

Abstract
Polycystic ovary syndrome (PCOS) is an endocrine-metabolic disorder associated with insulin resistance and compensatory hyperinsulinemia. Scarce information is available on the expression of molecules involved in the insulin pathway in endometria from women with PCOS. Therefore, we examined the protein levels of insulin-signaling molecules, like insulin receptor, insulin-receptor substrate (IRS)-1, pIRS-1Y612, Akt, AS160, pAS160T642 and GLUT4 in endometria from PCOS women with or without hyperinsulinemia. Protein levels were assessed by Western blot and immunohistochemistry in 21 proliferative-phase endometria from control women (CE = 7), normoinssulinemic PCOS women (PCOSE-NI = 7) and hyperinsulinemic PCOS women (PCOSE-HI = 7). The data show no differences in the expression of insulin receptor between all groups as assessed by Western blot; however, IRS-1 and pIRS-1Y612 were lower in PCOSE-HI than controls and PCOSE-NI (P < 0.05). AS160 was detected in all analyzed tissues with similar expression levels between groups. Importantly, PCOSE-HI exhibited lower levels of pAS160T642 (P < 0.05) and of GLUT4 (P < 0.05) compared with CE. The immunohistochemistry for insulin receptor, IRS-1, Akt, AS160 and GLUT4 showed epithelial and stromal localization; IRS-1 staining was lower in PCOSE-HI (P < 0.05). In conclusion, human endometrium has the machinery for glucose uptake mediated by insulin. The diminished expression of GLUT4, as well as the lower level of pIRS-1Y612 and pAS160T642 exhibited by PCOSE-HI, suggests a disruption in the translocation of vesicles with GLUT4 to the cell surface in these patients.
INTRODUCTION
Polycystic ovary syndrome (PCOS) is a steroid-related disorder of unknown etiology that affects 5%–10% of women during their reproductive years (1). The clinical characteristics include oligo- or anovulation, clinical or biochemical signs of hyperandrogenism and ultrasound evidence of polycystic ovaries (2). In addition, the Androgen Excess Society has recently reported that hyperandrogenism is necessary for the diagnosis of the syndrome (3). Up to 60% of PCOS women who show compensatory hyperinsulinemia exhibit insulin resistance ((1,4,5)).
It has been reported that resistance to insulin in PCOS women is characterized by a receptor postbinding failure in the action of this hormone (6). However, evidence shows that even in insulin-resistant patients without PCOS the molecules involved in the insulin pathway do not present a common pattern of molecular alterations in different tissues (1), as has been described in adipose and muscle tissues. For example, insulin- receptor substrate (IRS)-1 protein expression differs between adipocytes and muscle of women with PCOS compared with controls (6–8). Therefore, it is important to study the signal transduction implicated in glucose uptake in PCOS women, because it is possible that their hyperinsulinemia is associated with the development of the syndrome, as shown by the overproduction of androgens by the ovarian theca cells from hyperinsulinemic PCOS women (10).
Recently, it has been reported that androgens can modulate the insulin- dependent glucotransporter (GLUT4) content and translocation to plasma membrane, and this steroid hormone also induces changes in Akt and PKCζ/λ phosphorylation patterns in cultured skeletal muscle cells from neonate rats (11). Moreover, it was reported that in cultures of endometrial epithelial cells testosterone reduces protein expression of IRS-1 and GLUT4 (12). This evidence suggests that the hyperandrogenic condition present in PCOS patients could disrupt the insulin-signaling pathway in various tissues.
In addition to the alterations described in the ovarian function of PCOS women, the failure in their reproductive capacity involves other tissues, such as the endometrium. This tissue has a cyclic behavior regulated by ovarian steroids, and in PCOS endometrium we have observed overexpression of steroid receptors and coactivators (13,14) and deregulation of endometrial homeostasis (15–17). In addition, endometria from PCOS women exhibit alterations in the levels of molecules associated with uterine receptivity (18). However, little information is available about the regulatory role exerted by insulin in the endometria from control and PCOS women.
In the signaling pathway, the binding of insulin to its receptor provokes auto-phosphorylation in tyrosine residues and phosphorylation on IRS (IRS-1 and -2) (19,20). When the PI3K pathway is activated (1,21), phosphorylation of Akt is achieved in T308 and S473 residues ((19,22,23)). One of the several substrates that may be influenced by activated Akt is AS160, a Rab-GAP protein, which is phosphorylated in T642. This phosphorylated protein acts as a mediator for the translocation of the vesicle with GLUT4 to the cell surface, which results in glucose uptake by the cells (24–26). Recently, it was reported that muscle biopsy tissue from PCOS patients showed reduced insulin-stimulated AS160 phosphorylation compared with muscle tissue from control women (7).
