

Abstract
Iron oxides catalyze the conversion of hydrogen peroxide (H2O2) into oxidants capable of transforming recalcitrant contaminants. Unfortunately, the process is relatively inefficient at circumneutral pH values due to competing reactions that decompose H2O2 without producing oxidants. Silica- and alumina-containing iron oxides prepared by sol-gel processing of aqueous solutions containing Fe(ClO4)3, AlCl3 and tetraethyl orthosilicate efficiently catalyzed the decomposition of H2O2 into oxidants capable of transforming phenol at circumneutral pH values. Relative to hematite, goethite and amorphous FeOOH, the silica-iron oxide catalyst exhibited a stoichiometric efficiency, defined as the number of moles of phenol transformed per mole of H2O2 consumed, that was 10 to 40 times higher than that of the iron oxides. The silica-alumina-iron oxide catalyst had a stoichiometric efficiency that was 50 to 80 times higher than that of the iron oxides. The significant enhancement in oxidant production is attributable to the interaction of Fe with Al and Si in the mixed oxides, which alters the surface redox processes, favoring the production of strong oxidants during H2O2 decomposition.
Introduction
The activation of hydrogen peroxide (H2O2) by iron minerals (e.g., hematite, goethite, iron-containing clays and sands) and its application for contaminant oxidation has been intensively studied over the last two decades (1-6) and the process is being applied for in situ contaminant oxidation (5), as well as for wastewater treatment (6). The reaction offers significant advantages over Fenton’s reagent (mixture of Fe2+ and H2O2) because it does not generate iron sludge and is not restricted to acidic conditions. Unfortunately, the process is relatively slow and inefficient at circumneutral pH values because only a small fraction of the H2O2 is converted into oxidants that are capable of transforming recalcitrant contaminants (1, 7, 8). As a result, very large amounts of H2O2 are needed for in situ treatment, or the water must be acidified prior ex situ treatment (6).
To overcome these limitations, heterogeneous iron-containing catalysts have been synthesized using silica supports to change the chemical environment of iron (9-12). For example, Chou et al. (10) developed a catalyst consisting of iron oxide on crushed brick. This composite catalyst oxidized more benzoic acid per mole of H2O2 consumed than goethite. However, the results of this study are difficult to interpret because the test solutions were unbuffered and the pH decreased substantially during the experiments (from initial pH values of 3.2, 6.0 and 10.0 to 3.0, 4.3 and 5.8 respectively), and it is unclear how much of the enhanced efficiency was attributable to acidification of the solutions. In another study, iron oxide nanoparticles immobilized on alumina-coated mesoporous silica exhibited an ability to catalyze the transformation of a dye, Reactive Black 5, by H2O2 at pH 4.1 with an efficiency that was substantially greater than that of similar amounts of hematite and magnetite (9). Similar results have been reported for H2O2 activation by Fe and Al – pillared clay catalysts (13-15). However, like the studies discussed above, most experiments were performed either under acidic conditions, at elevated temperatures or in the presence of ultraviolet light.
While it appears that alumina and silica supports improve the performance of heterogeneous iron-containing catalysts, the mechanism through which this occurs is not well understood. Possible explanations for the higher efficiency of iron/silica catalysts include less efficient scavenging of hydroxyl radicals by silica relative to iron oxide surfaces (10) and more oxidant production due to the better dispersion of iron on the surface (9). In addition, alumina, as a Lewis acid, could facilitate the reduction of Fe(III) to Fe(II) by H2O2, usually the rate limiting step in the Fenton’s reagent chain reaction, and thus accelerate activation of H2O2 (9).
The objective of this study was to determine how the presence of silica and alumina in an iron-containing catalyst alters H2O2 activation and contaminant oxidation at neutral pH values. For this purpose, silica- and alumina-containing iron precipitates were prepared, characterized and assessed for catalytic activity relative to iron oxides. Phenol was selected as a model target contaminant because it is not significantly adsorbed on any of the oxides in the catalysts and has a well-characterized reaction with hydroxyl radical. Understanding the role of alumina and silica on H2O2 activation may lead to the development of more efficient catalysts that could be used for ex situ treatment and provide a mean of harnessing the heterogeneous Fenton process using naturally occurring or modified minerals in the subsurface.
Materials and Methods
Materials
All chemicals were reagent grade and were used without further purification. Phenol and ferric perchlorate were obtained from Aldrich. Ferric nitrate and aluminum chloride were obtained from Fisher. Tetraethyl orthosilicate (TEOS) was obtained from Alfa Aesar. All solutions were prepared using 18 MΩ Milli-Q water from a Millipore system.
Hematite was synthesized by aging freshly made ferrihydrite in a strongly alkaline solution at 90°C for 48 hours (16). The identity of hematite was verified by X-ray diffraction. Commercial goethite and amorphous FeOOH were obtained from Fluka and Aldrich, respectively.
FeSi-ox and FeAlSi-ox synthesis
Precipitates containing iron, silicon and aluminum were synthesized by a sol-gel process. Specifically, 100 mL of 1 M ethanol, 1 M TEOS and either 0.2 M Fe(ClO4)3.9H2O (FeSi-ox synthesis) or 0.2 M Fe(ClO4)3.9H2O and 0.2 M AlCl3 (FeAlSi-ox synthesis) aqueous solution were stirred and heated at 80°C for 2 hrs in a 250-mL Pyrex flask. To initiate precipitation, 100 mL of 1.5 M ammonium hydroxide and 50 mL of water were added dropwise simultaneously (over about 15 min). After stirring at 80°C for 2 hrs, the mixture was transferred to a 500-mL beaker and then dried at 110°C for 24 hrs. The resulting particles were then washed three times with deionized water and dried at 110°C for another 24 hrs.
