

Abstract
Retroviral DNA integration is mediated by the preintegration complex, a large nucleoprotein complex derived from the core of the infecting virion. We previously have used Mu-mediated PCR to probe the nucleoprotein organization of Moloney murine leukemia virus preintegration complexes. A region of protection spans several hundred base pairs at each end of the viral DNA, and strong enhancements are present near the termini. Here, we show that these footprints reflect a specific association between integrase and the viral DNA ends in functional preintegration complexes. Barrier-to-autointegration factor, a cellular protein that blocks autointegration of Moloney murine leukemia virus DNA, also plays an indirect role in generating the footprints at the ends of the viral DNA. We have exploited Mu-mediated PCR to examine the effect of mutations at the viral DNA termini on complex formation. We find that a replication competent mutant with a deletion at one end of the viral DNA still exhibits a strong enhancement about 20 bp from the terminus of the mutant DNA end. The site of the enhancement therefore appears to be at a fixed distance from the ends of the viral DNA. We also find that a mutation at one end of the viral DNA, which renders the virus incompetent for replication, abolishes the enhancements and protection at both the U3 and U5 ends. A pair of functional viral DNA ends therefore are required to interact before the chemical step of 3′ end processing.
Integration of retroviral DNA into a host chromosome is an essential step in the viral replication cycle. This process is mediated by the preintegration complex, a large nucleoprotein complex derived from the core of the infecting virion (1, 2). Preintegration complexes, isolated from cells after viral infection, efficiently integrate their DNA into a target DNA in vitro (3–8). This reaction exhibits the full fidelity of in vivo integration, in contrast to the high frequency of single-end integration events that occur with simplified in vitro systems using purified integrase. Preintegration complexes therefore provide a valuable tool to study aspects of retroviral DNA integration that have not yet been reproduced with these simplified in vitro systems.
In addition to the viral DNA, the preintegration complex includes integrase, the key viral enzyme that catalyzes the chemical steps of integration (9). Besides integrase, several other viral proteins have been reported to be associated with preintegration complexes, but their functional roles in viral DNA integration have not yet been well defined. A striking feature of in vitro reactions with both Moloney murine leukemia virus (MLV) (3, 10) and HIV (5, 6) preintegration complexes is the strong preference for intermolecular integration into a target DNA and avoidance of intramolecular integration into its own DNA, a reaction termed autointegration (11). In the case of MLV preintegration complexes, the protection against autointegration is conferred by a small host protein, termed barrier-to-autointegration factor (BAF) (12). BAF probably prevents autointegration by compacting the viral DNA, thereby making it inaccessible as a target for integration.
Little is known concerning the nucleoprotein organization of preintegration complexes. We previously have developed a footprinting technique [Mu-mediated PCR (MM-PCR)], based on the Mu transposition system, to probe the association of MLV DNA with protein factors (13). The ends of the viral DNA were found to form part of a large nucleoprotein structure, which we call the intasome to distinguish it from the entire preintegration complex. A hallmark of this structure is the presence of strong enhancements near both termini of the viral DNA, together with a region of protection that spans several hundred base pairs. Both the enhancements and protection could be abolished by treating the complexes with high salt and subsequently restored by incubation with a cytoplasmic extract of uninfected NIH 3T3 cells. These results suggested the involvement of both integrase and a host factor in the intasome, although the contribution of each protein to the observed footprint was uncertain. We now show that the host factor is the BAF, a cellular protein that protects the viral DNA against autointegration. However, we conclude that the role of BAF in generating the footprints is indirect and that the footprints are caused by a specific association of integrase with the viral DNA ends. We also show that intasome assembly requires a pair of functional viral DNA ends.
MATERIALS AND METHODS
Reagents.
Enzymes used for DNA manipulations were from New England Biolabs. Cells were grown in DMEM (GIBCO) supplemented with 10% (vol/vol) calf serum (ICN). 293T cells were kindly provided by Robert Gorelick (Science Applications International Corporation, Frederick, MD).
Proteins.