Mioni et al. (27) reported diminished levels of GLUT4 mRNA and protein in endometria from PCOS women who were normoinsulinemic compared with controls and that this decrease was even greater in PCOS women with hyperinsulinemia and obesity (27,28). Nevertheless, scarce information is available related to the expression of other molecules of the insulin signaling pathway. Therefore, based on this evidence, in the present investigation we examined whether the hyperinsulinemia and hyperandrogenemia present in PCOS modifies the protein levels of several molecules involved in the insulin pathway in endometria from PCOS women with (PCOS-HI) or without (PCOS-NI) hyperinsulinemia.
MATERIALS AND METHODS
Antibodies and Reagents
Monoclonal antibodies for insulin receptor β–subunit and β-actin were purchased from Biosource (Camarillo, CA, USA) and Sigma (St. Louis, MO, USA), respectively. Polyclonal antibodies for AS160 (Rab-GAP) were obtained from Millipore (Billerica, MA), pAS160T642 from Biosource, Akt1 from BD Pharmingen (Franklin Lakes, NJ, USA), IRS-1 and GLUT4 from Santa Cruz Biotechnolgy (Santa Cruz, CA, USA) and pIRS-1 from Abcam (Cambridge, UK). Secondary antibodies were purchased from Amersham Biosciences (Piscataway, NJ, USA). Protease-inhibitor cocktail was obtained from Roche Molecular Biochemicals (Mannheim, Germany), BCA protein assay kit from Pierce (Rockford, IL, USA) and labeled streptavidin biotin kit from Dako (Carpinteria, CA, USA). Hormone determinations were assayed by use of commercial kits: serum testosterone, estradiol and progesterone by solid-phase, competitive chemiluminescent enzyme immunoassay (Ortho-Clinical Diagnostics, Buckinghamshire, UK); androstenedione by radioimmunoassay (Siemens, Los Angeles, CA, USA); sex hormone–binding globulin (SHBG) concentration by Immulite and solid-phase chemiluminescent immunometric assay (Siemens, Surrey, UK).
Subjects
Human endometria were obtained with a Pipelle suction curette from the corpus of the uteri of women with PCOS. Glucose and insulin levels were evaluated by an oral glucose tolerance test with a 75-g load of glucose. To determine the hyperinsulinemic condition we measured plasma glucose and insulin levels at 2 h after administration of the load of glucose. The diagnosis of hyperinsulinemia was determined when levels of insulin were 2 standard deviations (SD) of insulin concentration over the mean of the control group, as in previous studies (13). The insulin values (mean ± SD) were: control group, 49.6 ± 11.6 μIU/mL; PCOS-NI, 34.4 ± 19.1 μIU/mL; PCOS-HI, 154.2 ± 46.3 μIU/mL. All women had normal glycemic values during the oral tolerance glucose test. This measurement was performed on each subject on the day that the endometrial sample was obtained. Specimens from the study groups were proliferative endometria from women with PCOS who presented with or without hyperinsulinemia (n = 7 PCOSE-HI and n = 7 PCOSE-NI, respectively).
The diagnosis of PCOS was made according to Rotterdam Consensus (Rotterdam European Society for Human Reproduction and Embryology/American Society for Reproductive Medicine, 2003) (2) and Androgen Excess Society criteria (3) for the definition of PCOS. The exclusion criteria were women who presented with hyperprolactinemia (prolactin >35 ng/mL), hypothyroidism (thyroid-stimulating hormone >5 UI/L), androgen-secreting tumors (total testosterone >2 ng/mL; DHEAS >3600 μg/mL), Cushing syndrome (urine cortisol concentration <150 μg/24 h and fasting plasma concentration of cortisol 5–25 μg/dL), congenital adrenal hyperplasia (17-OH progesterone >2,5 ng/mL) and women with diabetes or treatment with hormones and/or ovulation induction. The reference values are from the Endocrinology Laboratory of the University of Chile Clinical Hospital.
Control endometria (CE) were obtained from seven fertile healthy women during the proliferative phase of the menstrual cycle at the time of hysterectomy performed to treat benign uterus pathology. Controls were selected in the proliferative phase because of the similar morphology between proliferative endometrium and PCOSE. None of the women, neither controls nor those with PCOS, had received hormonal therapy within 3 months before recruitment into the study, and the endometria used in this investigation all showed normal morphology. The proliferative phase in CE and PCOSE was confirmed by an experienced pathologist on the basis of histological dating and classification according to Noyes criteria (29). The control women and the women with PCOS (PCOSE-NI and PCOSE-HI) were accrued prospectively, and each group was recruited independently.
This investigation was approved by the ethics committees from the San Borja-Arriarán Clinical Hospital and University of Chile Clinical Hospital, School of Medicine, University of Chile. Informed written consent was obtained from all subjects.
Western Blot
As previously reported (15,17), the endometrial tissue was homogenized in a lysis buffer (HEPES 20 mmol/L, EDTA 2 mmol/L, EGTA 2 mmol/L, Triton 1%, PMSF 5 μmol/L, Na3VO4 50 μmol/L) containing protease inhibitor cocktail (Roche Molecular Biochemicals). After centrifugation at 10,000g for 20 min at 4°C, protein concentration was determined by use of the BCA protein Assay kit (Pierce). Total proteins (50 μg) were denatured and fractionated by use of 7.5% one-dimensional SDS-PAGE and transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA, USA).