Characterization
The surface area of the solids was determined using N2 physisorption in a Micromeretics 2000 system using the 5 point BET (Brunauer–Emmett–Teller) method. X-ray diffraction (XRD) analysis was performed with Cu Kα radiation using a Panalytical 2000 diffractometer. The morphology of FeSi-ox and FeAlSi-ox was determined using a FEI Tecnai 12 transmission electron microscope (TEM) at 100 kV and a Hitachi S-5000 scanning electron microscope (SEM) at 10 kV. The distribution of elements on the surface was determined using a LEO 439 scanning electron microscope coupled with a Princeton Gamma-Tech energy dispersive X-ray spectrometer (SEM-EDX). The composition of the catalysts was measured by first dissolving particles in a concentrated solution of HCl, then measuring Fe and Al in the liquid phase using atomic absorption spectrophotometry.
Oxidation of Phenol
All oxidation experiments were carried out at room temperature (20 ± 2°C) in the dark in 50 mL of reaction solution. All the reactors were open to the atmosphere. The initial concentration of phenol was 0.5 mM. The initial solution pH was adjusted using 1 M NaOH or 0.5 M H2SO4. The pH of solutions was buffered with 1 mM piperazine-N,N’-bis(ethanesulfonic acid) (PIPES) for pH 7 or 1 mM borate for pH 8 – 9. Solutions with initial pH values of 5.5 were unbuffered. The pH was measured throughout each experiment, and the average pH value was calculated. The difference between the initial and final pH never exceeded 1.5 units in the experiments with pure iron oxides and 1 unit in the case of FeSi-ox and FeAlSi-ox.
The reactions were initiated by adding an aliquot of H2O2 stock solution to a pH-adjusted solution containing phenol and catalyst. In some experiments, 200 mM of tert-butanol (t-BuOH) was added as a hydroxyl radical scavenger. Samples were withdrawn at predetermined time intervals, filtered immediately through a 0.22-μm nylon filter and analyzed for phenol and H2O2. Experiments were carried out at least in triplicate and average values and standard deviations are presented.
Analytical Methods
Filtered samples were acidified to pH 2 and analyzed for phenol by high-performance liquid chromatography (HPLC) using a Waters Alliance HPLC system equipped with a 4.6 × 150 mm Waters Symmetry C18 5 μm column. A mobile phase consisting of 50% methanol and 50% water (pH 2) was used at a flow rate of 0.8 mL/min. Phenol was detected with UV absorbance detection at 270 nm. Prior to HPLC analysis, an excess amount of methanol (i.e., 50 μL) was added to 1 mL filtered aliquots to quench any additional oxidation reactions involving residual H2O2. H2O2 was analyzed spectrophotometrically by the titanium sulfate method (17). Total dissolved iron was quantified using the 1,10-phenanthroline method after adding hydroxylamine hydrochloride to the filtered samples (18).
Results
Catalyst properties
SEM and TEM images, along with XRD spectra of FeAlSi-ox (Figure 1) and FeSi-ox (Supporting Information (SI), Figure SI 1) show that these materials are amorphous xerogels, a typical product from sol-gel processing (19). The iron and aluminum content and BET surface areas of these materials are listed in Table 1.
Figure 1.
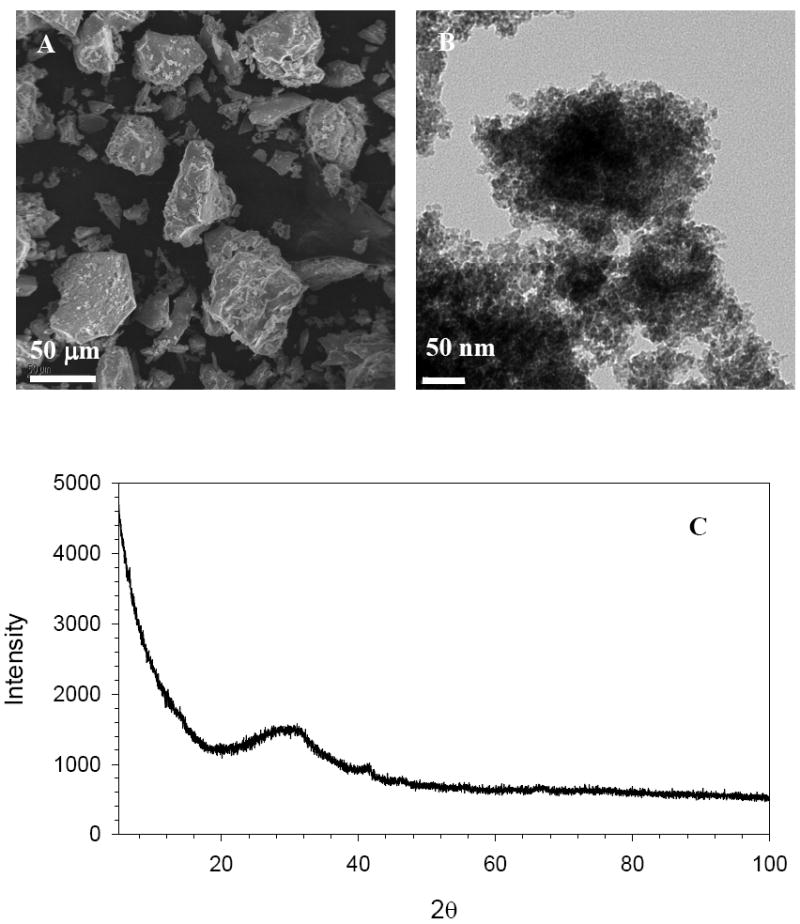
FeAlSi-ox obtained by sol – gel processing of aqueous mixture of Fe(ClO4)3, Al(NO3)3 and TEOS. (A) SEM. (B) TEM. (C) XRD.