BAF was expressed and purified from Escherichia coli as described (12). Murine high mobility group (HMG) Y was expressed from a construct generated by PCR cloning from an NIH 3T3 cell cDNA library (CLONTECH). Human HMG I(Y) was expressed from a construct provided by Raymond Reeves (Washington State University, Pullman). Both proteins were purified from E. coli as described (14). Human HMG1 was kindly provided by Tanya Paull (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health). The MLV integrase gene was cloned into pET15b, and the protein with an N-terminal His tag was purified from E. coli after induction with isopropyl β-d-thiogalactoside. The insoluble fraction of cell lysates was solubilized with 4 M urea in buffer containing 20 mM Hepes pH 7.6, 0.4 M potassium glutamate, 0.1% Nonidet P-40, 10% glycerol, 2 mM 2-mercaptoethanol. The protein was bound to chelating Sepharose (Pharmacia) charged with Ni2+ and eluted with a gradient of imidazole. Peak fractions were pooled, and the urea was removed by dialysis against 20 mM Hepes, pH 7.6/0.4 M potassium glutamate/0.1% Nonidet P-40/10% glycerol/5 mM DTT.
DNA.
Plasmid pJM1X1, carrying a MLV proviral clone with the dl8269–7 mutation (15), was a gift from John Murphy (Chiron). Plasmid pSQ70 carrying mutation dl8276–22 was constructed as follows. Plasmid pNCA (16) carrying the wild-type MLV genome was cut with NdeI to generate 8.2- and 3-kb DNA fragments. The larger fragment was self-ligated, and the smaller one was subcloned into pUC19. Both constructs, each carrying one copy of the long terminal repeat (LTR), then were used as templates to introduce the dl8276–22 mutation at the U3 region by using a QuikChange Site-Directed Mutagenesis Kit (Stratagene). Oligos used in the mutagenesis were GGG GAA TGA AAG ACC AGC TTA AGT AAC GCC A and its complement. The resulting constructs with the desired mutations were confirmed by DNA sequencing and were used to regenerate the full-length MLV DNA, carrying the dl8276–22 mutation at both U3 regions.
Preintegration Complexes.
Wild-type and integrase-minus preintegration complexes were prepared by coculturing as described (11). Preintegration complexes containing various mutations in the U3 regions of the viral DNA were made as follows. Culture supernatants containing viral particles were collected after transient transfection of the corresponding plasmid DNA into 293T cells by the calcium phosphate method using the Statagene mammalian transfection kit. NIH 3T3 cells were infected with the filtered supernatants for 2 hr in the presence of polybrene (8 μg/ml). Cytoplasmic extract containing the preintegration complexes were isolated 18 hr postinfection. All complexes were treated with 750 mM KCl and partially purified on a 10–50% Nycodenz gradient (13). Complexes used in the functional footprinting experiments were subjected to gel filtration on Sepharose 4B-CL (Pharmacia) columns equilibrated with 20 mM Hepes, pH 7.5/5 mM MgCl2/6 mM EDTA/750 mM KCl/1 mM DTT/6% sucrose/1 μg/ml BSA before the Nycodenz gradient.
Reconstitution Reactions.
Most reconstitution experiments were done directly on the Nycodenz gradient purified complexes (15 μl) in the presence of BSA (5 μg) and BAF (2 ng) or other proteins (1 μg). Reconstitution reactions in the functional footprinting assay were carried out immediately after the Mu insertion reaction in the same buffer conditions.
Footprinting of MLV Integrase on the Viral DNA End.
The viral DNA substrate was the 3-kb NdeI fragment from plasmid pNCA. It carries a copy of LTR with a precut U5 end. Binding was done with ≈0.5 ng DNA substrate and ≈200 ng MLV integrase in a total volume of 16 μl at room temperature for 20 min followed by MuA insertion reaction. The reaction buffer included 25 mM Hepes, pH 7.5/5 mM MgCl2/10 mM DTT/75 mM KCl/100 mM K glutamate/5% polyethylene glycol 8000/10% dimethyl sulfoxide/0.05% Nonidet P-40/0.3 μg/μl BSA/2.5% glycerol.
MM-PCR.
