

Abstract
Limitation in copper (Cu) leads to pathophysiology in developing brain. Cu deficiency impairs brain mitochondria and results in high brain lactate suggesting augmented anaerobic glycolysis. AMP activated protein kinase (AMPK) is a cellular energy “master-switch” that is thought to augment glycolysis through phosphorylation and activation phosphofructokinase 2 (PFK2) resulting in increases of the glycolytic stimulator fructose-2,6-bisphosphate (F2,6BP). Previously, Cu deficiency has been shown to augment cerebellar AMPK activation. Cerebella of Cu-adequate (Cu+) and Cu-deficient (Cu−) rat pups were assessed to evaluate if AMPK activation in Cu− cerebella functioned to enhance PFK2 activation and increase F2,BP concentration. Higher levels of pAMPK were detected in Cu− cerebella. However, PFK2 activity, mRNA, and protein abundance were not affected by Cu deficiency. Surprisingly, F2,6BP levels were markedly lower in Cu− cerebella. Lower F2,6BP may be due to inhibition of PFK2 by citrate, as citrate concentration was significantly higher in Cu− cerebella. Data suggest AMPK activation in Cu− cerebellum does not augment glycolysis through a PFK2 mechanism. Furthermore, other metabolite data suggest that glycolysis may actually be blunted, since levels of glucose and glucose-6-phosphate were higher in Cu− cerebella than controls.
Keywords: copper-deficient; rat; cerebellum; fructose-2,6-bisphosphate; AMPK; citrate
Introduction
Copper (Cu) is a cofactor for approximately a dozen important mammalian enzymes and is an essential micronutrient for mammalian development (1). Cu deficiency occurs in humans with Menkes syndrome, where a Cu deficient phenotype exists because of a defect in the Cu transporter ATP7A, and results in severe mental retardation and defective brain development, particularly in the cerebellum (2–4). Similar effects are observed following dietary Cu deficiency in rodent models of this disease (5). Cu deficiency blunts cerebellar growth and limits biosynthetic processes like myelination and synaptogenesis (5–9). However, the precise mechanisms responsible for the Cu deficient phenotype are still poorly understood.
Recently, a study of Cu deficient (Cu−) rat cerebella found that the energetic “master-switch” molecule AMP-activated protein kinase (AMPK) is activated during Cu deficiency (10). Phosphorylation of AMPK's physiological target acetyl CoA carboxylase, which regulates the first step of fatty acid biosynthesis and is inhibited by AMPK, was also found to be in greater abundance. These are the first data to suggest a potential mechanism accounting for blunted myelination and synaptogenesis in Cu− cerebella. AMPK functions to reduce energy expenditure and augment ATP generating processes through a complex network of signaling pathways (11). It is therefore likely that AMPK activation plays more than one role in Cu− cerebella. However, these facets of AMPK function in Cu− cerebella have yet to be explored.
One potential AMPK role is regulating brain glycolysis. Heart tissue AMPK has previously been shown to augment anaerobic glycolysis during conditions causing mitochondrial inhibition. In heart, AMPK activation leads to phosphorylation and activation of phosphofructokinase 2 (PFK2), which synthesizes fructose-2,6-bisphosphate (F2,6BP) (12). F2,6BP is a potent allosteric activator of phosphofructokinase 1 (PFK1), the pace-setter of glycolysis. Evaluation of brain AMPK in regulating glycolysis in vivo via PFK2 and F2,6BP has not been accomplished. In fact, as reviewed recently, current data are inconclusive (13).
Altered energy metabolism and mitochondrial inhibition are a marked phenomenon in Cu− cerebella and may be the underlying cause of Cu deficiency induced pathology. Cu is a co-factor for the mitochondrial cuproenzyme cytochrome c oxidase (CCO) and CCO deficiency is a hallmark of both Menkes syndrome and Cu−rodents (4, 14–16). In rodents Cu deficiency leads to mitochondrial and energetic impairment including mitochondrial structural abnormalities, high brain lactate levels, and elevated lactate/pyruvate ratios (10, 16, 17). Augmented anaerobic glycolysis may be one important mechanism to compensate for impaired mitochondrial function, but has never been thoroughly explored in context of Cu deficiency and induced AMPK activation. Therefore, our study investigated how Cu deficiency, which induces AMPK activation, impacted PFK2 activity, F2,6BP levels, and glucose metabolism in Cu− rat cerebellum.
Materials and Methods
Animal Care and Induction of Cu Deficiency
Holtzman sperm-positive rats were purchased commercially (Harlan Sprague Dawley, Indianapolis, IN) and received either Cu-adequate (Cu+) or Cu-deficient (Cu−) dietary treatment consisting of a Cu-deficient modified AIN-76A diet (Teklad Laboratories, Madison, WI) that contained 0.34 mg Cu/kg by analysis. Normal AIN-76A diet contains approximately 6 mg Cu/kg. All dams and offspring were fed the Cu− diet. Cu+ groups drank water supplemented with cupric sulfate, 20 mg Cu/L, and Cu− groups drank deionized water (14). Offspring in this perinatal model of Cu deficiency were sampled at postnatal age 24 (P24) (Experiment I). The dietary paradigm was totally repeated, Experiment II, and rats were sampled at P24 to confirm selected results in Experiment I. Each experiment sampled rats from a minimum of four separate litters of each treatment group. All animals were maintained at 24°C with 55% relative humidity on a 12-h light cycle (0700–1900-h). All protocols were formally approved by the University of Minnesota Animal Care Committee.