Table 1.
Properties of different Fe-containing materials.
| Type of material | BET surface area (m2/g) | Fe content (weight %) | Al content (weight %) |
|---|---|---|---|
| Hematite | 35.9 | 70 (a) | - |
| Goethite | 13 | 35 (b) | - |
| Amorphous FeOOH | 165.8 | 62.9 (a) | - |
| FeSi-ox | 521 | 12.3 | - |
| FeAlSi-ox | 423 | 10.9 | 4.95 |
theoretical value.
value reported by the manufacturer
Catalytic performance toward H2O2 decomposition and phenol oxidation
The oxidation of phenol catalyzed by FeAlSi-ox (4.9 wt % Al, 10.9 wt % Fe) is a pH dependent process, with a reaction rate that decreases with increasing pH. After 8 hours over 90% of the phenol was transformed at pH 5.3, 30%-35% at pH 6.9 and 23%-25% at pH 8.5 (Figure 2). The concentration of phenol decreased by less than 15% in the presence of 200 mM t-BuOH at all three pH values (Figure 2). Control experiments (data not shown) showed that adsorption accounted for less than 3% of the total phenol loss and thus can be neglected compared to losses due to oxidation. A gradual increase of total dissolved iron ([FeTOT]) was observed during the experiments conducted in the absence of t-BuOH (inset of Figure 2).
Figure 2.
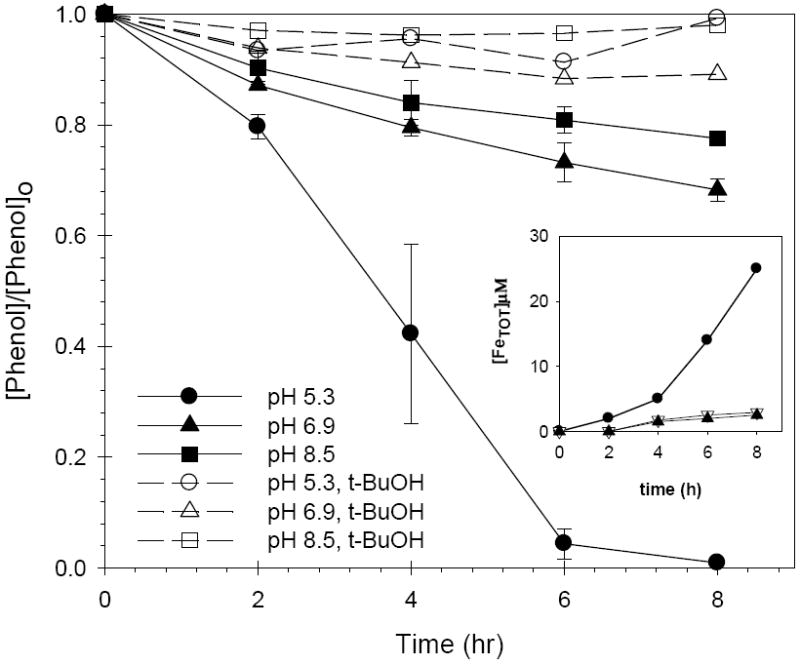
Effect of pH on phenol loss in the FeAlSi-ox/H2O2 system in the absence (solid lines) and presence (dashed lines) of t-BuOH; [phenol]o = 0.5 mM; [H2O2] = 50 mM; [FeAlSi-ox ] = 3 g/L; [t-BuOH] = 200 mM. [FeTOT] as a function of time (inset): (●) pH 5.3; (∇) pH 6.9; (▲) pH 8.5. For the purpose of clarity, error bars were eliminated from the data in the inset. In all cases, the pH decreased by less than 1 unit during the reaction.
While t-BuOH decreased phenol loss at all pH values, H2O2 decomposition was retarded by t-BuOH only at pH 5.3 (Figure 3). At pH 5.3, approximately 30% of the initial H2O2 was decomposed over 8 hours in the t-BuOH – free system, while less than 5% of the H2O2 decomposed in the presence of 200 mM t-BuOH.
Figure 3.