Membranes were blocked at room temperature for 2 h in Tris-buffered saline/Tween 20 (TBST) (20 mmol/L Tris, pH 7.6; 137 mmol/L NaCl; 0.1% Tween 20) containing 10% nonfat dry milk (for all markers). Subsequently, the membranes were washed three times for 7 min each in TBST and then incubated at 4°C with rocking overnight with antibodies against IRS-1 (1:500), pIRS-1Y612 (1:300), AS160 (1: 2000), pAS160T642 (1:500) and GLUT4 (1:250), and then were incubated room temperature for 2 h and 1 h, respectively, with antibodies against human insulin receptor β (1:250) and β-actin (1:15,000). The membranes were then washed three times for 7 min each with TBST, followed by incubation for 30 min at room temperature with antimouse IgG, peroxidase-conjugated species-specific (1:2500 for insulin receptor β, and 1:5000 for β-actin) or 1 h with antirabbit IgG peroxidase-conjugated species-specific (1:3000 for IRS-1 and pIRS-1Y612; 1:8000 for AS160; 1:1500 for pAS160 T642 and 1:7000 for GLUT4), while being rocked. Then, after three washings of 7 min each with TBST, the bound antibodies were detected with an enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ, USA). Band intensities were quantified by scanning densitometry using the UnSCAN-IT software Automated Digitizing System, version 5.1, normalized relative to β-actin and expressed as arbitrary units (AU).
Immunohistochemistry
Immunostaining for insulin receptor, IRS-1, Akt, AS160 and GLUT4 was performed on 5-μm sections of formalin-fixed, paraffin-embedded endometrial biopsies. Tissue sections were deparaffinized in xylene and hydrated gradually through graded alcohols. The sections were incubated in antigen retrieval solution (100 mmol/L Tris buffer, pH 9.5) at 100ºC for 20 min. Endogenous peroxidase activity was prevented by incubating the samples in 0.3% hydrogen peroxide in phosphate-buffered saline for 30 min. Nonspecific antibody binding was prevented with specific blocker of a Histostain Bulk kit for 1 h. Different dilutions for primary antibodies were used: insulin receptor β subunit (1:200), IRS-1 (1:750), Akt (1:600), AS160 (1:2000), GLUT4 (1:1800). Negative controls were analyzed on adjacent sections and incubated without primary antibody and with nonimmune species-specific antisera.
The secondary antibody was a biotinylated antimouse/antirabbit immunoglobulin. The reaction was developed by the streptavidin-peroxidase system, and 3,3′ diaminobenzidine was used as the chromogen; counterstain was carried out with hematoxylin. We performed immunohistochemical evaluation for each protein by use of the HScore (histochemical score), a semiquantitative analysis described by Lessey et al. (30) and validated in our laboratory (15). The HScore corresponds to: [P] (i + 1)/100, where [P] is the percentage of positively stained cells and i is the intensity of the staining on a scale of 1–3 (1 = low intensity, 2 = mid intensity and 3 = higher intensity). Each protein was evaluated in the functional layer by three independent observers blinded to patient category, and the positive staining was assessed in at least 3000 cells per sample. In all the cases, the antigen studied was evaluated in a Nikon optical microscope (Nikon, Melville, NY, USA).
Statistics
The number of subjects in this study was calculated assuming α = 0.05, β = 0.20 and a difference between means of 0.25 and SDs of 0.2, according to our previous studies (31). Because the distribution of the data was not parametric (assessed by Kolmogorov-Smirnov test), we used multiple comparisons Kruskal-Wallis test followed by a Dunn posttest. P-values < 0.05 were considered significant. Statistical tests were performed by using Graphpad Prism for Windows version 5.0 Software.
RESULTS
Clinical, Endocrine and Metabolic Characteristics
The ages of the women in the CE group were higher than those of PCOS women because the CE women belonged to a group undergoing hysterectomy; the higher body mass index in patients with PCOS and with hyperinsulinemia is inherent to the syndrome (Table 1). In addition, in women with PCOS, an increase in plasma testosterone levels and a diminution of SHBG plasma levels lead to a higher free-androgen index (Table 1). The levels of insulin after load of glucose in each group are described in Methodology section.
Table 1.