Tissue Collection
To prevent changes in metabolite concentrations induced by anesthetics, animals were decapitated without anesthesia. To minimize stress experienced by animals from handling before decapitation, rats were handled daily from P0 to day of tissue collection. Upon decapitation, cerebella were immediately removed from the skull and frozen in liquid nitrogen. To minimize possible AMPK activation resulting from anoxia, only cerebella frozen in under 15 seconds from the time of decapitation were analyzed. Cerebellar extraction time did not differ between Cu+ and Cu− animals. Fast frozen cerebella were stored at −75°C until analysis. Remaining brain and a piece of liver tissue were collected for total Cu analysis.
Biochemical and Metabolite Analyses
Brains, without cerebella, were wet-digested with HNO3 (Trace Metal grade, Fisher Scientific, Pittsburgh, PA) and samples were analyzed for total Cu content by flame atomic absorption spectroscopy (Model 1100B, Perkin-Elmer, Norwalk, CT). Protein content of tissue samples was determined using a modified version of the Lowry method (18).
For measurement of cerebellar citrate, glucose, and glucose-6-phosphate (G6P) concentrations, cerebellar metabolite extracts were prepared and analyzed according to Lowry et al. with some modifications (19). Briefly, fast frozen tissues were powdered under liquid nitrogen and then transferred to tubes, chilled on dry ice, and mixed with 5 volumes of 0.7 M HClO4. Tubes were then transferred to a −8°C alcohol bath, where samples and acid were mixed. This mixture was then rapidly homogenized using a tissue probe cooled to 4°C. Homogenates were spun, supernatant neutralized with KHCO3 and metabolite concentrations determined using enzymatic analyses as previously described (19). For F2,6BP analysis, frozen tissue was processed and extracts analyzed as described elsewhere (20). Fructose-6-phosphate phosphotransferase, the enzyme necessary for F2,6BP determination, was isolated from potatoes as previously described (20).
Plasma and cerebellar glucose was evaluated using enzyme coupled assays.
Enzymatic Analyses
To determine total PFK2 activity, cerebella were homogenized in 6 volumes of 50 mM Tris pH 7.5 buffer containing 50 mM NaF, 5 mM sodium pyrophosphate, 250 mM sucrose, 1 mM EDTA, 0.5% Triton X-100, and protease inhibitors (Protease Inhibitor Cocktail, Sigma, St. Louis, MO). Homogenates were spun at 15,000 × g for 15 minutes at 4°C and supernatants were assessed for PFK2 activity as described elsewhere (21). Total cerebellar hexokinase was assayed spectrophotometrically at 340 nm using methods described elsewhere with minor modifications (22). The reaction buffer was 50 mM Tris pH 8.0 containing 0.5 mM dithiothreitol. Cerebellar glucose 6-phosphate dehydrogenase was assayed in 50 mM Tris pH 7.8 containing 3 mM MgCl2 (Worthington Biochemical Corp. Manual). Substrate concentrations were 2 mM NADP+ and 3.33 mM glucose. NADPH formation was monitored at 340 nm.
Analysis of PFK2 mRNA Expression in Rat Pup Cerebellum
Total rat cerebellar RNA was isolated and purity established spectrophotometrically and by RNA gels (23). cDNA was synthesized using Omniscript Reverse Transcripase (Qiagen) and amplified with a Roche SYBR Green I kit. The copy number of mRNAs stemming from different PFK2 genes and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA content, used as a control, was determined by qRT-PCR. Primers for analysis of total brain/placental PFK2 mRNA expression were designed using published sequences of rat brain PFKFB3 transcripts (24). PFKFB3 mRNA splice variants, either containing or missing segment B, were quantified individually using two sets of primers. The forward primer for both sets was 5′-TGC CCA CTT CAC ACA GTC CTG AAA-3′. Reverse primer for detection of transcripts containing segment B was 5′-GCG GAC TCC ACA GAC AGA AAG TCA-3′ and for transcripts missing segment B was 5′-TCT TTG CAT CCT CTG ACC TCT CCC-3′, respectively. Primers specific to PFKFB2, heart-type PFK2 mRNA splice variant lacking the AMPK phosphorylation target sequence (MRRNS), were designed using published sequences (25, 26). The forward and reverse primers were 5′-CGT GAC AAG CCA ACT GAA GTG GAG-3′ and 5′-AGG GAT GCT GGC ATG CCA AGC TT-3′, respectively. Agarose gel analysis showed each primer pair to yield a single band of the expected size. Resultant products were purified, quantified using absorbance at 260 nm, and used in standard curves for qRT-PCR. Each reaction contained cDNA synthesized from 100 ng total RNA in a 10 μl total reaction volume. The mRNA copy numbers of PFKFB3 splice variants and the splice variant of PFKFB2 lacking the AMPK phosphorylation site were estimated by parallel real-time PCR analysis of purified PCR products. Forward and reverse primers used for GAPDH analysis were 5′-TTC CTA CCC CCA ATG TAT CCG-3′ and 5′-ACC ACC CTG TTG CTG TAG CCA-3′, respectively. Total brain/placental PFK2 mRNA expression in an individual rat brain cerebellum was determined by combining copy numbers for the two cerebellar PFKFB3 gene product splice variant groups analyzed.
Western Blot Analysis
Samples for Western blot analysis of copper chaperone for superoxide dismutase (CCS), CCO subunit IV (COX IV), and AMPK were prepared by homogenizing fast frozen cerebella in 6 volumes of a 50 mM Tris pH 7.5 buffer containing 50 mM NaF, 5 mM sodium pyrophosphate, 250 mM sucrose, 1 mM EDTA, 0.5% Triton X-100, and protease inhibitors (Protease Inhibitor Cocktail, Sigma, St. Louis, MO). Homogenates were spun at 15,000 × g for 15 minutes at 4°C and supernatant protein samples saved for analysis. Membrane protein extracts used to detect glucose transporters Glut1 and Glut3 were prepared as described elsewhere (27). Protein samples were stored at −75°C until Western blot analysis. Fractionation was carried out by loading 40 μg of protein for AMPK analysis and 10 μg of membrane protein for glucose transporter analysis on 10% SDS-PAGE gels. CCS and COX IV protein analysis was carried out by fractionation of 24 μg protein on a 15% SDS-PAGE gel. Proteins were transferred to 0.2 μm nitrocellulose membranes and processed for immunoblotting as described elsewhere (28). Some membranes were reprobed after incubation of membranes with buffer containing 2-mercaptoethanol and SDS at 55°C.