The technique was carried out as described (13) for assembly of MuA complexes and insertion reactions. PCR detection was done with oligos SW1 and SW2 for the U3 end and SW4 and SW5 for the U5 end of the viral DNA. Exceptions are for the functional footprinting experiments: (i) undiluted MuA complexes (17 μl) were added directly to 50 μl of retroviral complexes for the Mu insertion reactions; and (ii) oligos SW1 and SW40 (5′-TCC CTG GGC AGG GGT CTC CCG ATC CCG GA) were used for the U3 end detection.
Reverse Transcriptase (RT) and Intermolecular Integration Activity Assays.
RT activity from viral particles and the intermolecular integration activity of the preintegration complexes were assayed as described (11, 17). To check the replication of various mutant MLVs, RT assays were performed on the culture supernatants 18 hr postinfection. Most intermolecular integration reactions were done with φX174 replicative form DNA as the target except that in the Mu interference experiments a biotinylated DNA fragment (≈180 bp) served as the target.
Selection for Biotinylated DNA.
Biotinylated DNA in the functional footprinting experiment was isolated as previously described (13).
RESULTS
BAF Reveals the Footprint of Integrase on the Viral DNA Ends Within the Intasome by Masking Dissociated Viral DNA.
Using the footprinting technique MM-PCR (13), we have demonstrated that several hundred base pairs at the ends of the viral DNA within the preintegration complex are organized in a large nucleoprotein structure. We call this complex the intasome to distinguish it from the entire preintegration complex. Footprinting of the viral DNA within the intasome, compared with naked viral DNA, reveals highly preferred sites for Mu insertion near the ends of the viral DNA together with an extensive region of protection that spans several hundred base pairs. The strong terminal enhancements appear to be a hallmark of proper intasome assembly. Both the strong enhancements and protection are abolished by treatment with high salt and subsequently can be reconstituted by incubation with an extract of uninfected NIH 3T3 cells.
Elimination of the enhancements and protection at the ends of the viral DNA by treatment with high salt, and their reconstitution by incubation with a host cell extract, closely parallels the loss and reconstitution of the barrier to autointegration (12). We therefore tested whether the BAF is also the host factor responsible for reconstitution of the enhancements and footprint after stripping complexes with high salt. Incubation of salt-stripped complexes with BAF reconstituted both the enhancements and protection (Fig. 1A), demonstrating that the host factor involved in the footprinting is indeed BAF. As BAF is titrated in reconstitution reactions, the enhancements appear first, followed by both enhancements and protection at the higher concentrations of BAF. Several other host proteins have been implicated in stimulating in vitro integration reactions with either purified integrase protein or preintegration complexes (14, 18). We therefore tested whether any of these proteins altered the footprint on salt-stripped complexes (Fig. 1B). Even at concentrations 500 times greater than that used for BAF, none of these proteins restored the enhancements (Fig. 1B). Although BAF restores the enhancements at the ends of the viral DNA, these enhancements do not result from binding of BAF alone. They are not seen when BAF is incubated with naked viral DNA or with salt-stripped preintegration complexes that lack integrase (Fig. 1C).
Figure 1.

Reconstitution of the footprints on the viral DNA U3 end. All preintegration complexes were treated with 750 mM KCl before separation from free proteins on a 10–50% Nycodenz gradient. Reconstitution reactions then were carried out on the gradient purified complexes, followed by MM-PCR footprinting (see Materials and Methods for details). End-labeled primer SW2 (13) was used for PCR. Numbers on the left side of each panel indicate the distance from the very end of the viral DNA. Distinctive enhancements are located at ≈20 bp and ≈100 bp from the U3 end. (A) Reconstitution with BAF protein. Samples are naked viral DNA (lane 1) and MLV preintegration complexes (lane 2). Reconstitution reactions included: lane 3, buffer only control, and lanes 4–7, BAF (0.2, 0.6, 2, and 6 ng, respectively). (B) Reconstitution with various protein factors. Reconstitution reactions included: lane 1, buffer only control; lane 2, BAF (2 ng); lane 3, murine HMG Y; lane 4, human HMG I(Y); and lane 5, human HMG1. Purified protein factors shown in lanes 3–5 have been tested in a range of 0.5 to 5 μg, and no differences were observed in this range (data not shown); therefore only reactions with 1 μg of protein are shown. (C) Enhancements are observed only with complexes containing integrase. Reconstitution reactions with BAF were carried out with (a) salt-stripped MLV preintegration complexes, (b) naked viral DNA, and (c) salt-stripped integrase-minus MLV preintegration complexes. Lanes 1 and 2 included 0 and 2 ng BAF, respectively.