Clinical and endocrine characteristics of control women (CE) and women with PCOS with (PCOSE-HI) or without hyperinsulinemia (PCOSE-NI).
| Parameters | CE | PCOSE-NI | PCOSE-HI |
|---|---|---|---|
| Age, y | 39.3 ± 1.8 | 25.2 ± 1.1 | 28.4 ± 0.9 |
| Body mass index | 26 ± 2.3 | 28.7 ± 2.1 | 33.9 ± 1.1a |
| Estradiol, pg/mL | 73.8 ± 22.4 | 63.4 ± 7.9 | 59 ± 4.2 |
| Progesterone, ng/mL | 0.6 ± 0.2 | 0.6 ± 0.1 | 0.5 ± 0.1 |
| Androstenedione, ng/mL | 1.6 ± 0.4 | 3.1 ± 0.4a | 2.5 ± 0.7 |
| Testosterone, ng/mL | 0.3 ± 0.1 | 0.8 ± 0.1a | 0.8 ± 0.1a |
| SHBG, nmol/mL | 51.7 ± 9.5 | 25.8 ± 2.3 | 22.8 ± 4.1 |
| Free-androgen index | 3.1 ± 1.1 | 10.3 ± 2.0a | 10.7 ± 2.0a |
P < 0.05 compared with CE.
Endometrial Histological Characteristics
All endometrial samples from PCOS women showed morphological characteristics comparable to the proliferative phase observed in CE (Figure 1A, B, C), with tubular and straight glands, compact stroma, pseudostratificated epithelium nuclei and cell mitosis as established by Noyes criteria (29).
Figure 1.

Immunohistochemistry for proteins of the insulin-signaling pathway. Representative image from immunohistochemical detection of insulin receptor, IRS-1, Akt, AS160 and GLUT4 in paraffin wax sections of proliferative endometria obtained from CE women (n = 7) (left panel), women with PCOS without (PCOSE-NI, n = 7) (middle panel) and with hyperinsulinemia (PCOSE-HI, n = 7) (right panel). The hematoxylin/eosin staining showed comparable architecture in the three analyzed groups (A, B, C). Positive staining was detected in epithelial and stromal cells of all studied endometria for all antigens. A similar positive staining for insulin receptor was observed in all groups (D, E, F). (G, H, I) Immunostaining for IRS-1, which is less intense in the PCOSE-HI group (I). For Akt-1 (J, K, L), the immunostaining was more intense in the epithelia of PCOSE-HI compared with CE. The three analyzed groups showed positive staining for AS160 in a comparable manner (M, N, O). GLUT4 (P, Q, R) was immunodetected in all groups with less staining intensity in PCOSE-NI and PCOSE-HI compared with CE. Magnification 400× in all panels. Inserts show negative controls for all immunohistochemical assays. Scale bars represent 50 μm.
Protein Levels of Molecules Involved in the Insulin-Signaling Pathway
The immunohistochemical analysis showed that the positive staining for insulin receptor was detected in the cytoplasm of cell compartments, epithelium and stroma from all studied groups, particularly at the epithelial level, with an HScore similar between all groups (CE, 2.2 ± 0.2; PCOSE-NI, 1.8 ± 0.2; PCOSE-HI, 2.2 ± 1.3) (Figure 1D, E, F). These results were confirmed by Western blot analysis (Table 2).
Table 2.
Western blot analysis for insulin receptor, IRS-1 and AS160 protein in proliferative endometria from control and PCOS women.
| Protein | CE | PCOSE-NI | PCOSE-HI |
|---|---|---|---|
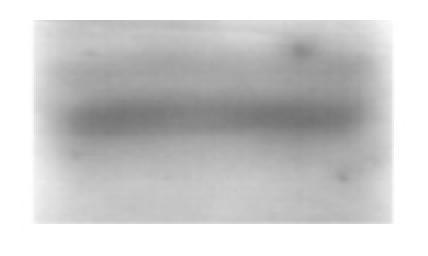
| Insulin Receptor (95 kDa) |  |
 |
 |
| IRS-1 (110 kDa) |  |
 |
 |
| AS160 (160 kDa) |  |
 |
 |
| β-actin (42 kDa) |  |
 |
 |
| Protein | CE (n = 7) | PCOSE-NI (n = 7) | PCOSE-HI (n = 7) |
|---|---|---|---|
| Insulin receptor, AU | 1.54 ± 0.14 | 1.46 ± 0.20 | 1.55 ± 0.26 |
| IRS-1, AU | 0.43 ± 0.18 | 0.43 ± 0.09 | 0.22 ± 0.02a |
| AS160, AU | 1.15 ± 0.30 | 1.21 ± 0.20 | 1.41 ± 0.36 |
P < 0.05 compared with PCOSE-NI.
We also studied the levels and immunolocalization of some proteins involved in the insulin-signaling pathway downstream insulin receptor. We observed lower protein levels for IRS-1 in PCOSE-HI compared with PCOSE-NI and CE in the Western blot analysis (49%, P < 0.05) (Table 2). The immunohistochemical study showed the detection of IRS-1 in stromal and epithelial compartments from all studied endometria. Interestingly, the intensity of the staining was lower in PCOSE-HI than in PCOSE-NI (P < 0,05) (HScore values: CE, 2.7 ± 0.2; PCOSE-NI, 3.0 ± 0.1; PCOSE-HI, 2.1 ± 0.2) (Figure 1G, H, I). Considering that IRS-1 corresponds to the insulin substrate isoform that is related to the metabolic effects of insulin, particularly glucose uptake, we assessed the IRS-1 activating tyrosine phosphorylation (Y612) by western blot and we found a lower pIRS-1Y612/IRS-1 ratio in PCOSE-HI compared with CE (P < 0.05) (Figure 2).