Cerebellar protein levels of CCS were evaluated using affinity purified rabbit anti-hCCS characterized previously, at a 1:1000 dilution (29). COX IV protein levels were analyzed using mouse monoclonal anti-COX IV, at 1:4000 dilution (Molecular Probes, Eugene, OR). Detection of total AMPK (α1 and α2) and phospho-AMPK (α1 and α2 Thr172) was achieved using antibodies purchased from Cell Signaling Technology, Inc. and used at 1:1000 dilution, according to manufacturer instructions. Antibodies for glial fibrillary acidic protein (GFAP) and brain/placental PFK2 were purchased from Dako and Santa Cruz respectively. Antibodies used to quantify Glut 1 and 3 have been previously characterized (30, 31). All membranes were blocked for at least an hour using 5% (w/v) albumin/Tris buffered saline (TBS) containing 0.1% Triton X-100, and incubated overnight with primary antibodies at 4°C. All secondary species-specific antibodies were diluted 1:10,000. Super-Signal West Pico chemiluminescent substrate (Pierce, Rockford, IL) was used for detection. Chemiluminescence was captured using high speed blue X-ray film (Lake Superior X Ray Inc., Duluth, MN) and densitometry was carried out using the FluorChem™ system (Alpha Innotech, San Leandro, CA).
Statistical Analysis
Means ± SEM were calculated. Student's unpaired two-tailed t test was used to compare data between the two dietary treatments, α = 0.05. Variance equality was evaluated by F-test. All data were handled using Microsoft Excele™. For comparative purposes immunoblot data was normalized. A value of 1.0 was assigned to the mean pixel density of Cu+ samples; then all, both Cu+ and Cu−, individual densities were recalculated before graphing.
Results
Confirmation of Copper Deficiency, Mitochondrial Impairment, and AMPK Activation
Following perinatal Cu deficiency, P24 Cu− rat pups exhibited lower body weight as compared to controls (Fig. 1). Cu deficiency also resulted in 93% lower Cu levels in the liver and 81% lower Cu levels in the brain of Cu− rat pups (Fig. 1), establishing a close similarity between Cu deficiency induced in these rats with previous work with this model (14). Cerebellar Cu deficiency was confirmed by evaluation of Western blots of cerebellar homogenates probed for the Cu deficiency marker copper chaperone for superoxide dismutase (CCS). CCS increases in concentration when cellular copper concentration is reduced (32). Protein levels of CCS were 86% higher in P24 rat Cu− cerebellum, consistent with cerebellar Cu deficiency (Fig. 2).
Figure 1.
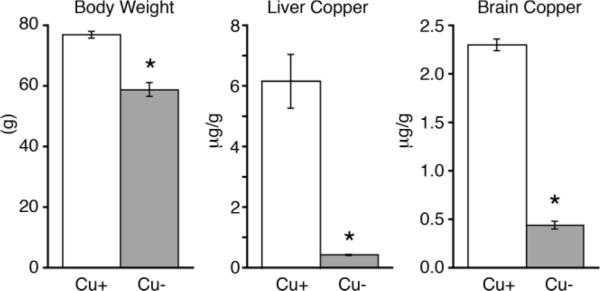
Characterization of induced Cu deficiency in P24 rat pups. Cu+ and Cu− P24 rat pups were assessed for copper status (n = 5). Body weight, a reflection of Cu deficiency, was reduced in Cu− rat pups compared to controls. Tissue Cu content in both liver and brain were also significantly lower in Cu− animals, confirming Cu deficiency. Values are means ± SEM; * P < 0.01.
Figure 2.

Evaluation of cerebellar Cu status in P24 rat pups. Cu deficiency in cerebellum was evaluated by Western blot using the Cu deficiency marker CCS, which was significantly increased in Cu− (−) cerebellar homogenates compared to Cu+ controls (+). Mitochondrial impairment in Cu− cerebella was confirmed via Western blot of cerebellar homogenates showing lower CCO subunit IV (COX IV) protein content. Protein loading, 24 μg per lane, in a 15% gel was evaluated using Ponceau S stain. Values are means ± SEM; * P < 0.01.
Previous work with a perinatal Cu deficient rat model has established the presence of mitochondrial impairment in brain tissues stemming from reduced CCO activity and protein levels (15, 16). To confirm an impact of Cu deficiency on cerebellar mitochondria in this experiment, total protein extracts were probed for levels of mitochondrial CCO subunit IV (COX IV). Compared to Cu+ rats, levels of COX IV in Cu− cerebella were lower by 88% (Fig. 2).
Phosphorylation of catalytic subunit α of AMPK at Thr172 increases AMPK's kinase activity (33, 34). AMPK activation, through augmented phosphorylation of AMPK, was confirmed in Cu− P24 rat cerebellum. Protein extracts from rapidly frozen cerebella were probed. Rapid freezing is achieved by extracting and freezing cerebella within 15 seconds of decapitation in order to reduce anoxia induced AMPK activation. Western blots confirmed 86% higher levels of phospho-AMPK (pAMPK) in Cu− P24 rat cerebella compared to controls, while overall levels of AMPK remained the same (Fig. 3).
Figure 3.