We initially interpreted these results to indicate that both integrase and BAF are required for intasome assembly. However, we now consider that the DNA binding activity of BAF, which is responsible for blocking autointegration, plays an indirect role in generating the footprints on the viral DNA ends. The most compelling reason for reaching this conclusion is that identical enhancements can be obtained by incubating DNA corresponding to the U5 end of the MLV LTR with purified MLV integrase (Fig. 2). We suggest that BAF masks the contribution to the footprint of viral DNA that is either fully or partially dissociated from integrase. The result is to reveal the footprint on viral DNA that is properly associated with integrase. Some dissociation of integrase from the viral DNA may occur during salt stripping. Dissociated viral DNA would be expected to be a much better target for Mu insertion, and this signal could dominate the footprint in the absence of BAF. This will be discussed more fully later.
Figure 2.
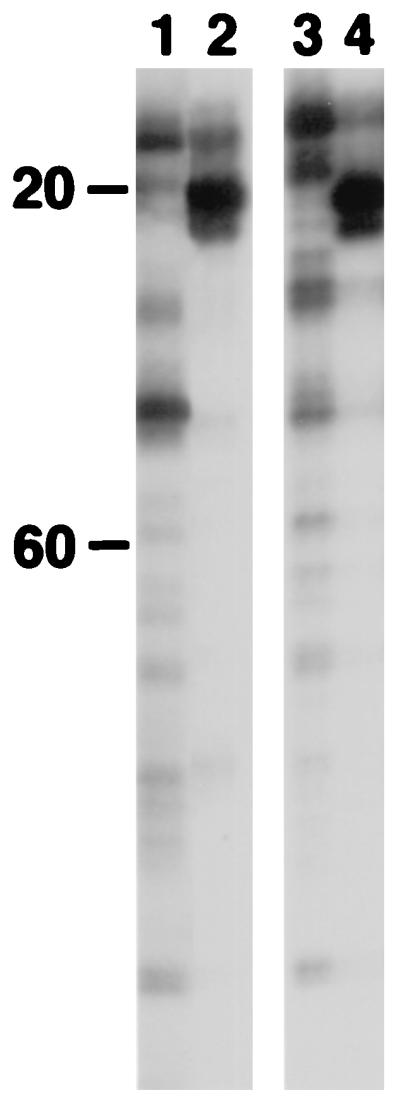
Footprinting of MLV integrase on terminal viral DNA. Footprinting was carried out as described in Materials and Methods. End-labeled primer SW6 (13) was used for PCR to detect the U5 viral DNA end. Strong enhancements are located at ≈20 bp from the tip of the U5 end. Controls are with naked viral DNA (lane 1) and MLV preintegration complexes (lane 2). Footprinting in the absence (lane 3) or presence (lane 4) of purified integrase is shown.
Salt-Stripped and Native Preintegration Complexes Exhibit a Similar Functional Footprint.
We previously have used Mu footprinting combined with a functional integration assay to demonstrate that several hundred base pairs at the ends of the viral DNA are functionally important for intermolecular DNA integration (13). When Mu was inserted into the DNA within preintegration complexes before the retroviral DNA integration reaction, Mu insertions within several hundred base pairs of the ends of the viral DNA were conspicuously absent in the integration products.
To test the hypothesis that salt-stripped and native MLV preintegration complexes have a similar intasome structure, Mu insertion was performed on salt-stripped complexes, followed by reconstitution of the autointegration barrier with BAF and assay for intermolecular integration activity. DNA from complexes that underwent intermolecular integration was separated from the pool of total DNA by using the biotin tag on the target DNA (Fig. 3A). We observed a striking absence of Mu insertions within ≈300 bp from the ends of the viral DNA in the pool of active complexes (Fig. 3B, compare lanes 5 and 6). This result can be interpreted as either an absence of Mu insertions into this region of viral DNA in active complexes or inactivation of complexes that have Mu insertions within a few hundred base pairs of the ends of the viral DNA. This extensive protection of the ends of the viral DNA in functional footprinting is essentially identical to that previously observed with native MLV complexes. Although the relative contributions of protection from Mu insertion and inactivation by Mu insertion are uncertain, the results are consistent with a similar nucleoprotein organization at the ends of the viral DNA in both native and salt-stripped complexes.