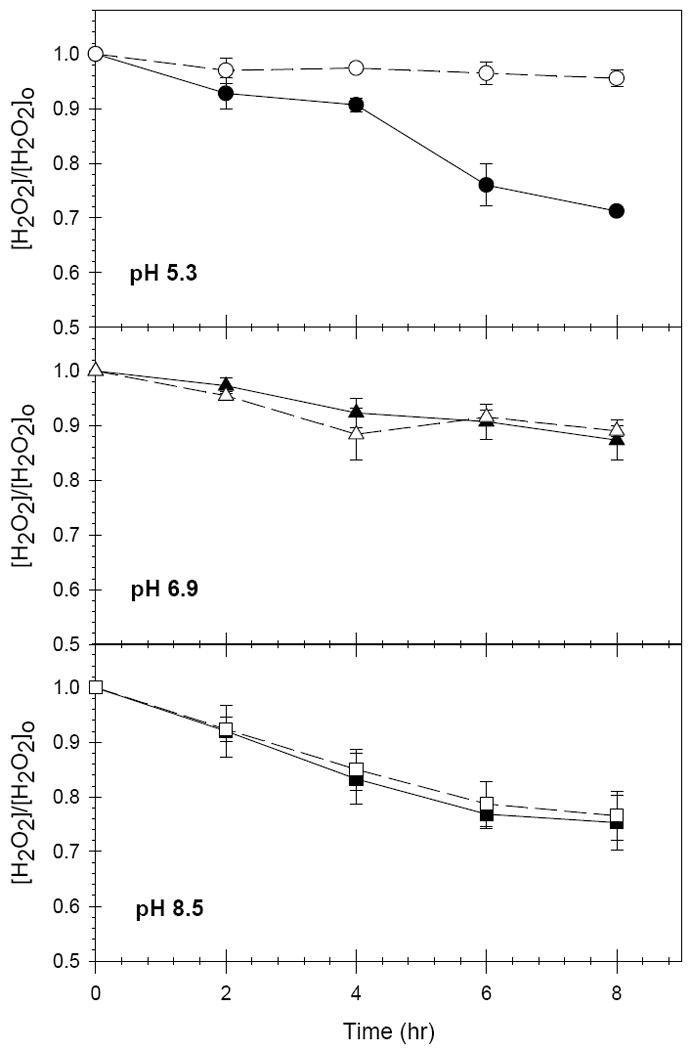
H2O2 loss in the FeAlSi-ox/H2O2 system in the absence (solid lines) and presence (dashed lines) of t-BuOH; [phenol]o = 0.5 mM; [H2O2] = 50 mM; [FeAlSi-ox ] = 3 g/L; [t-BuOH] = 200 mM.
The catalytic performance of the FeAlSi-ox catalyst was compared to the alumina-free analog, FeSi-ox (12.3 wt % Fe) at pH 6.9. The alumina-free catalyst resulted in faster H2O2 decomposition and phenol transformation (Figure 4).
Figure 4.
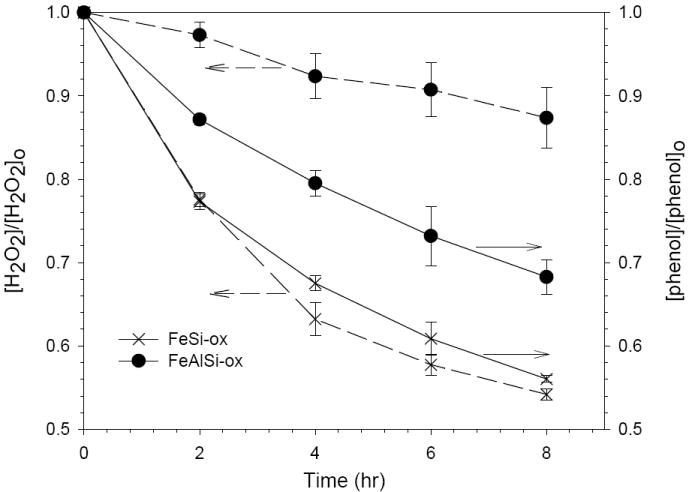
Phenol and H2O2 loss in the FeSi-ox/H2O2 and FeAlSi-ox/H2O2 systems; [phenol]o = 0.5 mM; [H2O2] = 50 mM; [FeAlSi-ox ] = [FeSi-ox ] = 3 g/L; pH = 6.9. The pH decreased by less than 0.3 units during the reaction.
The rate of H2O2 decomposition and transformation of phenol catalyzed by iron oxides was also investigated over pH values ranging pH values of 5.5 – 8.8. After 8 hours, approximately 20 to 35% of the initial H2O2 was decomposed in the hematite/H2O2 system, whereas all of the H2O2 was decomposed in the presence of amorphous FeOOH and commercial goethite (Figure SI 2). However, phenol transformation catalyzed by iron oxides was very low: less than 1% phenol loss was observed for amorphous FeOOH and commercial goethite. For hematite, the concentration of phenol decreased by approximately 5% to 7% mainly due to surface adsorption.
The stoichiometric efficiency, defined as the amount of phenol decomposed per mole of H2O2 consumed (i.e., ) was used to compare the performance of the catalysts. For each experiment, we calculated the stoichiometric efficiency after 25% of the phenol was transformed to assure that the comparisons were valid. This is necessary because the products of phenol transformation (e.g., hydroquinone) could react with oxidants and decrease the apparent efficiency. Conversely, measuring the stoichiometric efficiency early in the reaction (i.e., when less than 10% of the phenol was transformed) could result in reduced precision due to difficulties in detecting small losses of phenol. In the iron oxide/H2O2 systems, phenol loss was always less than 25% and in these cases, Δphenol values at the end of the experiments were used to determine stoichiometric efficiency. The stoichiometric efficiency of the FeAlSi-ox catalyst was 3 to 4 times greater than that of the FeSi-ox catalyst and approximately 50 to 80 times greater than that of the iron oxides over the pH range studied (Figure 5).
Figure 5.