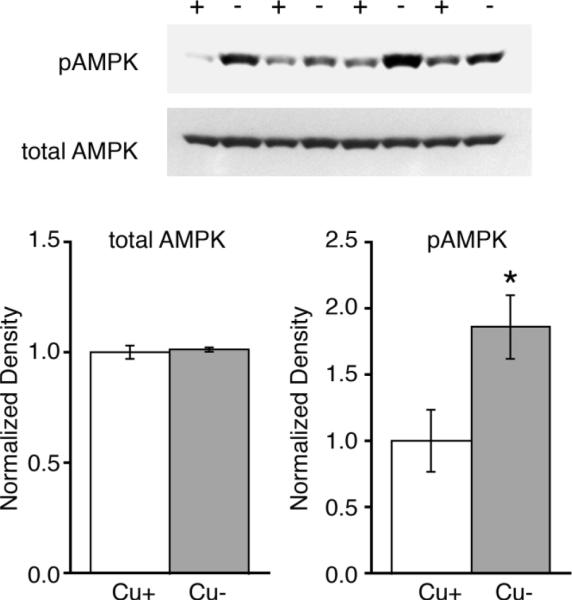
AMPK is activated in Cu− P24 rat pup cerebellum. Western blots of cerebellar homogenates demonstrate that Cu− cerebella (−) of P24 rat pups have higher levels of phosphorylated AMPK (pAMPK) than Cu+ pups (+) (n = 4). Total AMPK remained unchanged by Cu deficiency. Each lane was loaded with 40 μg protein. Values are means ± SEM; * P < 0.05.
PFK2 Activity Is Unchanged and F2,6BP Concentrations Are Reduced in Cu− Cerebellum
Antibodies against phospho-PFK2 were not available at the time of this study. To determine if one function of AMPK activation in Cu− cerebella was to modulate glycolysis through PFK2 activation, total PFK2 activity and F2,6BP concentration were analyzed in extracts from rapidly frozen cerebella. Total PFK2 activity measured in vitro, under optimal conditions, was not impacted by Cu deficiency (Fig. 4A). Furthermore, F2,6BP levels were surprisingly reduced by more than 39% in cerebella of Cu− rats compared to controls (Experiment I, Fig. 4B). To confirm this unexpected result, cerebellar F2,6BP levels were evaluated in a repeat experiment with Cu− rats that had a similar degree of brain Cu deficiency. Experiment II P24 Cu− rats had 83% lower brain Cu levels than Cu+ (P < 0.01) (data not shown). Cu− cerebellar F2,6BP levels were reduced by 50% in Experiment II (Fig. 4B).
Figure 4.

Cerebellar PFK2 evaluation. A) Analysis of cerebellar homogenates revealed that Cu− cerebellar total PFK2 activity (n = 4) was not altered by Cu deficiency. B) Cerebellar fructose-2,6-bisphosphate levels (F2,6BP) were evaluated in Cu+ and Cu− rats. In two independent experiments, Experiment I (n = 4) and II (n = 5), F2,6BP concentration was found to be markedly lower. C) Astocytes are thought to have a high F2,6BP content. However, astrocytic content was not different between Cu+ and Cu− cerebella, as assessed by astrocytic marker GFAP abundance (n = 4). Values are means ± SEM; * P < 0.01.
Cell culture comparisons of F2,6BP concentrations between neurons and astrocytes have shown astrocytes contain 4–9 fold higher levels than neurons (35, 36). To eliminate the possibility that astrocytic cell loss in the cerebellum could account for reduced F2,6BP levels observed in Cu− cerebellum, Western blots of cerebellar homogenates were probed for the astrocytic marker glial fibrillary acidic protein (GFAP) (Fig. 4C). Western blots revealed no changes in overall GFAP levels in Cu− cerebella as compared to controls, suggesting overall astrocytic volume/number was not altered in the cerebellum by Cu deficiency and thus did not account for reduction in cerebellar F2,6BP concentration.
PFK2 Isozyme mRNA Expression and Protein Concentration Unchanged in Cu− Cerebella
Unchanged PFK2 activity and low F2,6BP levels in Cu− cerebella did not reflect the activation status of AMPK in Cu− cerebellum. Potential reasons responsible for these observations were investigated.
In rats, as in humans, the PFK2 enzyme is encoded by four different genes that give rise to four types of enzymes, termed as the liver, heart, testes, and brain/placental PFK2, named for organs in which they were discovered to be expressed in relatively high abundance (37). However, the expression of the four PFK2 genes is not limited to their namesake organ (25, 38). The brain expresses both the brain/placental and heart PFK2 genes (24, 26). The brain/placental PFK2 isozyme contains an AMPK phosphorylation consensus sequence (MRRNS) (24, 37) and is subject to AMPK induced activation (39). The heart-type PFK2 expressed by brain lacks the AMPK phosphorylation consensus sequence making the heart PFK2 isozyme insensitive to AMPK (26). Total brain PFK2 activity is presumably a combination of activities of both isoenzymes.
The absence of a measurable increase in PFK2 activity in Cu− cerebella may have been due to an increase in the ratio between heart and brain type PFK2 isozyme expression in Cu− cerebella. To test this possibility mRNA content of both isozymes was evaluated in Cu− and Cu+ cerebella. Using real-time PCR we determined copy numbers of the brain and heart PFK2 gene products in Cu+ and Cu− cerebella, normalized to GAPDH expression (Fig. 5A). Cu deficiency did not influence the expression of either heart or brain type PFK2 mRNAs. Interestingly, our analysis indicated that in cerebellum transcripts of the AMPK insensitive heart-type PFK2 significantly outnumbered those of the AMPK sensitive brain/placenta PFK2. For Cu+ samples the heart-type:brain/placenta PFK2 mRNA abundance was 80% greater (Fig. 5A).
Figure 5.

