fig. 1c, dot-plots). Expression of CD3 and CD8 is also decreased on T cells in Tgfb1−/−Cnab+/− mice (Fig. 1c, histograms). These data suggest that while TGFβ1 deficiency affects the balance in T cell homeostasis towards an inflammatory phenotype CNAβ deficiency counteracts it, at least partially. However, CNAβ deficiency does not prevent down-modulation of CD3 and CD8 on TGFβ1-deficient T cells which exhibit an activated phenotype.
Fig. 1.
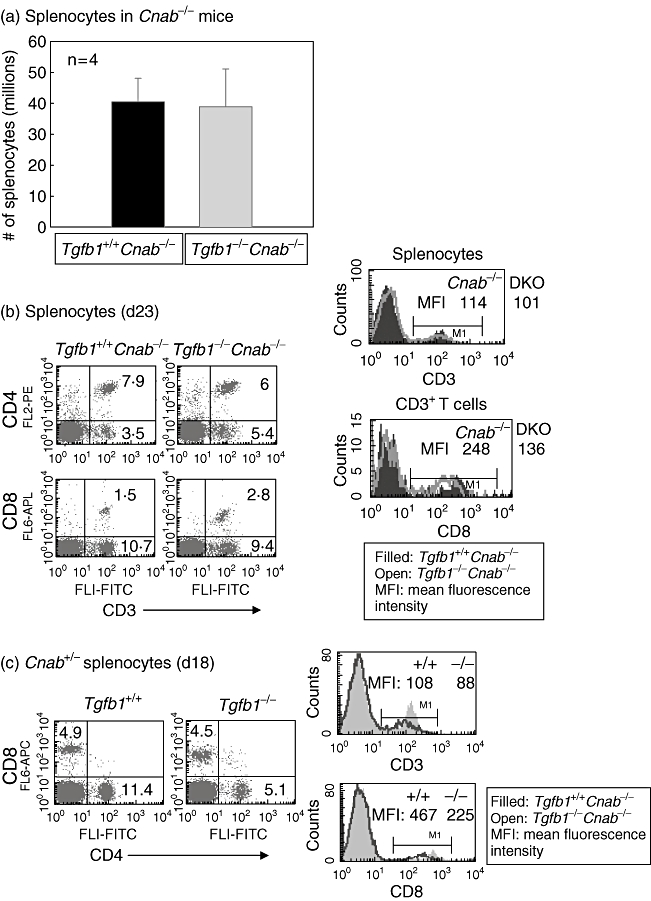
Splenocytes and T cell subsets are unaltered in double knock-out (DKO) mice, but their surface CD3 and CD8 are down-regulated. Total splenocytes were prepared, counted and stained for surface expression of CD3, CD4 and CD8, as described in the Methods section. Total splenocyte numbers in Cnab−/− and DKO mice are shown (a). Flow cytometric analyses of splenocytes expressing CD3 and CD4 or CD8 from Cnab−/− and DKO mice (b) and Tgfb1+/+ and Tgfb1−/− mice on Cnab+/− background (c) are shown in dot plots. Histogram overlays of CD3 expression on splenocytes [upper panel in (b) and (c)] and CD8 expression on CD3+ gated splenocytes [lower panel in (b)] or total splenocytes [lower panel in (c)] from the above mice.
CNAβ deficiency decreases inflammation in Tgfb1−/− mice
Because DKO mice do not live longer, they were analysed for inflammatory lesions. Contrary to expectations, the moderate to severe inflammation found in Tgfb1−/−Cnab+/− mice, especially in heart and lung (Fig. 2a), is reduced to mild inflammation in those organs in Tgfb1−/−Cnab−/− mice (Fig. 2b) on the same genetic background. Diaphragm and quadriceps muscle are usually not affected in DKO mice. Surprisingly, there is no significant reduction in severity of inflammation in pancreas, although there is a reduction in the number of DKO mice (50%, Fig. 2b) that have pancreatic inflammatory lesions (compare to 77% of Tgfb1−/−Cnab+/−, Fig. 2a). Analysis of the organ specificity of inflammation also suggests that only two to four organs are affected in DKO mice, usually liver, lung and pancreas. H&E staining of liver, heart and lung sections from Tgfb1+/+Cnab−/− (Fig. 3, top panels), Tgfb1−/−Cnab−/− (Fig. 3, middle panels) and Tgfb1−/− mice (Fig. 3, bottom panels) are shown for comparison and demonstrate that there is a severe reduction in inflammation in DKO mice compared to Tgfb1−/− mice. However, we do not know whether such mild inflammation is sufficient to cause death in DKO mice.