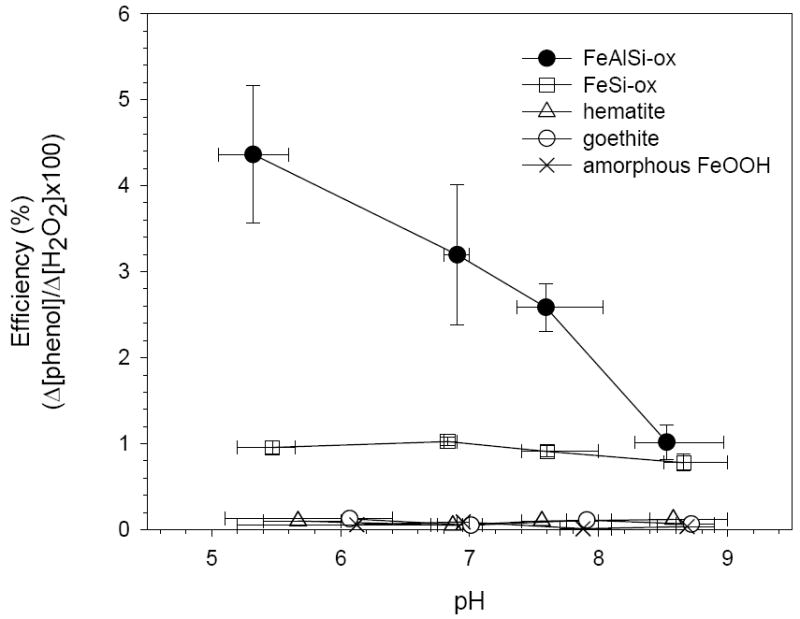
Stoichiometric efficiency (Δ[phenol]/Δ[H2O2] × 100%) as function of pH. Data collected when Δ[phenol] = 23-27% [phenol]o. [Phenol]o = 0.5 mM; [H2O2]o = 50 mM; [oxide] = 3 g/L.
Discussion
Activation of H2O2 by iron oxides
The decomposition of H2O2 by pure iron oxides (e.g., goethite, hematite) has been studied over a wide pH range. Under acidic conditions, the process appears to be controlled by redox cycling of both surface and dissolved iron (i.e., Fe[II]/Fe[III]), the latter resulting from dissolution of the iron oxides (20, 21). At circumneutral pH values, the contribution of dissolved iron to H2O2 activation should be minimal because Fe(III) is sparingly soluble (22). Therefore, the decomposition of H2O2 under circumneutral pH conditions is likely a surface-catalyzed process.
It has been suggested that the surface-initiated H2O2 decomposition proceeds through a chain reaction that is analogous to the Fe3+ - initiated decomposition of H2O2 that was initially described by Haber and Weiss under acidic conditions (23-25). The oxidation of organic contaminants during H2O2 decomposition has been attributed to hydroxyl radical (•OH) production from the reaction of H2O2 with reduced surface iron (i.e., ≡FeII) (scheme 1 and reaction 2 in table 2).
Scheme 1.
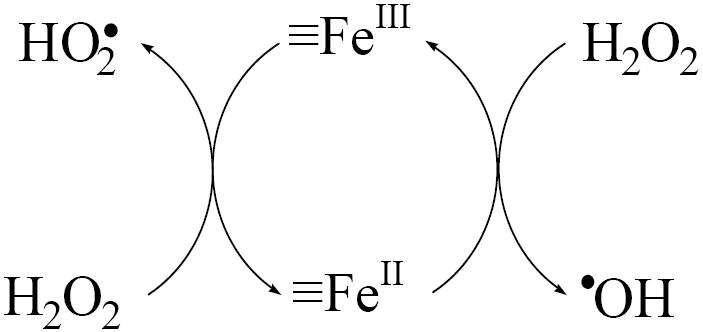
Haber – Weiss mechanism
Table 2.
Mechanism of surface-initiated H2O2 decomposition.
| Haber – Weiss mechanism (23, 25) | |
| reaction | |
| ≡Fe(III) + H2O2 → ≡Fe(II) + HO2•(O2•-) + H+ (2H+) | (1) |
| ≡Fe(II) + H2O2 → ≡Fe(III) +•OH + OH- | (2) |
| ≡Fe(III) + HO2• (O2•-) → ≡Fe(II) + O2 (+ H+) | (3) |
| HO2•⇔ H+ + O2•- | (4) |
| •OH + H2O2→ H2O + HO2• | (5) |
| •OH + ≡Fe(II) → ≡Fe(III) + HO- | (6) |
| •OH + HO2• (O2•-) → O2 + H2O (+ OH-) | (7) |
| HO2•+ HO2• → H2O2 + O2 | (8) |
| A possible non-radical mechanism | |
| reaction | |
| ≡Fe(III) + H2O2→ ≡Fe(II) + HO2• + H+ | (1) |
| ≡Fe(II) + H2O2 → ≡Fe(IV) + 2OH- | (9) |
| ≡Fe(IV) + H2O2 → ≡Fe(II) + O2 + 2H+ | (10) |
| ≡Fe(IV) + ≡Fe(II) → 2≡Fe(III) | (11) |
| ≡Fe(III) + HO2• (O2•-) → ≡Fe(II) + O2 (+ H+) | (3) |
| HO2•+ HO2• → H2O2 + O2 | (8) |
The application of the iron oxide/H2O2 systems for oxidation of contaminants has been limited by the extremely low stoichiometric efficiency of oxidant production at neutral pH values (e.g., (1, 7) and Figure 5). The low efficiency is often attributed to the generation of •OH in areas on the oxide surface that are inaccessible to the contaminants (e.g., •OH is scavenged by the iron oxide surface (1)). Alternatively, the activation of H2O2 by iron oxides could produce oxidants such as high-valent iron species (i.e., ≡Fe[IV]) (26). While little is known about the exact structure and reactivity of such surface-bound oxidants, solution phase Fe[IV] species are less reactive than •OH and do not react with aromatic compounds to an appreciable extent (27). Some investigators also have suggested that the decomposition of H2O2 on the surface of iron oxides may proceed mainly through a non-radical mechanism that converts H2O2 directly into O2 and H2O by a series of 2e- transfer reactions (e.g., by the presence of oxygen vacancies on the surface (28) or the cycling of ≡Fe[IV]/≡Fe[II] as proposed in scheme 2 and reactions 9 – 11 in table 2). The principal net reaction in these pathways is the conversion of H2O2 into H2O and O2 without the production of •OH.