Heart-type and brain/placental PFK2 mRNA expression, brain/placental PFK2 protein concentration, and citrate concentration in Cu+ and Cu− cerebellum. Analysis of cerebellar total mRNA (Panel A) showed that a splice variant mRNA for an AMPK insensitive heart-type PFK2 outnumbered mRNA transcripts of the AMPK sensitive brain/placental splice variants (n = 5). Cu deficiency did not impact gene expression. Unlike superscripts are statistically different, P < 0.01, factorial ANOVA. Analysis of cerebellar homogenates revealed that Cu− cerebellar levels of brain/placental PFK2 protein (Panel B; n = 4) were not different than Cu+ controls. Analysis of cerebellar extracts revealed a significantly higher concentration of the PFK2 inhibitor citrate in Cu− cerebella (Panel C; n = 5). Values are means ± SEM; * P < 0.01.
To test the possibility that translation of brain/placental PFK2 was reduced in Cu− cerebella, cerebellar homogenates were analyzed by Western blot for brain/placental PFK2 protein content (Fig. 5B). Cu deficiency did not change brain/placental PFK2 concentrations in rat cerebella.
Citrate Levels Augmented in Cu− Cerebellum
PFK2 activity, altered expression of PFK2 isoenzymes, or protein levels of brain/placental PFK2 isozyme did not readily explain reductions in F2,6BP concentrations observed in Cu− cerebella. However, in vitro assays of total cerebellar PFK2 activity could not assess allosteric modifiers of enzyme activity that occur in vivo. We therefore investigated whether possible allosteric inhibition of brain PFK2 may be responsible for the observed F2,6BP reduction in concentration. Citrate is a powerful inhibitor of brain PFK2 activity and increased citrate in brain is thought to reduce brain tissue levels of F2,6BP (40). Metabolite analyses confirmed that citrate concentrations were 42% higher in Cu− cerebella compared to Cu+ values, providing a potential mechanism for F2,6BP reduction through citrate inhibition of PFK2 (Fig. 5C).
Lower F2,6BP Associated with Evidence of Altered Glucose Metabolism in Cu− Cerebellum
Because F2,6BP levels modulate the rate of glycolysis by activating the glycolytic enzyme PFK1, reduced F2,6BP levels in Cu− cerebellum may blunt cerebellar glycolysis. Buildup of glucose and the glycolytic intermediate glucose-6-phosphate (G6P) occurs during glycolytic inhibition (41–44). Therefore, concentrations of these metabolites were measured as markers of cerebellar glycolytic inhibition. Both glucose and G6P concentrations were significantly higher in Cu− cerebella, by 45% and 85% respectively, as compared to Cu+ (Fig. 6A).
Figure 6.
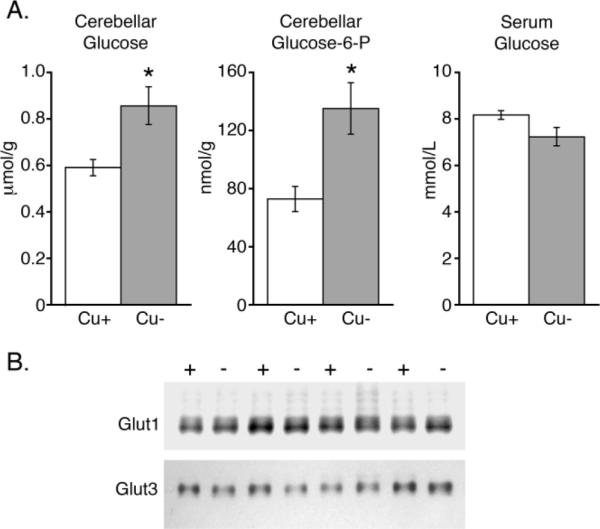
Glucose metabolism in Cu− cerebellum. Cerebellar concentrations of glucose (n = 5) and glucose-6-phosphate (G6P) (n = 5) were found to be higher in Cu− rats without a corresponding increase in serum glucose (n = 5) (Panel A). Levels of cerebellar glucose transporters Glut1 and Glut3 were not altered in Cu− rats (−) compared to Cu+ controls (+) (Panel B; n = 4). Values are means ± SEM; * P < 0.05.
Serum glucose levels were analyzed and found to be unaltered in Cu− rat pups, ruling out the possibility that increased brain glucose was due to augmented serum glucose concentrations in Cu− animals (Fig. 6A). Furthermore, changes in brain glucose transporters Glut1 and Glut3 were not detected in Cu− cerebella and thus were also unlikely to contribute to changes in cerebellar glucose and G6P concentrations (Fig. 6B). Changes in glucose and G6P concentrations are also unlikely to have stemmed from changes in cerebellar hexokinase or glucose 6-phosphate dehydrogenase (G6PDH), as activity was not altered by dietary Cu deficiency: for hexokinase means ± SEM (n = 4) for Cu+, 0.108 ± 0.012, μmol/min × mg protein, were not different from Cu−, 0.113 ± 0.011. For glucose 6-phosphate dehydrogenase: Cu+ was 0.0157 ± 0.0009 compared to Cu− 0.0180 ± 0.0027.
Discussion
One goal of our investigation was to determine whether AMPK activation in Cu− cerebella functions to modulate cerebellar glycolysis by increasing PFK2 enzyme activity and augmenting cerebellar F2,6BP levels. Perinatal Cu deficiency was induced in rats and AMPK activation in Cu− cerebellum was confirmed through increased levels of phospho-AMPK. However, AMPK activation in Cu− cerebellum did not coincide with increased PFK2 activity or higher levels of its product F2,6BP. We found that Cu deficiency did not impact cerebellar total PFK2 activity. Moreover, F2,6BP concentrations in Cu− cerebella were half of those found in age matched Cu+ controls.