Fig. 2.
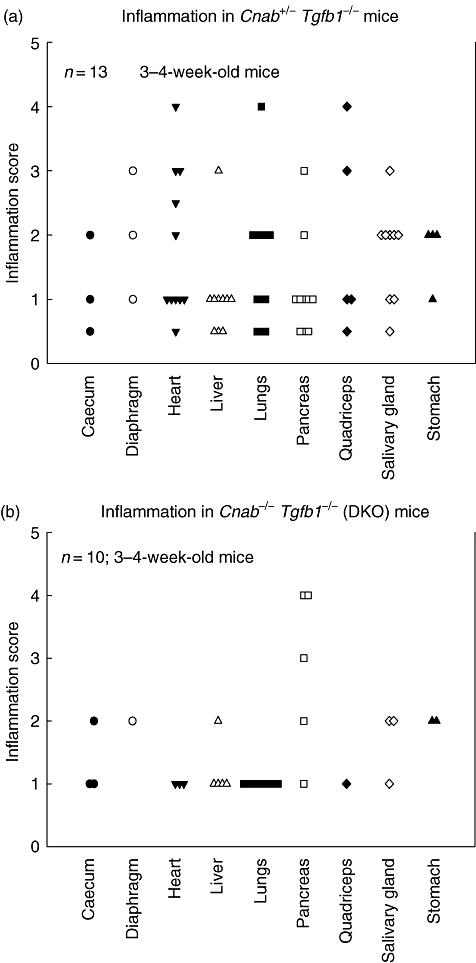
Calcineurin Aβ (CNAβ) deficiency decreases inflammation in transforming growth factor (TGF) β1 knock-out mice. Mice were killed and immersed in 10% neutral buffered formalin (NBF) for fixation after the lungs and gut were profused with 10% NBF. Formalin-fixed tissues were embedded in paraffin blocks; 5–10 µm sections were cut and haematoxylin and eosin-stained and analysed for inflammatory lesions by a pathologist. Inflammation score was determined as described in the text. Inflammation score in various organs of Tgfb1−/−Cnab+/− (a) and Tgfb1−/−Cnab−/− mice (b). Each mouse was represented by one symbol. Organs with no inflammation are not shown.
Fig. 3.
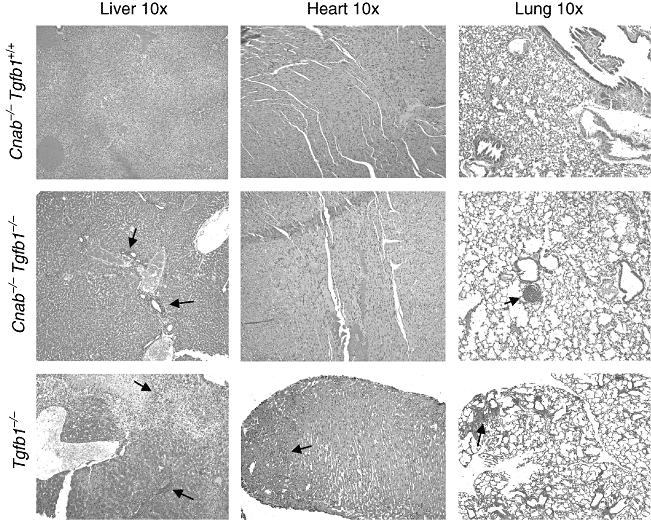
Reduced inflammation in double knock-out (DKO) mice. Formalin-fixed tissues were embedded in paraffin blocks; 5–10-µm sections were cut and haematoxylin and eosin-stained and analysed for inflammatory lesions by a pathologist. Representative sections of liver (left), heart (middle) and lung (right) from Cnab−/− (upper), DKO (middle) and Tgfb1−/− (lower) mice are shown. Inflammation lesions are indicated by a black arrow.
CNAβ deficiency does not prevent activation of Tgfb1−/− T cells
Flow cytometric analysis of splenocytes from Tgfb1−/−Cnab−/− and littermate Tgfb1+/+Cnab−/− mice suggests that CD62L (Fig. 4a, centre row) and CD3 and CD8 (Fig. 1b, histograms) are down-regulated, and LFA-1 and CD44 (Fig. 4a, top and bottom rows) and CD49d (not shown) are up-regulated in Tgfb1−/−Cnab−/− T cells. Analysis of CD69 also suggests that the CD69+ population (activated T cells) is increased in DKO mice (Fig. 4b). These data suggest that although CNAβ-deficiency drastically reduces inflammation, CNAβ is not essential for activation of self-reactive T cells in the absence of TGFβ1 [10,18].