Figure 2.

Ratio between pIRS-1Y612 and IRS-1 in proliferative endometria from CE and PCOSE-HI women, assessed by Western blotting. Equal amounts of protein were loaded in each lane, and pIRS-1Y612 and IRS-1 band intensities were quantified by scanning densitometry and normalized to intensities observed for β-actin as internal control. A representative image of the media of bands obtained from 7 CE and 7 PCOSE-HI endometrial is shown. The results are expressed as AU and the values shown are mean ± SEM in CE and PCOSE-HI. a = P < 0.05 in PCOSE-HI compared with CE.
As shown in Figure 1 (J, K, L), Akt staining was positive in all the analyzed endometria in both epithelial and stromal compartments, particularly in epithelial cells from PCOSE-HI, which was similar to the other studied groups (HS values = CE, 2.3 ± 0.2; PCOSE-NI, 2.1 ± 0.6; PCOSE-HI, 2.9 ± 0.3) (Figure 1J, K, L).
We also observed that the staining for AS160 was positive in all groups of analyzed endometria (Figure 1M, N, O). HS values were similar in control and PCOS patients (HS values: CE, 2.3 ± 0.4; PCOSE-NI, 2.3 ± 0.2; PCOSE-HI:, 2.6 ± 0.3). By Western blot, we found similar levels of AS160 in PCOSE-NI, PCOSE-HI and CE (Table 2). As reported, the phosphorylation of AS160 protein at T642 constitutes an important site for GLUT4 translocation. As determined by Western blot, pAS160T642 protein levels in PCOSE-NI were similar to those found for CE (CE, 0.53 ± 0.16; PCOSE-NI, 0.38 ± 0.05). However, a decrease in the levels of pAS160T642 was obtained in PCOSE-HI compared with CE (57% 0.53 ± 0.16; PCOSE-HI, 0.19 ± 0.06, P < 0.05). Furthermore, the ratio between pAS160T642 and AS160 significantly diminished in PCOSE-HI related to CE (63%; P < 0.05) (Figure 3), and no significant differences were obtained between PCOSE-NI and CE (0.45 ± 0.13 versus 0.29 ± 0.05 CE and PCOSE-NI, respectively).
Figure 3.

Ratio between pAS160T642 and AS160 in proliferative endometria from CE and PCOSE-HI women, assessed by Western blotting. Equal amounts of protein were loaded in each lane, and pAS160T642 and AS160 band intensities were quantified by scanning densitometry and normalized to intensities observed for β-actin as internal control. A representative image of the media of bands obtained from 7 CE and 7 PCOSE-HI is shown. The results are expressed as AU and the values shown are mean ± SEM in CE and PCOSE-HI. a = P < 0.05 in PCOSE-HI compared with CE.
Likewise, the data presented in Figure 4 show 59% less protein content of GLUT4 in PCOSE-HI compared with CE (P < 0.05), as assessed by Western blot. In the case of PCOSE-NI no differences were observed (0.82 ± 0.24 arbitrary units [AU] versus 0.48 ± 0.09 AU in CE compared with PCOSE-NI, respectively). The immunohistochemical analysis for GLUT4 revealed a positive staining in all the analyzed endometria (Figure 1P, Q, R), in epithelial and stromal compartments, of granular type distributed homogenously in the cytoplasm.
Figure 4.

Western blot analysis of protein levels of GLUT4 in proliferative endometria from CE and PCOSE-HI women. Equal amounts of protein were loaded in each lane. GLUT4 was detected as a band with a molecular mass of 46 kDa. Band intensities were quantified by scanning densitometry and normalized to intensities observed for β-actin as internal control. A representative image of the media of bands obtained from 7 CE and 7 PCOSE-HI is shown. The results are expressed as AU and the values shown are means ± SEM in CE and PCOSE-HI. a = P < 0.05 in PCOSE-HI compared with CE.
DISCUSSION
It is well established that in PCOS the overproduction of androgens by the ovary, in addition to the hyperinsulinemia present in a high percentage of PCOS women, may affect the function of several tissues (32), including the endometrium. Previously, our group has shown changes in the expression of certain molecules related to tissue homeostasis, intracellular steroid bioavailability ((17,33,31)) and uterine receptivity (18) in PCOS endometrial tissue. In the present study we assessed the endometrial expression of some proteins involved in the pathway of insulin, the PI3K-Akt route, which is known to participate in glucose uptake mediated by insulin in the cells (21,34). Moreover, it is important to note that to our knowledge this is the first study to demonstrate in both normal and PCOS endometria the presence and cell distribution of AS160, as well as its phosphorylation rate in a key site for the GLUT4 vesicle translocation to the cell surface. This is an important issue because glucose has been proposed as the main energy source of the endometrial cell and high levels of energy are needed to fulfill endometrial functions. Therefore, the knowledge about the levels of molecules involved in the insulin pathway in endometria from PCOS women, particularly those with hyperinsulinemia, is of importance in the understanding, at least in part, of the reproduction failure observed in a great number of these patients (35). Interestingly, in the present study we found lower levels in several studied molecules in the endometria from PCOS women with hyperinsulinemia compared with the other two studied groups.