Rat brain expresses two types of PFK2s: a heart-type and the brain/placental PFK2. While the brain/placental isozyme contains an AMPK activation site, the heart-type PFK2 is a splice variant that does not (24–26). Decreased expression of the brain/placental PFK2 and increased expression of heart-type PFK2 was one possibility that could have accounted for unchanged PFK2 activity in Cu− cerebellum. However, we found that Cu deficiency did not alter the expression of either PFK2 isoenzymes or brain/placental PFK2 protein levels. Together, these results suggest that AMPK in Cu− cerebellum does not play a direct role in augmenting glycolysis through PFK2 activation.
One possible explanation for the failure of AMPK activation to increase PFK2 activity in the cerebellum in the current experiments is cell-specific separation of AMPK and brain/placental PFK2. In vitro data suggest that brain/placental PFK2 expression is concentrated in astrocytes (35). However, most in vivo expression of AMPK occurs in neurons (45). Unactivated astrocytes express very little if any AMPK in vivo (45). This suggests that AMPK dependent brain/placental PFK2 activation may not occur in vivo (13). This supposition is further supported by studies of brain anoxia, a condition that activates AMPK. Unlike heart, where AMPK dependent PFK2 activation was first described, brain anoxia does not result in increased F2,6BP (12, 40). Our evaluation of PFK2 in Cu− cerebellum is the first in vivo evaluation of the relationship between AMPK and PFK2 in any rat brain region during a pathological condition. Our data are consistent with the cell separation hypothesis.
Another goal of this study was to characterize glucose metabolism in the mitochondrially inhibited Cu− cerebellum. F2,6BP is key to regulating anaerobic glucose metabolism and we investigated the potential causes and consequences of a drop in F2,6BP concentration in Cu− cerebellum. Brain F2,6BP concentrations are remarkably stable even during serious metabolic insults like starvation and short-term ischemia (40). Conditions that reduce brain F2,6BP concentrations by the same magnitude observed in the current studies are hypoglycemic coma and 10 minutes of ischemia, two extreme physiological challenges (21, 40).
In hypoglycemia F2,6BP loss stems from a decrease in precursors of F2,6BP synthesis: brain glucose and fructose-6-phosphate (21). Cu− pups had serum glucose levels that were unchanged and brain glucose concentrations that were significantly higher than controls, arguing that substrate shortage for F2,6BP synthesis is unlikely.
A rat model of diabetes demonstrated a modest reduction in brain F2,6BP concentration (40). This streptozotocin induced diabetes increased brain citrate and is a likely mechanism for lowered brain F2,6BP because citrate is a powerful inhibitor of PFK2 (40). We found that citrate was significantly higher in Cu− cerebellum. This is possibly a reflection of the increased NADH/NAD+ ratio and prolonged inhibition of the TCA cycle in Cu− mitochondria. Since neither loss of F2,6BP rich astrocytes or reduced total cerebellar PFK2 activity account for the drop in F2,6BP concentration, high citrate levels are a reasonable explanation for lower F2,6BP levels in Cu− cerebellum.
A possible consequence of lower F2,6BP levels in Cu− cerebella is reduced cerebellar glycolysis. Levels of Cu− cerebellar glucose and G6P are both augmented while total hexokinase and glucose-6-phosphate dehydrogenase activity remained unchanged. Others have shown that elevated glucose and glucose-6-phosphate accompany glycolytic inhibition (44, 46). This suggests potential glycolytic inhibition in the current study. Our results are also consistent with NMR studies of brain glucose metabolism in humans with mitochondrial disorders that show, in some cases, brain glucose consumption is actually reduced (47).
Further study is needed to confirm glycolytic inhibition, as other factors may account for our observations. Higher G6P levels could stem from citrate inhibition of PFK1, though this is unlikely. Taking the mitochondrial to cytoplasmic citrate ratio into account, estimated Cu− cerebellar cytoplasmic citrate would be approximately 15 μM (48). This citrate concentration, while inhibiting rat brain PFK2 activity by more than 80%, would have little impact on rat brain PFK1 activity (40, 49). Higher G6P levels in Cu− tissue may also be due to a potential rise in NADPH/NADP+, which has not been evaluated in Cu− cerebellum. Furthermore, glycolytic inhibition can be also be caused by tissue lactate buildup (42). Lactate accumulation is a well-described phenomenon in Cu deficiency and merits further investigation in this regard. Further work is needed to determine the extent to which lower F2,6BP is involved in this potential glycolytic reduction.
The possibility that Cu− cerebella reduce their reliance on anaerobic glycolysis during mitochondrial inhibition raises questions how the cerebellum continues to function and sustain adenine nucleotide energy charge. AMPK phosphorylation of ACC inhibits its enzymatic activity, which may serve as one temporary energy-saving measure by attenuating fatty acid biosynthesis (10). It is possible that at earlier time points glycolysis in Cu− cerebella is augmented to compensate for dysfunctional mitochondria. Also mitochondrial oxidative phosphorylation may be partially functional despite the marked loss of CCO (16, 50, 51). Thus, multiple mechanisms may exist to preserve energy charge during brain Cu deficiency. As mitochondrial inhibition and lactate accumulation mount with age, increased brain citrate, reduced brain F2,6BP, and lower glycolytic rates may be symptoms of energetic failure.
Taken together the data in these experiments reveal that AMPK activation in Cu− cerebellum occurs without activation of PFK2 or an increase in F2,6BP, suggesting that AMPK activation in Cu− cerebellum does not function to augment glycolysis through this mechanism. Rather, F2,6BP concentration in Cu− cerebellum is lower than in control rats, consistent with citrate inhibition of PFK2. In fact Cu− cerebellum shows signs of glycolytic inhibition. More studies on Cu deficiency are needed to determine whether cerebellar glucose utilization is indeed blunted and whether this is proportional to the degree of Cu deficiency and mitochondrial inhibition. Also additional studies are needed to determine the mechanisms for maintenance of energy charge in the Cu− brain.
Acknowledgments
We thank Margaret Boderius and Joshua Pyatskowit for their excellent technical assistance.