Fig. 4.
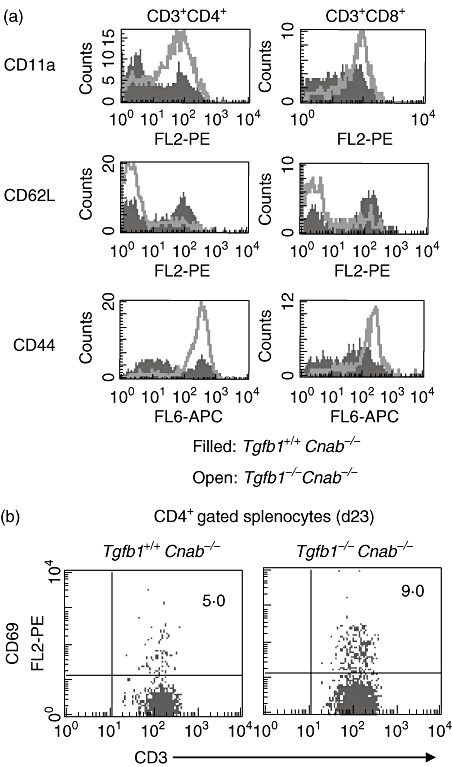
T cells are activated in double knock-out (DKO) mice. Splenocytes were prepared from control and DKO mice and stained for CD3, CD4, CD8 and activation markers CD11a lymphocyte function-associated antigen-1 (LFA-1), CD44, CD62L using fluorochrome-conjugated antibodies after blocking with Fc blocking antibodies in fluorescence activated cell sorter (FACS) staining buffer at 4°C in the dark. Cells were washed from unbound antibodies and fixed in 2% paraformaldehyde and acquired using a BD-liquid silicone rubber (LSR) flow cytometer. Data are analysed using CellQuest software. Histogram overlays of CD11a LFA-1, CD44 and CD62L on CD4+ (left panels) and CD8+ gated (right panels) T cells are shown in (a). CD69 expression on CD4+ T cells is shown in dot plots (b).
CN deficiency does not enhance the survival of Tgfb1−/− mice
Analysis of 12 Tgfb1 Cnab DKO mice revealed little difference in survival compared to Tgfb1−/−Cnab+/− mice on the same genetic background. Fifty per cent survival was at about 27 days for Tgfb1−/− mice and about 22 days for DKO mice (Fig. 5). However, all the mice in both groups died by the end of 5 weeks, suggesting that CNAβ deficiency does not alter significantly the survival of TGFβ1 KO mice.
Fig. 5.
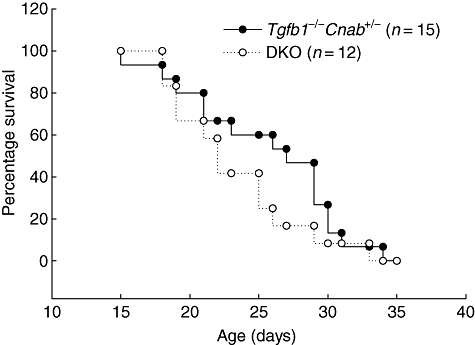
Calcineurin Aβ (CNAβ)-deficiency does not extend the survival of Tgfb1−/− mice. Tgfb1−/−Cnab+/− (n = 15) and Tgfb1−/−Cnab−/−[double knock-out (DKO); n = 12] mice were monitored until they were either moribund or they were euthanized for tissue collection when they started losing weight.
We have analysed activation of T cells from Cnab−/− and littermate control mice before and after in vitro stimulation using multi-colour flow cytometry. Ex vivo analysis of splenic T cells revealed an increase in the proportion of CD4+ T cells that are spontaneously activated (CD62L-CD44+) in Cnab−/− compared to control mice. This activation is similar whether the Cnab−/− mice are on Tgfb1+/+ or Tgfb1+/− backgrounds (Fig. 6a). However, T cells from DKO mice (3 weeks old) are highly activated, resembling the phenotype of Tgfb1−/− T cells (Fig. 4). Analysis of CD25 and FOXP3 expression revealed a similar proportion of FOXP3+ Tregs in both groups, albeit a decrease in total T cells and Treg numbers was found in Cnab−/− mice (Fig. 6b, data not shown). To test whether CNAβ signalling is required for T cell activation and response, we stimulated T cells in vitro from Cnab−/− and control mice. The expression of CD25 is up-regulated upon anti-CD3 and anti-CD28 stimulation on control and Cnab−/− T cells, suggesting that T cell activation is independent of CNAβ expression (Fig. 6c). Analysis of cytokines secreted by splenocytes in these cultures indicates that Cnab−/− T cells produce elevated levels of interferon (IFN)-γ and interleukin (IL)-6 upon anti-TCR stimulation compared to control cultures (Figs 6d and e).