Scarce literature exists about the presence and activity of molecules involved in the metabolic insulin signaling in the endometrium. Experimental evidence shows that the binding of insulin to its receptor may be regulated by ovarian steroids (36), which is an important observation for our study model in PCOS women. Furthermore, Dunaif et al. (1) have reported that in PCOS, the hyperinsulinemia could be triggered by a defect in the expression or activity of molecules located downstream from the insulin receptor (1,21). The activated insulin receptor phosphorylates intracellular substrates, with the IRS proteins being the first to be phosphorylated in tyrosine residues. IRS-1 has a greater impact on insulin signal transduction in muscle and fat, IRS-2 is more relevant in liver and pancreatic β cells and IRS-3 and IRS-4 (37) appear to contribute modestly to the metabolic effects of insulin action. Therefore, in this study we focused on IRS-1 because this isoform is involved in peripheral glucose uptake by insulin and GLUT4 translocation (37). Previous reports on adipose tissue from patients with PCOS and hyperinsulinemia have shown a lower content of IRS-1 in this group compared with control women (38), which is in agreement with the results obtained in the present study in endometrial tissue. Undoubtedly, the lower content of IRS-1 that we found in endometria from PCOS patients with hyperinsulinemia could affect insulin signal transduction. In fact, the lower expression of pIRS-1Y612 in PCOSE-HI further supports this hypothesis.
The Akt protein, another intermediary in the insulin pathway, is a point of convergence and divergence at the same time in different signaling pathways. Recent findings from our laboratory showed an increase in pAktSer473 in PCOSE (39), and in the present study we examined the immunodetection of total Akt (phosphorylated protein at S473 and/or T308 residues and nonphosphorylated protein). The results showed positive staining in all studied groups, particularly in the epithelial compartment of PCOSE with hyperinsulinemia. As described, the activation of Akt may induce the phosphorylation of several substrates (40); one of them is AS160. This protein was immunodetected in all analyzed endometrial, and the phosphorylated AS160 protein in T642 (41–43) was diminished in the endometria from PCOS women and hyperinsulinemia. This observation suggests a lower translocation of GLUT4 vesicles to the periphery of the cell, compromising the availability of GLUT4 in the plasma membrane and leading to impaired glucose uptake by the cell. In agreement with these results, decreased phosphorylation of AS160, and lower glucose entrance in cells was observed in muscle tissue obtained from women with PCOS who are also insulin-resistant (7).
In addition, the amount of GLUT4 protein within the cell is relevant for the success of glucose uptake. This molecule is the final effector of the insulin-signaling pathway, and it is the only glucose transporter known to play a role in exposing the cell surface in response to insulin (25,28). In the present investigation we found that PCOSE-HI exhibited a significant decrease of GLUT4 protein levels compared with control endometria. This observation is in agreement with previous reports by Mozzanega and Mioni (27,28), who showed differences in endometrial GLUT4 level between nonhyperinsulinemic and hyperinsulinemic women with PCOS. Moreover, this difference was more drastic in obese women with hyperinsulinemia compared with lean patients with high insulin levels. From these data, Mioni et al. (27) proposed obesity as a determinant factor for reduced levels of GLUT4 in the cells. In our work, all of the PCOS patients were overweight and most of the PCOS women with hyperinsulinemia were obese; therefore we did not separate lean from obese women. Importantly, the significant decrease in the glucotransporter observed in the endometria from PCOS women and hyperinsulinemia may account for impairment in glucose metabolism and homeostasis at the endometrial level.
Likewise, the hyperandrogenic environment could reduce protein expression of IRS-1 and GLUT4 in endometrial epithelial cells, as previously described (12); in addition, androgens have been shown to reduce Akt and AS160 phosphorylation in skeletal muscle from PCOS women (7).
In conclusion, normal as well as PCOS endometria have the capacity for the uptake of glucose. To our knowledge, this is the first study to show a defect in the insulin-signaling pathway at the level of IRS-1, AS160 and GLUT4 in PCOS endometria from patients with hyperinsulinemia, which can lead to impairment in glucose uptake. Nevertheless, the experimental design and the data of the present study do not allow us to conclude that hyperandrogenism can affect the protein levels of the studied molecules. Several reports mentioned above point out the involvement of androgens in the molecular defects of the insulin pathway.