This research was supported by NIH HD-039708.
References
- 1.Prohaska JR. Copper. In: Bowman BA, Russell RM, editors. Present Knowledge in Nutrition. International Life Sciences Institute; Washington, DC: 2006. pp. 458–470. [Google Scholar]
- 2.Danks DM, Campbell PE, Stevens BJ, Mayne V, Cartwright E. Menkes's kinky hair syndrome. An inherited defect in copper absorption with widespread effects. Pediatrics. 1972;50:188–201. [PubMed] [Google Scholar]
- 3.Menkes JH, Alter M, Steigleder GK, Weakley DR, Sung JH. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–779. [PubMed] [Google Scholar]
- 4.Mercer JF. Menkes syndrome and animal models. Am J Clin Nutr. 1998;67:1022S–1028S. doi: 10.1093/ajcn/67.5.1022S. [DOI] [PubMed] [Google Scholar]
- 5.Prohaska JR, Wells WW. Copper deficiency in the developing rat brain: a possible model for Menkes' steely-hair disease. J Neurochem. 1974;23:91–98. doi: 10.1111/j.1471-4159.1974.tb06920.x. [DOI] [PubMed] [Google Scholar]
- 6.Everson GJ, Tsai HC, Wang TI. Copper deficiency in the guinea pig. J Nutr. 1967;93:533–540. doi: 10.1093/jn/93.4.533. [DOI] [PubMed] [Google Scholar]
- 7.El Meskini R, Crabtree KL, Cline LB, Mains RE, Eipper BA, Ronnett GV. ATP7A (Menkes protein) functions in axonal targeting and synaptogenesis. Mol Cell Neurosci. 2007;34:409–421. doi: 10.1016/j.mcn.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prohaska JR. Changes in brain enzymes accompanying deficiencies of the trace elements, copper, selenium, or zinc. In: Howell JM, Gawthorne JM, White CL, editors. Trace Element Metabolism in Man and Animals (TEMA-4) Australian Academy of Science; Canberra: 1981. pp. 275–282. [Google Scholar]
- 9.Zimmerman AW, Matthieu JM, Quarles RH, Brady RO, Hsu JM. Hypomyelination in copper-deficient rats. Prenatal and postnatal copper replacement. Arch Neurol. 1976;33:111–119. doi: 10.1001/archneur.1976.00500020039007. [DOI] [PubMed] [Google Scholar]
- 10.Gybina AA, Prohaska JR. Copper deficiency results in AMP-activated protein kinase activation and acetyCoA carboxylase phosphorylation in rat cerebellum. Brain Res. 2008;1204:69–76. doi: 10.1016/j.brainres.2008.01.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 13.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J Physiol. 2006;574:85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gybina AA, Prohaska JR. Increased rat brain cytochrome c correlates with degree of perinatal copper deficiency rather than apoptosis. J Nutr. 2003;133:3361–3368. doi: 10.1093/jn/133.11.3361. [DOI] [PubMed] [Google Scholar]
- 15.Gybina AA, Prohaska JR. Variable response of selected cuproproteins in rat choroid plexus and cerebellum following perinatal copper deficiency. Genes Nutr. 2006;1:51–60. doi: 10.1007/BF02829936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prohaska JR, Wells WW. Copper deficiency in the developing rat brain: evidence for abnormal mitochondria. J Neurochem. 1975;25:221–228. doi: 10.1111/j.1471-4159.1975.tb06956.x. [DOI] [PubMed] [Google Scholar]
- 17.Rusinko N, Prohaska JR. Adenine nucleotide and lactate levels in organs from copper-deficient mice and brindled mice. J Nutr. 1985;115:936–943. doi: 10.1093/jn/115.7.936. [DOI] [PubMed] [Google Scholar]
- 18.Markwell MA, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 19.Lowry OH, Passonneau JV. A Flexible System of Enzymatic Analysis. Academic Press, Inc.; New York: 1972. [Google Scholar]
- 20.Van Schaftingen E, Lederer B, Bartrons R, Hers HG. A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose-2,6-bisphosphate. Eur J Biochem. 1982;129:191–195. doi: 10.1111/j.1432-1033.1982.tb07039.x. [DOI] [PubMed] [Google Scholar]
- 21.Ambrosio S, Ventura F, Rosa JL, Bartons R. Fructose-2,6-bisphosphate in hypoglycemic rat brain. J Neurochem. 1991;57:200–203. doi: 10.1111/j.1471-4159.1991.tb02116.x. [DOI] [PubMed] [Google Scholar]
- 22.Knull HR, Taylor WF, Wells WW. Effects of energy metabolism on in vivo distribution of hexokinase in brain. J Biol Chem. 1973;248:5414–5417. [PubMed] [Google Scholar]
- 23.Prohaska JR, Brokate B. Copper deficiency alters rat dopamine beta-monooxygenase mRNA and activity. J Nutr. 1999;129:2147–2153. doi: 10.1093/jn/129.12.2147. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe F, Sakai A, Furuya E. Novel isoforms of rat brain fructose 6-phosphate 2-kinase/fructose-2,6-bisphosphatase are generated by tissue-specific alternative splicing. J Neurochem. 1997;69:1–9. doi: 10.1046/j.1471-4159.1997.69010001.x. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe F, Furuya E. Alternative splicing of novel exons of rat heart-type fructose-6-phosphate 2-kinase/fructose-2,6-bisphosphatase gene. Biochem Biophys Res Commun. 2001;282:803–810. doi: 10.1006/bbrc.2001.4648. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe F, Sakai A, Furuya E, Uyeda K. Molecular cloning and tissue specific expression of fructose 6-phosphate,2-kinase:fructose-2,6-bisphosphatase of rat brain. Biochem Biophys Res Commun. 1994;198:335–340. doi: 10.1006/bbrc.1994.1047. [DOI] [PubMed] [Google Scholar]