Scheme 2.
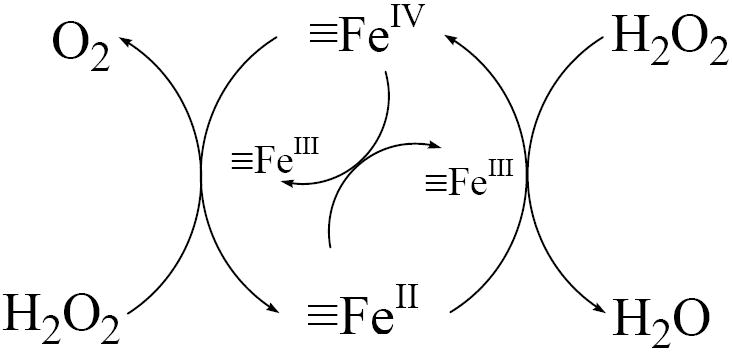
Non-radical mechanism
Efficiency enhancement with FeAlSi-ox and FeSi-ox
The stoichiometric efficiency of FeSi-ox and FeAlSi-ox is much higher than that of iron oxides (Figure 5). In the FeAlSi-ox/H2O2 system, the rate of phenol transformation decreases dramatically with increasing pH (Figure 2) while the rate of H2O2 loss only varies by about 15% with a minimum at pH 6.9 (Figure 3). The decreased rate of phenol loss at higher pH values appears to be attributable to a decrease in the production of oxidants capable of reacting with phenol. The oxidation of phenol in this system is most likely due to •OH because upon addition of t-BuOH phenol transformation rate decreased significantly (Figure 2).
As mentioned previously, Fe(III) is sparingly soluble at circumneutral pH values. The concentration of soluble Fe(III) is expected to range from 0.001 μM to 0.1 μM over pH values ranging from 5.5 to 9, assuming that the system is at equilibrium with 2-line ferrihydrite (22). As the reaction proceeded, however, the concentration of dissolved iron increased to 20 ± 10 μM at pH 5.3 and 2 ± 1 μM at pH 6.9 – 8.5 (inset of Figure 2). This dissolution of Fe(III) was attributable to the interaction of surface iron and intermediate oxidation products of phenol (e.g., hydroquinones, organic acids) that enhance iron solubility via complexation and reductive dissolution (20). This hypothesis was supported by the fact that iron leaching and phenol oxidation were not observed upon addition of t-BuOH, as little intermediates were formed.
In a previous study (21), the activation of H2O2 by dissolved iron was observed at [Fe(III)] as low as 0.42 μM. Consequently, to determine whether the higher stoichiometric efficiency of FeAlSi-ox/H2O2 system was due to better H2O2 activation by the FeAlSi-ox surface or to activation of H2O2 by dissolved iron, we investigated the proportions of homogeneous and heterogeneous reaction using filtration to isolate the solution-phase reaction (Figure SI 3). Following filtration, at pH 6.9 and 8.5, the phenol concentration decreased by less than 3% during a 16-hour period, indicating that dissolved iron is unimportant to phenol transformation compared to surface catalyzed reactions. At pH 5.3, the phenol concentration decreased significantly after filtration (Figure SI 4), albeit less than in the presence of FeAlSi-ox. Phenol transformation at this pH value therefore was attributable to the production of oxidants from both homogeneous and surface-catalyzed reaction.
Role of silica and alumina
SiO2 and Al2O3 do not catalyze the decomposition of H2O2 at circumneutral pH values (less than 1% of the initial H2O2 was lost in the presence of just either SiO2 or Al2O3). Therefore, the significant enhancement in H2O2 activation by FeSi-ox and FeAlSi-ox at circumneutral pH values relative to pure iron oxides is attributable to the interaction of iron with alumina and silica in the mixed catalyst. There are several possible explanations for this phenomenon. First, the dispersion of the iron oxide phase within the silica and alumina matrix might prevent the iron from aggregating into clusters, resulting in changes in the number and properties of the reactive surface sites. These structural differences can alter the relative proximity of reactive sites, which in turn may affect the reactions between the surface and the reactant (i.e., H2O2). The role of steric position of reactive sites on redox processes has been speculated to be important in the reduction of carbon tetrachloride by Fe(II) associated with goethite, where the steric position of the latter can enhance multiple electron transfer reactions (29). In a similar way, iron dispersion within the silica and alumina matrix might favor the radical mechanism (series of 1e- transfer steps) over the non-radical mechanisms (2e- transfer step), leading to more •OH production during the decomposition of H2O2.