ACKNOWLEDGMENTS
The authors thank MD Armando Cortinez and E Soto (University of Chile Medical School) for their role in the recruitment of subjects and K Bacallao and M Lepez for laboratory assistance. We are also grateful to the women who donated tissue. This study was supported by grant nos. 1050098 and 1095127 from the Fondo Nacional de Desarrollo Científico y Tecnológico, Chile.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare they that have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this article.
REFERENCES
- 1.Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocrine Rev. 1997;18:774–800. doi: 10.1210/edrv.18.6.0318. [DOI] [PubMed] [Google Scholar]
- 2.Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Azziz R, et al. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91:4237–45. doi: 10.1210/jc.2006-0178. [DOI] [PubMed] [Google Scholar]
- 4.DeUgarte CM, Bartolucci AA, Azziz R. Prevalence of insulin resistance in the polycystic ovary syndrome using the homeostasis model assessment. Fertil Steril. 2005;83:1454–60. doi: 10.1016/j.fertnstert.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 5.Legro RS, Castracane VD, Kauffman RP. Detecting insulin resistance in polycystic ovary syndrome: purposes and pitfalls. Obstet Gynecol Surv. 2004;59:141–54. doi: 10.1097/01.OGX.0000109523.25076.E2. [DOI] [PubMed] [Google Scholar]
- 6.Corbould A. Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol. 2007;192:585–94. doi: 10.1677/joe.1.07070. [DOI] [PubMed] [Google Scholar]
- 7.H⊘jlund K, et al. Impaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. Diabetes. 2008;57:357–66. doi: 10.2337/db07-0706. [DOI] [PubMed] [Google Scholar]
- 8.Corbould A, Dunaif A. The adipose cell lineage is not intrinsically insulin resistant in polycystic ovary syndrome. Metabolism. 2007;56:716–22. doi: 10.1016/j.metabol.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corbould A, et al. Insulin resistance in the skeletal muscle of women with PCOS involves intrinsic and acquired defects in insulin signaling. Am J Physiol Endocrinol Metab. 2005;288:E1047–54. doi: 10.1152/ajpendo.00361.2004. [DOI] [PubMed] [Google Scholar]
- 10.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 11.Sato K, Iemitsu M, Aizawa K, Ajisaka R. Testosterone and DHEA activate the glucose metabolism-related signaling pathway in skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E961–8. doi: 10.1152/ajpendo.00678.2007. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Liao Q. Effects of testosterone and metformin on glucose metabolism in endometrium. Fertil Steril. 2009 2009 Mar 26; doi: 10.1016/j.fertnstert.2009.01.096. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 13.Maliqueo M, et al. Proinsulin serum concentrations in women with polycystic ovary syndrome: a marker of beta-cell dysfunction? Hum Reprod. 2003;18:2683–8. doi: 10.1093/humrep/deg482. [DOI] [PubMed] [Google Scholar]
- 14.Maliqueo M, et al. Sex hormone-binding globulin expression in the endometria of women with polycystic ovary syndrome. Fertil Steril. 2007;87:321–8. doi: 10.1016/j.fertnstert.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 15.Villavicencio A, et al. Androgen and estrogen receptors and co-regulators levels in endometria from patients with polycystic ovarian syndrome with and without endometrial hyperplasia. Gynecol Oncol. 2006;103:307–14. doi: 10.1016/j.ygyno.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 16.Villavicencio A, et al. Deregulation of tissue homeostasis in endometria from patients with polycystic ovarian syndrome with and without endometrial hyperplasia. Gynecol Oncol. 2007;104:290–5. doi: 10.1016/j.ygyno.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Avellaira C, et al. Expression of molecules associated with tissue homeostasis in secretory endometria from untreated women with polycystic ovary syndrome. Hum Reprod. 2006;21:3116–21. doi: 10.1093/humrep/del183. [DOI] [PubMed] [Google Scholar]
- 18.Quezada S, et al. Evaluation of steroid receptors, coregulators, and molecules associated with uterine receptivity in secretory endometria from untreated women with polycystic ovary syndrome. Fertil Steril. 2006;85:1017–26. doi: 10.1016/j.fertnstert.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 19.Fiory F, et al. Tyrosine phosphorylation of phosphoinositide-dependent kinase 1 by the insulin receptor is necessary for insulin metabolic signaling. Mol Cell Biol. 2005;25:10803–14. doi: 10.1128/MCB.25.24.10803-10814.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunaif A. Insulin resistance in women with polycystic ovary syndrome. Fertil Steril. 2006;86(Suppl 1):S13–4. doi: 10.1016/j.fertnstert.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 21.Schinner S, Scherbaum WA, Bornstein SR, Barthel A. Molecular mechanisms of insulin resistance. Diabet Med. 2005;22:674–82. doi: 10.1111/j.1464-5491.2005.01566.x. [DOI] [PubMed] [Google Scholar]