- 27.Prohaska JR, Gybina AA. Rat brain iron concentration is lower following perinatal copper deficiency. J Neurochem. 2005;93:698–705. doi: 10.1111/j.1471-4159.2005.03091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prohaska JR, Brokate B. Dietary copper deficiency alters protein levels of rat dopamine beta-monooxygenase and tyrosine monooxygenase. Exp Biol Med (Maywood) 2001;226:199–207. doi: 10.1177/153537020122600307. [DOI] [PubMed] [Google Scholar]
- 29.West EC, Prohaska JR. Cu,Zn-superoxide dismutase is lower and copper chaperone CCS is higher in erythrocytes of copper-deficient rats and mice. Exp Biol Med (Maywood) 2004;229:756–764. doi: 10.1177/153537020422900807. [DOI] [PubMed] [Google Scholar]
- 30.Leino RL, Gerhart DZ, van Bueren AM, McCall AL, Drewes LR. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J Neurosci Res. 1997;49:617–626. doi: 10.1002/(SICI)1097-4547(19970901)49:5<617::AID-JNR12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 31.Van Bueren AM, Moholt-Siebert M, Begley DE, McCall AL. An immunization method for generation of high affinity antisera against glucose transporters useful in immunohistochemistry. Biochem Biophys Res Commun. 1993;197:1492–1498. doi: 10.1006/bbrc.1993.2645. [DOI] [PubMed] [Google Scholar]
- 32.Prohaska JR, Broderius M, Brokate B. Metallochaperone for Cu,Zn-superoxide dismutase (CCS) protein but not mRNA is higher in organs from copper-deficient mice and rats. Arch Biochem Biophys. 2003;417:227–234. doi: 10.1016/s0003-9861(03)00364-3. [DOI] [PubMed] [Google Scholar]
- 33.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345(Pt 3):437–443. [PMC free article] [PubMed] [Google Scholar]
- 35.Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 36.Pauwels PJ, Trouet A. Role of fructose-2,6-bisphosphate in the regulation of glycolysis in various types of cultivated brain cell. Neurosci Lett. 1984;46:173–177. doi: 10.1016/0304-3940(84)90437-3. [DOI] [PubMed] [Google Scholar]
- 37.Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381:561–579. doi: 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe F, Furuya E. Tissue-specific alternative splicing of rat brain fructose 6-phosphate 2-kinase/fructose 2,6-bisphosphatase. FEBS Lett. 1999;458:304–308. doi: 10.1016/s0014-5793(99)01174-6. [DOI] [PubMed] [Google Scholar]
- 39.Marsin AS, Bouzin C, Bertrand L, Hue L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J Biol Chem. 2002;277:30778–30783. doi: 10.1074/jbc.M205213200. [DOI] [PubMed] [Google Scholar]
- 40.Ventura F, Rosa JL, Ambrosio S, Gil J, Bartrons R. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in rat brain. Biochem J. 1991;276(Pt 2):455–460. doi: 10.1042/bj2760455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.John SA, Ottolia M, Weiss JN, Ribalet B. Dynamic modulation of intracellular glucose imaged in single cells using a FRET-based glucose nanosensor. Pflugers Arch. 2007 doi: 10.1007/s00424-007-0395-z. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rovetto MJ, Lamberton WF, Neely JR. Mechanisms of glycolytic inhibition in ischemic rat hearts. Circ Res. 1975;37:742–751. doi: 10.1161/01.res.37.6.742. [DOI] [PubMed] [Google Scholar]
- 43.Newsholme EA, Sugden PH, Williams T. Effect of citrate on the activities of 6-phosphofructokinase from nervous and muscle tissues from different animals and its relationships to the regulation of glycolysis. Biochem J. 1977;166:123–129. doi: 10.1042/bj1660123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruderman NB, Ross PS, Berger M, Goodman MN. Regulation of glucose and ketone-body metabolism in brain of anaesthetized rats. Biochem J. 1974;138:1–10. doi: 10.1042/bj1380001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 46.John SA, Ottolia M, Weiss JN, Ribalet B. Dynamic modulation of intracellular glucose imaged in single cells using a FRET-based glucose nanosensor. Pflugers Arch. 2008;456:307–322. doi: 10.1007/s00424-007-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Molnar MJ, Valikovics A, Molnar S, Tron L, Dioszeghy P, Mechler F, Gulyas B. Cerebral blood flow and glucose metabolism in mitochondrial disorders. Neurology. 2000;55:544–548. doi: 10.1212/wnl.55.4.544. [DOI] [PubMed] [Google Scholar]
- 48.Kauppinen RA, Hiltunen JK, Hassinen IE. Compartmentation of citrate in relation to the regulation of glycolysis and the mitochondrial transmembrane proton electrochemical potential gradient in isolated perfused rat heart. Biochim Biophys Acta. 1982;681:286–291. doi: 10.1016/0005-2728(82)90033-0. [DOI] [PubMed] [Google Scholar]
- 49.Vora S, Oskam R, Staal GE. Isoenzymes of phosphofructokinase in the rat. Demonstration of the three non-identical subunits by biochemical, immunochemical and kinetic studies. Biochem J. 1985;229:333–341. doi: 10.1042/bj2290333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chao JC, Medeiros DM, Altschuld RA, Hohl CM. Cardiac nucleotide levels and mitochondrial respiration in copper-deficient rats. Comp Biochem Physiol. 1993;104:163–168. doi: 10.1016/0300-9629(93)90024-x. [DOI] [PubMed] [Google Scholar]
- 51.Matz JM, Saari JT, Bode AM. Functional aspects of oxidative phosphorylation and electron transport in cardiac mitochondria of copper-deficient rats. J Nutr Biochem. 1995;6:644–652. [Google Scholar]