Figure 3.

Extensive regions at the ends of the viral DNA are required for integration activity. (A) Schematic depiction of the functional footprinting assay. (B) Functional footprints on the viral DNA U3 end. Lanes 1 and 2 are controls of standard MM-PCR footprinting with salt-stripped MLV preintegration complexes reconstituted with 0 or 2 ng BAF, respectively. Reconstitution reactions in the functional footprinting experiments contained 0 (lanes 3 and 4) or 20 ng BAF (lanes 5 and 6). DNA samples before and after biotin selection are marked with B and A, respectively. In the absence of reconstitution of the autointegration barrier with BAF, intermolecular integration was very inefficient, and therefore little signal is seen in lane 4.
The Terminal Enhancement Requires a Pair of Viral DNA Ends.
The footprints on the U3 and U5 ends of the viral DNA in preintegration complexes are strikingly similar. Both exhibit protection and a strong terminal enhancement (13). To investigate the importance of viral DNA sequence in assembly of the intasome, we used MM-PCR to footprint preintegration complexes containing viral DNA with mutations at the U3 end (Fig. 4). Mutant dl8269–7 has a deletion at one end of the viral DNA. This mutant previously has been demonstrated to be defective in viral replication because of a block in 3′ processing at both ends of the viral DNA (15). Mutant dl8276–22, which bears a different deletion at the U3 end, is fully active and behaves just like the wild type (17). Because different preparations of complexes exhibit quantitative differences in the degree of protection and the strength of enhancements, we compared complexes after salt stripping and reconstitution with BAF; wild-type complexes from different preparations give very similar degrees of enhancement after this treatment (Fig. 1), presumably caused by masking of dissociated complexes. Both wild-type and mutant complexes gave a similar footprinting pattern after salt stripping, and this pattern resembled that of naked DNA (Fig. 5, lanes 1). Upon reconstitution with BAF, wild-type and the active dl8276–22 mutant complexes showed the distinctive enhancements at the U3 end of the viral DNA, while dl8269–7 did not (Fig. 5A, lanes 2). Interestingly, the enhancement is absent at the U5 end of the inactive mutant d18269–7, even though this end has the wild-type sequence (Fig. 5Bd). This finding indicates that a pair of viral DNA ends are required to assemble the nucleoprotein structure responsible for the terminal enhancement.
Figure 4.

Summary of MLV U3 end mutants used in this study. Deletions shown in the boxes are at the U3 ends of both LTRs. ▴ indicates the position of the deletion, and • indicates the strong enhancement observed at ≈20 bp from the U3 end. Viral replication was determined by measuring the reverse transcriptase activity in culture supernatants. Integration activity was assayed with preintegration complexes in the corresponding cytoplasmic extracts. MM-PCR was used to assay for the presence of the characteristic enhancements (see Fig. 5).
Figure 5.

Footprints on the viral DNA ends of various MLV mutants. All complexes were salt-stripped and reconstituted with BAF followed by MM-PCR. Characteristic enhancements are located at ≈20 bp and ≈100 bp from the U3 end, and ≈20 bp from the U5 end. Shown are footprints on the U3 (A) and U5 (B) viral DNA ends. (a) Control MLV preintegration complexes made by coculturing clone 4 with NIH 3T3 cells. (b-d) Preintegration complexes obtained by transient transfection of 293T cells with plasmid (b) pNCA (wild-type MLV); (c) pSQ70 (active dl8276–22 MLV); and (d) pJM1X1 (inactive dl8269–7 MLV). Reconstitution was done in the absence of BAF in lanes 1 and with 2 ng BAF in lanes 2.
The Position of the Terminal Enhancement Is Measured from the Tip of the Viral DNA.