- 22.Toyofuku A, et al. Cyclic and characteristic expression of phosphorylated Akt in human endometrium and decidual cells in vivo and in vitro. Hum Reprod. 2006;21:1122–8. doi: 10.1093/humrep/dei454. [DOI] [PubMed] [Google Scholar]
- 23.Watson RT, Pessin JE. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem Sci. 2006;31:215–22. doi: 10.1016/j.tibs.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Eguez L, et al. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–72. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 25.Van Dam EM, Govers R, James DE. Akt activation is required at a late stage of insulin- induced GLUT4 translocation to the plasma membrane. Mol Endocrinol. 2005;19:1067–77. doi: 10.1210/me.2004-0413. [DOI] [PubMed] [Google Scholar]
- 26.Thong F, Dugani C, Klip A. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology. 2005;20:271–84. doi: 10.1152/physiol.00017.2005. [DOI] [PubMed] [Google Scholar]
- 27.Mioni R, et al. Evidence for the presence of glucose transporter 4 in the endometrium and its regulation in polycystic ovary syndrome patients. J Clin Endocrinol Metab. 2004;89:4089–96. doi: 10.1210/jc.2003-032028. [DOI] [PubMed] [Google Scholar]
- 28.Mozzanega B, et al. Obesity reduces the expression of GLUT4 in the endometrium of nor-moinsulinemic women affected by the polycystic ovary syndrome. Ann N Y Acad Sci. 2004;1034:364–74. doi: 10.1196/annals.1335.038. [DOI] [PubMed] [Google Scholar]
- 29.Noyes RW, Hertig AT, Rock J. Dating the endometrial biopsy. Am J Obstet Gynecol. 1975;122:262–3. doi: 10.1016/s0002-9378(16)33500-1. [DOI] [PubMed] [Google Scholar]
- 30.Lessey BA, et al. Immunohistochemical analysis of human uterine estrogen and progesterone receptors throughout the menstrual cycle. J Clin Endocrinol Metab. 1988;67:334–40. doi: 10.1210/jcem-67-2-334. [DOI] [PubMed] [Google Scholar]
- 31.Bacallao K, et al. In situ estrogen metabolism in proliferative endometria from untreated women with polycystic ovarian syndrome with and without endometrial hyperplasia. J Steroid Biochem Mol Biol. 2008;110:163–9. doi: 10.1016/j.jsbmb.2008.03.031. [DOI] [PubMed] [Google Scholar]
- 32.Diamanti-Kandarakis E, Papavassiliou AG. Molecular mechanisms of insulin resistance in polycystic ovary syndrome. Trends Mol Med. 2006;12:324–32. doi: 10.1016/j.molmed.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Leon L, et al. Activities of steroid metabolic enzymes in secretory endometria from untreated women with polycystic ovary syndrome. Steroids. 2008;73:88–95. doi: 10.1016/j.steroids.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 34.Chakraborty C. Biochemical and molecular basis of insulin resistance. Curr Protein Pept Sci. 2006;7:113–21. doi: 10.2174/138920306776359759. [DOI] [PubMed] [Google Scholar]
- 35.Homburg R. Pregnancy complications in PCOS. Best Pract Res Clin Endocrinol Metab. 2006;20:281–92. doi: 10.1016/j.beem.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Strowitzki T, von Eye HC, Kellerer M, Häring HU. Tyrosine kinase activity of insulin-like growth factor I and insulin receptors in human endometrium during the menstrual cycle: cyclic variation of insulin receptor expression. Fertil Steril. 1993;59:315–22. doi: 10.1016/s0015-0282(16)55674-x. [DOI] [PubMed] [Google Scholar]
- 37.Patel N, Huang C, Klip A. Cellular location of insulin-triggered signals and implications for glucose uptake. Pflugers Arch. 2006;451:499–510. doi: 10.1007/s00424-005-1475-6. [DOI] [PubMed] [Google Scholar]
- 38.Chu YL, Sun YY, Qiu HY, Li HF. Tyrosine phosphorylation and protein expression of insulin receptor substrate-1 in the patients with polycystic ovary syndrome [in Chinese] Zhonghua Fu Chan Ke Za Zhi. 2004;39:176–9. [PubMed] [Google Scholar]
- 39.Villavicencio A, et al. Involvement of Akt, Ras and cell cycle regulators in the potential development of endometrial hyperplasia in women with polycystic ovarian syndrome. Gynecol Oncol. 2009;115(1):102–7. doi: 10.1016/j.ygyno.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welsh GI, et al. Role of protein kinase B in insulin-regulated glucose uptake. Biochem Soc Trans. 2005;33:346–9. doi: 10.1042/BST0330346. [DOI] [PubMed] [Google Scholar]
- 41.Sano H, et al. Insulin- stimulated phosphorylation of a Rab GTPase- activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 42.Larance M, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–13. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 43.Barros RPA, Machado UF, Warner M, Gustafsson JA. Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc Natl Acad Sci U S A. 2006;103:1605–8. doi: 10.1073/pnas.0510391103. [DOI] [PMC free article] [PubMed] [Google Scholar]