The higher efficacy of FeAlSi-ox and FeSi-ox compared with iron oxides may also arise from the difference in electronic properties of iron because silica, alumina and iron oxides exhibit different points of zero charge (pzc) values. The pzc of SiO2 (i.e., 2 – 5) is much lower than that of iron oxides and alumina (i.e., 7.5 – 9) (30). At circumneutral pH values, it is expected that the surface of FeSi-ox and FeAlSi-ox will be negatively charged because SiO2 is the predominant component in these materials (see the Energy Dispersive X-ray spectra data, Figure SI 5) whereas iron oxide surfaces will be positively charged, or will have a much less negative charge. In addition to altering the electronic properties of reactive sites, the negative surface charge of FeAlSi-ox may also affect the sorption of H2O2 on the surface, which was suggested to be the rate limiting step in H2O2 decomposition (2). While the mechanism and kinetics of H2O2 interactions with surfaces have not been well studied, H2O2 forms strong hydrogen bonds with the oxygen in siloxane bridges, Si-O-Si (31). It is possible that such interactions with the silica-containing catalyst may alter the reactions of H2O2 with iron on the catalyst surface.
It is interesting to note that although the decomposition of phenol and H2O2 occurred at a faster rate when catalyzed by FeSi-ox, a higher stoichiometric efficiency was obtained with FeAlSi-ox. The mechanism through which alumina alters the efficiency is unclear. Lim et al. (9) postulated that alumina facilitates the reduction of Fe(III) by H2O2 because alumina, a Lewis acid, can attract electron density from iron and thus raise the oxidation potential of the Fe(III) center. This explanation seems unlikely because the reactions were slower for the Al-containing catalyst (i.e., FeAlSi-ox). On the basis of the iron content and surface area (Table 1), we hypothesize that faster reactions observed with FeSi-ox are related to its higher surface and iron content. However, this cannot explain the higher H2O2 utilization efficiency of the FeAlSi-ox catalyst. Additional research is needed to characterize the role that Al plays in the catalyst.
Environmental implications
The silica- and alumina-containing iron oxide catalyst has the potential to be more effective in the oxidative treatment of industrial waste and contaminated water at circumneutral pH values than iron oxides studied previously for this application. While over 90% of the H2O2 that was lost in the presence of the catalyst does not produce oxidants capable of transforming aromatic compounds, the absence of a pH adjustment step, minimal waste production and low potential for production of toxic byproducts may provide advantages over other approaches. Additional research is needed to further enhance the efficiency of the catalyst and assess the scale up of the treatment systems employing the catalyst.
This study also has important implications for the design and operation of in situ remediation systems that use H2O2 for oxidation of contaminants. Previous studies on the mechanism of H2O2 reduction by pure iron oxides indicated that iron oxides can activate H2O2 into species capable of oxidizing contaminants. Researchers studying pure iron oxides suggested that iron-containing minerals in the subsurface could be exploited to activate H2O2 for in situ remediation. The present study suggests that iron oxides associated with alumina and silica may behave differently from pure iron oxides. The activity of iron associated with aluminosilicates and silica-containing minerals may help to explain differences in the production of oxidants observed during H2O2 decomposition in soils (24). Additional research on the stoichiometric efficiency of aquifer materials may lead to better predictions of the efficacy of H2O2-based in situ remediation systems.
Supplementary Material
Acknowledgments
This research was funded by the U.S. National Institute for Environmental Health Sciences (NIEHS) Superfund Basic Research Program (Grant P42 ES004705). A.L.P was supported in part by Vietnam Education Foundation (VEF).
Footnotes
Brief. Higher H2O2 utilization efficiency in heterogeneous Fenton reaction at neutral pH values was observed in the presence of silica and alumina.
Literature cited
- 1.Valentine RL, Wang HCA. Iron oxide surface catalyzed oxidation of quinoline by hydrogen peroxide. Journal of Environmental Engineering. 1998;124(1):31–38. [Google Scholar]
- 2.Kwan WP, Voelker BM. Rates of hydroxyl radical generation and organic compound oxidation in mineral-catalyzed Fenton-like systems. Environmental Science & Technology. 2003;37(6):1150–1158. doi: 10.1021/es020874g. [DOI] [PubMed] [Google Scholar]
- 3.Ravikumar JX, Gurol MD. Chemical oxidation of chlorinated organics by hydrogen peroxide in the presence of sand. Environmental Science & Technology. 1994;28(3):394–400. doi: 10.1021/es00052a009. [DOI] [PubMed] [Google Scholar]
- 4.Miller CM, Valentine RL. Mechanistic studies of surface catalyzed H2O2 decomposition and contaminant degradation in the presence of sand. Water Research. 1999;33(12):2805–2816. [Google Scholar]
- 5.Huling SG, Pivetz BE. In-situ chemical oxidation. EPA Engineering Issue. 2006 Available at : http://www.epa.gov/ada/topics/oxidation_pubs.html.