Because the terminal enhancement requires a pair of functional viral DNA ends and the presence of integrase in the preintegration complex, it presumably reflects a structural feature of the intasome. Consistent with this view, the active deletion mutant dl8276–22 also exhibits an enhancement approximately 20 bp from the tip of the viral DNA (Fig. 5A, compare a-c), even though the DNA sequence at this position is different from that of wild-type viral DNA (Fig. 4).
DISCUSSION
Enhancements and Protection at the Ends of the Viral DNA Are Caused by Integrase.
Reconstitution of the footprints on the ends of the viral DNA in salt-stripped MLV preintegration complexes by a host cell extract led us to implicate the involvement of both integrase and a host factor in the footprints on the ends of the viral DNA (13). Although this appeared to be the simplest interpretation of the footprinting data, some results were not easily reconciled with this interpretation, and our new data argue strongly against this view. First, because salt-stripped MLV preintegration complexes efficiently autointegrate, the viral DNA ends must remain functionally associated with integrase. It is therefore rather surprising that the Mu insertion pattern into the viral DNA within salt-stripped complexes resembles that of naked DNA. Second, the finding that the host factor implicated in footprinting is the BAF led us to consider alternative interpretations. BAF is a nonspecific DNA binding protein that bridges together double-stranded DNA molecules (12). Our current model is that BAF blocks autointegration by compacting the viral DNA, thereby making it less accessible as a target for integration. A direct role for BAF in assembly of the intasome would require that BAF has a second function in MLV preintegration complexes that is quite separate from its function in blocking autointegration. Third, the degree of enhancement and protection at the ends of viral DNA varies among different preparations of MLV preintegration complexes, but very strong enhancements and protection can always be seen after incubation with BAF. However, the percentage of complexes that are competent for integration in vitro is not increased by incubation with BAF (data not shown), as might be expected if addition of BAF reconstitutes additional functional complexes; BAF appears to simply block autointegration and promote intermolecular integration. Finally, the enhancements near the ends of the viral DNA can be reproduced with purified MLV integrase and viral DNA substrate.
These observations can be easily unified by the hypothesis that the enhancements and protection are caused by integrase alone, and the DNA binding activity of BAF masks the contribution to the footprint of viral DNA ends that are not properly associated with integrase within preintegration complexes. Salt stripping may dissociate integrase from a small fraction of complexes. Because the dissociated DNA is likely to be a better target for Mu insertion, the signal from this DNA could dominate the footprint. We propose that BAF binds to this dissociated DNA, making it inaccessible as a target for Mu insertion, whereas the regions of enhancement within the intasome are unable to bind BAF and remain accessible. The feasibility of this mechanism was tested by footprinting MLV preintegration complexes in the presence of added naked viral DNA. At a concentration of naked DNA sufficient to dominate the footprint, addition of BAF did indeed restore the characteristic enhancements of the preintegration complexes (data not shown). We note that, among the DNA binding proteins we have tested, only BAF is able to mask the signal from viral DNA ends that are not properly associated with integrase. The precise mechanism of this masking remains to be understood.
DNA Requirements for Assembly of the Intasome.
Several hundred base pairs at each end of the viral DNA are organized in a higher order nucleoprotein complex with integrase. Although host factors may participate in this intasome, our current data can be explained without invoking their direct involvement. Particularly striking features of the intasome footprint are the strong enhancements near the ends of the viral DNA. These enhancements, together with extensive regions of protection, provide an assay for assembly of a complex between integrase and the ends of the viral DNA before the catalytic steps of 3′ end processing and DNA strand transfer. We have used this footprinting assay to investigate some of the DNA requirements for formation of this complex. The footprints on complexes with deletions at the U3 end of the viral DNA were compared with that of wild type. One of the deletion mutants (dl8276–22) previously had been reported to be replication competent (17). This mutant, like wild-type MLV DNA, exhibited a strong enhancement about 20 bp from the U3 and U5 end. Because the U3 sequences at the position of the enhancement differ between the mutant and wild type (Fig. 4), this finding indicates that the position of the enhancement is dictated by the distance from the end of the viral DNA and does not depend on any particular viral DNA sequence at the site of the enhancement. The enhancement therefore is likely to reflect a structural feature of the viral DNA associated with integrase within the intasome. In contrast to the wild-type and active deletion mutant, viral DNA within the inactive deletion mutant complexes (dl8269–7) did not exhibit an enhancement at the mutated U3 end. Strikingly, the U5 end of this viral DNA, which is wild type in sequence, also did not show the enhancement. It previously has been reported that this mutant does not undergo 3′ processing at either end of the viral DNA (15). Our present study indicates that the defect manifests itself at the stage of assembling a proper complex. We conclude that a pair of functional viral DNA ends are required to interact before the chemical step of 3′ end processing. This may serve to ensure that a pair of viral DNA ends function in concert in vivo as is required for the proper integration reaction.