Fig. 6.
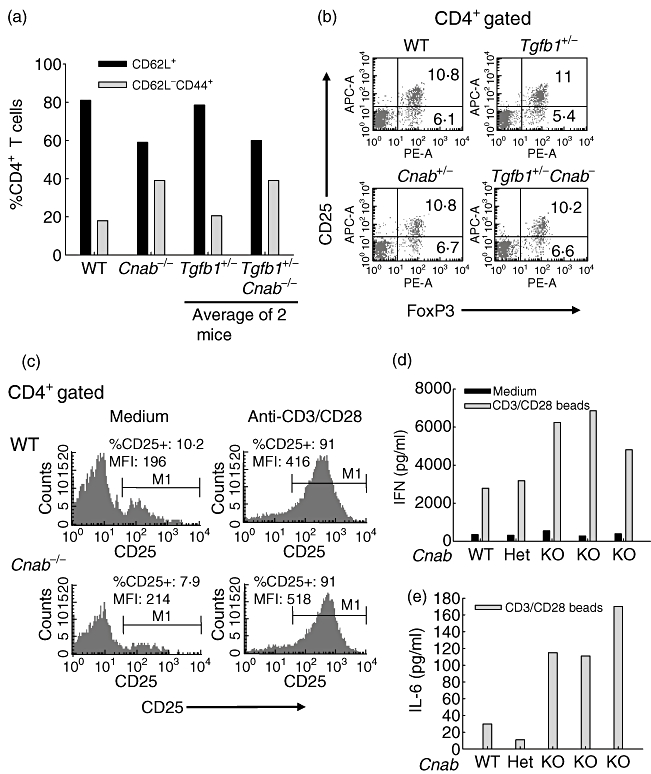
Calcineurin Aβ (CNAβ) deficiency increases spontaneous activation of T cells in vivo and effector differentiation in vitro. (a, b) Splenocytes from 2-month-old Cnab−/−, Cnab−/−Tgfb1+/− and control mice were prepared and surface-stained for CD4, CD8, CD62L and CD44 and analysed as described in Fig. 4. For forkhead box P3 (FOXP3) staining, cells were surface-stained with CD4 and CD25 antibodies and then fixed/permeabilized and stained with FOXP3 antibody, as described in the Methods. For in vitro stimulation experiments, 1 × 106 splenocytes from 7-week-old Cnab−/− and control mice were cultured in the presence of anti-CD3/CD28 microbeads from Invitrogen (T cell expander beads) for 3 days and cells were stained for CD4 and CD25 as described above and analysed by flow cytometry (c). Supernatants were collected and assayed for cytokines by sandwich enzyme-linked immunosorbent assay using kits from BD-Biosciences (d, e). Interleukin-6 levels in unstimulated (medium) cultures were below detection levels.
Discussion
Tgfb1−/− mice die around weaning age due to multi-focal autoimmune disease mediated primarily by autoreactive T cells [18]. TGFβ1-deficient T cells are hyperresponsive to stimulation due to increased [Ca2+]i-CN signalling [9,10]. Because CNAβ-deficient mice exhibit a defect in T cell maturation, and because mature T cells are hyporesponsive to stimulation, we have generated DKO mice in the hope of decreasing T cell activation. As expected, we found a significant reduction of inflammatory lesions in DKO mice. However, DKO mice die within 5 weeks after birth, due possibly to T cell activation through CNAα. In our preliminary studies we have observed that CNAα is expressed in the thymus, as well as splenocytes in Cnab−/− and control mice. Our in vitro stimulation experiments confirm the hypothesis that CNAβ is dispensable for T cell activation and that absence of CNAβ signalling actually increases spontaneous activation of T cells in vivo, and production of inflammatory cytokines upon stimulation in vitro. As TGFβ1 is required for induction of FOXP3 to induce the conversion of CD4+CD25- T cells into adaptive Tregs, we believe that a defect in the generation of adaptive Tregs or inducible Tregs (iTregs) in both KO mouse strains (Cnab−/− and Tgfb1−/−) could also be a cause of the activation of T cells in DKO mice. We are currently studying the role of TGFβ1 and CNAβ in the generation of iTregs. These studies will be published elsewhere.