- 6.Pignatello JJ, Oliveros E, MacKay A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Critical Reviews in Environmental Science and Technology. 2006;36(1):1–84. [Google Scholar]
- 7.Huang H-H, Lu M-C, Chen J-N. Catalytic decomposition of hydrogen peroxide and 2-chlorophenol with iron oxides. Water Research. 2001;35(9):2291–2299. doi: 10.1016/s0043-1354(00)00496-6. [DOI] [PubMed] [Google Scholar]
- 8.Yeh CK-J, Chen W-S, Chen W-Y. Production of hydroxyl radicals from the decomposition of hydrogen peroxide catalyzed by various iron oxides at pH 7. Practice Periodical of Hazardous, Toxic, and Radioactive Waste Management. 2004;8(3):161–165. [Google Scholar]
- 9.Lim H, Lee J, Jin S, Kim J, Yoon J, Hyeon T. Highly active heterogeneous Fenton catalyst using iron oxide nanoparticles immobilized in alumina coated mesoporous silica. Chemical Communications. 2006;4:463–465. doi: 10.1039/b513517f. [DOI] [PubMed] [Google Scholar]
- 10.Chou S, Huang C. Application of a supported iron oxyhydroxide catalyst in oxidation of benzoic acid by hydrogen peroxide. Chemosphere. 1999;38(12):2719–2731. [Google Scholar]
- 11.Crowther N, Larachi F. Iron-containing silicalites for phenol catalytic wet peroxidation. Applied Catalysis B: Environmental. 2003;46(2):293–305. [Google Scholar]
- 12.Calleja G, Melero JA, Martínez F, Molina R. Activity and resistance of iron-containing amorphous, zeolitic and mesostructured materials for wet peroxide oxidation of phenol. Water Research. 2005;39(9):1741–1750. doi: 10.1016/j.watres.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Barrault J, Abdellaoui M, Bouchoule C, Majesté A, Tatibouët JM, Louloudi A, Papayannakos N, Gangas NH. Catalytic wet peroxide oxidation over mixed (Al-Fe) pillared clays. Applied Catalysis B: Environmental. 2000;27(4):L225–L230. [Google Scholar]
- 14.Cheng M, Song W, Ma W, Chen C, Zhao J, Lin J, Zhu H. Catalytic activity of iron species in layered clays for photodegradation of organic dyes under visible irradiation. Applied Catalysis B: Environmental. 2008;77(34):355–363. [Google Scholar]
- 15.Luo M, Bowden D, Brimblecombe P. Catalytic property of Fe-Al pillared clay for Fenton oxidation of phenol by H2O2. Applied Catalysis B: Environmental. 2009;85(34):201–206. [Google Scholar]
- 16.Schwertmann U, Cornell RM. Iron oxides in the laboratory: preparation and characterization. Wiley-VCH Publishers; 2000. [Google Scholar]
- 17.Eisenberg G. Colorimetric determination of hydrogen peroxide. Industrial & Engineering Chemistry Analytical Edition. 1943;15(5):327–328. [Google Scholar]
- 18.Tamura H, Goto K, Yotsuyanagi T, Nagayama M. Spectrophotometric determination of iron(II) with 1,10-phenanthroline in the presence of large amounts of iron(III) Talanta. 1974;21(4):314–318. doi: 10.1016/0039-9140(74)80012-3. [DOI] [PubMed] [Google Scholar]
- 19.Corma A. From microporous to mesoporous molecular Sieve materials and their use in catalysis. Chemical Reviews. 1997;97(6):2373–2420. doi: 10.1021/cr960406n. [DOI] [PubMed] [Google Scholar]
- 20.Pera-Titus M, García-Molina V, Baños MA, Giménez J, Esplugas S. Degradation of chlorophenols by means of advanced oxidation processes: a general review. Applied Catalysis B: Environmental. 2004;47(4):219–256. [Google Scholar]
- 21.Kwan WP, Voelker BM. Decomposition of hydrogen peroxide and organic compounds in the presence of dissolved iron and ferrihydrite. Environmental Science & Technology. 2002;36(7):1467–1476. doi: 10.1021/es011109p. [DOI] [PubMed] [Google Scholar]
- 22.Stefansson A. Iron(III) hydrolysis and solubility at 25°C. Environmental Science & Technology. 2007;41(17):6117–6123. doi: 10.1021/es070174h. [DOI] [PubMed] [Google Scholar]
- 23.Lin SS, Gurol MD. Catalytic decomposition of hydrogen peroxide on iron oxide: kinetics, mechanism, and implications. Environmental Science & Technology. 1998;32(10):1417–1423. [Google Scholar]
- 24.Petigara BR, Blough NV, Mignerey AC. Mechanisms of hydrogen peroxide decomposition in soils. Environmental Science & Technology. 2002;36(4):639–645. doi: 10.1021/es001726y. [DOI] [PubMed] [Google Scholar]
- 25.Haber F, Weiss J. The Catalytic Decomposition of hydrogen peroxide by iron salts. Proceeding of The Royal Society A. 1934;147:332–351. [Google Scholar]
- 26.Voegelin A, Hug SJ. Catalyzed oxidation of arsenic(III) by hydrogen peroxide on the surface of ferrihydrite: an in situ ATR-FTIR study. Environmental Science & Technology. 2003;37(5):972–978. doi: 10.1021/es025845k. [DOI] [PubMed] [Google Scholar]
- 27.Keenan CR, Sedlak DL. Factors affecting the yield of oxidants from the reaction of nanoparticulate zero-valent iron and oxygen. Environmental Science & Technology. 2008;42(4):1262–1267. doi: 10.1021/es7025664. [DOI] [PubMed] [Google Scholar]
- 28.Lee YN, Lago RM, Fierro JLG, González J. Hydrogen peroxide decomposition over Ln1-xAxMnO3 (Ln = La or Nd and A = K or Sr) perovskites. Applied Catalysis A: General. 2001;215(12):245–256. [Google Scholar]
- 29.Amonette JE, Workman DJ, Kennedy DW, Fruchter JS, Gorby YA. Dechlorination of carbon tetrachloride by Fe(II) associated with goethite. Environmental Science & Technology. 2000;34(21):4606–4613. [Google Scholar]
- 30.Esshington ME. Soil and water chemistry: an integrative approach. CRC Press; 2004. [Google Scholar]
- 31.Zeglínski J, Piotrowski GP, Piekós R. A study of interaction between hydrogen peroxide and silica gel by FTIR spectroscopy and quantum chemistry. Journal of Molecular Structure. 2006;794(13):83–91. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.