Acknowledgments
We thank Tanya Baker, Dominic Esposito, Gary Felsenfeld, and Tanya Paull for comments on the manuscript. This work was supported in part by the National Institutes of Health Intramural AIDS Targeted Antiviral Program.
ABBREVIATIONS
- BAF
barrier-to-autointegration factor
- MLV
Moloney murine leukemia virus
- MM-PCR
Mu-mediated PCR
- HMG
high mobility group
- LTR
long terminal repeat
References
- 1.Varmus H, Brown P O. In: Mobile DNA. Berg D E, Howe M M, editors. Washington, DC: Am. Soc. Microbiol.; 1989. pp. 53–108. [Google Scholar]
- 2.Goff S P. Annu Rev Genet. 1992;26:527–544. doi: 10.1146/annurev.ge.26.120192.002523. [DOI] [PubMed] [Google Scholar]
- 3.Brown P O, Bowerman B, Varmus H E, Bishop J M. Cell. 1987;49:347–356. doi: 10.1016/0092-8674(87)90287-x. [DOI] [PubMed] [Google Scholar]
- 4.Bowerman B, Brown P O, Bishop J M, Varmus H E. Genes Dev. 1989;3:469–478. doi: 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- 5.Ellison V, Abrams H, Roe T, Lifson J, Brown P. J Virol. 1990;64:2711–2715. doi: 10.1128/jvi.64.6.2711-2715.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farnet C M, Haseltine W A. Proc Natl Acad Sci USA. 1990;87:4164–4168. doi: 10.1073/pnas.87.11.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y M, Coffin J M. J Virol. 1990;64:5958–5965. doi: 10.1128/jvi.64.12.5958-5965.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee Y M, Coffin J M. Mol Cell Biol. 1991;11:1419–1430. doi: 10.1128/mcb.11.3.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katz R A, Skalka A M. Annu Rev Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 10.Fujiwara T, Mizuuchi K. Cell. 1988;54:497–504. doi: 10.1016/0092-8674(88)90071-2. [DOI] [PubMed] [Google Scholar]
- 11.Lee M S, Craigie R. Proc Natl Acad Sci USA. 1994;91:9823–9827. doi: 10.1073/pnas.91.21.9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee M S, Craigie R. Proc Natl Acad Sci USA. 1998;95:1528–1533. doi: 10.1073/pnas.95.4.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei S Q, Mizuuchi K, Craigie R. EMBO J. 1997;16:7511–7520. doi: 10.1093/emboj/16.24.7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farnet C M, Bushman F D. Cell. 1997;88:483–492. doi: 10.1016/s0092-8674(00)81888-7. [DOI] [PubMed] [Google Scholar]
- 15.Murphy J E, Goff S P. J Virol. 1992;66:5092–5095. doi: 10.1128/jvi.66.8.5092-5095.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colicelli J, Goff S P. J Mol Biol. 1988;199:47–59. doi: 10.1016/0022-2836(88)90378-6. [DOI] [PubMed] [Google Scholar]
- 17.Murphy J E, De Los Santos T, Goff S P. Virology. 1993;195:432–440. doi: 10.1006/viro.1993.1393. [DOI] [PubMed] [Google Scholar]
- 18.Aiyar A, Hindmarsh P, Skalka A M, Leis J. J Virol. 1996;70:3571–3580. doi: 10.1128/jvi.70.6.3571-3580.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]