Because elimination of either CD4+ or CD8+ T cells alone does not reduce the severity of inflammation in Tgfb1−/− mice [19], the mild inflammation in DKO mice is probably not due to a decrease in peripheral T cells but rather to a defect in their effector function [2]. This is consistent with the original observation in Cnab−/− mice that they do not mount vigorous anti-tumour responses against allogenic tumours [2]. The data also suggest that a milder form of inflammation may be sufficient to cause early lethality if T cell regulation is diminished. It has been shown that administration of CsA, an inhibitor of CN phosphatase activity, to new-born BALB/c mice induces organ-specific autoimmunity [7] and also inhibits FOXP3 expression and Treg generation, both in vitro and in vivo[8,20]. The CsA effect on Treg generation could be reversed by IL-2, suggesting that CsA affects Treg generation through inhibition of IL-2 production [21–24]. Recently, it was reported that FK506 activates nuclear factor (NF)-κB and tumour necrosis factor (TNF)-α expression in macrophages, and this effect was shown to be through its inhibition of CN activity [25]. This suggests that innate immune responses may be up-regulated in Cnab−/− and DKO mice leading to increased proinflammatory cytokine production such as TNF-α, which can contribute to early lethality of DKO mice.
The above data leave open the possibility that there could be unanticipated phenotypes resulting from metabolic or other defects that present in DKO but not in Tgfb1 KO mice. Because T cells can become activated in older Cnab−/− mice, the inherent response of T cells could cause unknown phenotypes through soluble factors such as IL-1, IFN-γ, TNF-α and IL-6. We are particularly interested in the proinflammatory cytokines IL-6 and IFN-γ, because we know that Tgfb1−/− mice live longer on Il6−/− and Ifng−/− backgrounds, although Tgfb1−/−Il6−/− mice live much longer than Tgfb1−/−Ifng−/−mice [26,27]. Consistent with these data, we observed increased production of IFN-γ and IL-6 cytokines by Cnab−/− T cells upon stimulation, suggesting that proinflammatory cytokine secretion is regulated by CNAβ. Another possibility is that under such an environment DKO T cells utilize some CN signalling through the remaining CNAα isoform and undergo activation in the absence of TGFβ1. In our preliminary studies we have observed that Cnab−/− mice die after 8 months due to metastatic B cell lymphomas. Studies are under way to address whether CNAβ-deficiency contributes to early lethality in an inflammation-independent manner.
Acknowledgments
The authors thank Leena Pathak for PCR genotyping of the mice. The authors also thank Sandy Schwemberger for expert assistance in flow cytometry. This study was supported by NIH AI067903, ES06096 and CA84291 to TD, IRG7400128 to RB and by a grant from Shriners of North America to GFB.
Disclosure
The authors have no financial conflict of interest.
References
- 1.Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–70. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- 2.Bueno OF, Brandt EB, Rothenberg ME, Molkentin JD. Defective T cell development and function in calcineurin A beta-deficient mice. Proc Natl Acad Sci USA. 2002;99:9398–403. doi: 10.1073/pnas.152665399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–7. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 4.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 5.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–47. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y, Borde M, Heissmeyer V, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi S, Sakaguchi N. Organ-specific autoimmune disease induced in mice by elimination of T cell subsets. V. Neonatal administration of cyclosporin A causes autoimmune disease. J Immunol. 1989;142:471–80. [PubMed] [Google Scholar]
- 8.Mantel PY, Ouaked N, Ruckert B, et al. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. 2006;176:3593–602. doi: 10.4049/jimmunol.176.6.3593. [DOI] [PubMed] [Google Scholar]
- 9.Bommireddy R, Ormsby I, Yin M, Boivin GP, Babcock GF, Doetschman T. TGFbeta1 inhibits Ca2+-calcineurin-mediated activation in thymocytes. J Immunol. 2003;170:3645–52. doi: 10.4049/jimmunol.170.7.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bommireddy R, Saxena V, Ormsby I, et al. TGF-beta1 regulates lymphocyte homeostasis by preventing activation and subsequent apoptosis of peripheral lymphocytes. J Immunol. 2003;170:4612–22. doi: 10.4049/jimmunol.170.9.4612. [DOI] [PubMed] [Google Scholar]
- 11.Christ M, McCartney-Francis NL, Kulkarni AB, et al. Immune dysregulation in TGF-beta 1-deficient mice. J Immunol. 1994;153:1936–46. [PubMed] [Google Scholar]
- 12.Chen W, Jin W, Tian H, et al. Requirement for transforming growth factor beta1 in controlling t cell apoptosis. J Exp Med. 2001;194:439–53. doi: 10.1084/jem.194.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKarns SC, Schwartz RH. Distinct effects of TGF-beta 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: a role for T cell intrinsic Smad3. J Immunol. 2005;174:2071–83. doi: 10.4049/jimmunol.174.4.2071. [DOI] [PubMed] [Google Scholar]
- 14.Chen CH, Seguin-Devaux C, Burke NA, et al. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J Exp Med. 2003;197:1689–99. doi: 10.1084/jem.20021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bommireddy R, Babcock GF, Singh RR, Doetschman T. TGFbeta1 deficiency does not affect the generation and maintenance of CD4(+)CD25(+)FOXP3(+) putative T(reg) cells, but causes their numerical inadequacy and loss of regulatory function. Clin Immunol. 2008;127:206–13. doi: 10.1016/j.clim.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin–cyclosporin A and FKBP–FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 17.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–86. [PubMed] [Google Scholar]
- 18.Bommireddy R, Pathak LJ, Martin J, et al. Self-antigen recognition by TGFbeta1-deficient T cells causes their activation and systemic inflammation. Lab Invest. 2006;86:1008–19. doi: 10.1038/labinvest.3700460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bommireddy R, Engle SJ, Ormsby I, Boivin GP, Babcock GF, Doetschman T. Elimination of both CD4(+) and CD8(+) T cells but not B cells eliminates inflammation and prolongs the survival of TGFbeta1-deficient mice. Cell Immunol. 2004;232:96–104. doi: 10.1016/j.cellimm.2005.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeiser RS, Nguyen VH, Beilhack A, et al. Inhibition of CD4+CD25+ regulatory T cell function by calcineurin dependent interleukin-2 production. Blood. 2006;108:390–9. doi: 10.1182/blood-2006-01-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai M, Kitade H, Mathieu C, Waer M, Pirenne J. Inhibitory and stimulatory effects of cyclosporine A on the development of regulatory T cells in vivo. Transplantation. 2005;79:1073–7. doi: 10.1097/01.tp.0000153505.73700.32. [DOI] [PubMed] [Google Scholar]
- 22.Smiley ST, Csizmadia V, Gao W, Turka LA, Hancock WW. Differential effects of cyclosporine A, methylprednisolone, mycophenolate, and rapamycin on CD154 induction and requirement for NFkappaB: implications for tolerance induction. Transplantation. 2000;70:415–9. doi: 10.1097/00007890-200008150-00005. [DOI] [PubMed] [Google Scholar]
- 23.Smith CR, Mohanakumar T, Shimizu Y, et al. Brief cyclosporine treatment prevents intrathymic (IT) tolerance induction and precipitates acute rejection in an IT rat cardiac allograft model. Transplantation. 2000;69:294–9. doi: 10.1097/00007890-200001270-00016. [DOI] [PubMed] [Google Scholar]
- 24.Gao EK, Lo D, Cheney R, Kanagawa O, Sprent J. Abnormal differentiation of thymocytes in mice treated with cyclosporin A. Nature. 1988;336:176–9. doi: 10.1038/336176a0. [DOI] [PubMed] [Google Scholar]
- 25.Kang YJ, Kusler B, Otsuka M, et al. Calcineurin negatively regulates TLR-mediated activation pathways. J Immunol. 2007;179:4598–607. doi: 10.4049/jimmunol.179.7.4598. [DOI] [PubMed] [Google Scholar]
- 26.Gorham JD, Lin JT, Sung JL, Rudner LA, French MA. Genetic regulation of autoimmune disease: BALB/c background TGF-beta 1-deficient mice develop necroinflammatory IFN-gamma-dependent hepatitis. J Immunol. 2001;166:6413–22. doi: 10.4049/jimmunol.166.10.6413. [DOI] [PubMed] [Google Scholar]
- 27.Bommireddy R, Doetschman T. TGFbeta1 and T(reg) cells: alliance for tolerance. Trends Mol Med. 2007;13:492–501. doi: 10.1016/j.molmed.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]