

Abstract
Rho family small GTPases serve as molecular switches in the regulation of diverse cellular functions, including actin cytoskeleton remodeling, cell migration, gene transcription, and cell proliferation. Importantly, Rho overexpression is frequently seen in many carcinomas. However, published studies have almost invariably used immortal or tumorigenic cell lines to study Rho GTPase functions and there are no studies on the potential of Rho small GTPase to overcome senescence checkpoints and induce preneoplastic transformation of human mammary epithelial cells (hMEC). We show here that ectopic expression of wild-type (WT) RhoA as well as a constitutively active RhoA mutant (G14V) in two independent primary hMEC strains led to their immortalization and preneoplastic transformation. These cells have continued to grow over 300 population doublings (PD) with no signs of senescence, whereas cells expressing the vector or dominant-negative RhoA mutant (T19N) senesced after 20 PDs. Significantly, RhoA-T37A mutant, known to be incapable of interacting with many well-known Rho effectors including Rho kinase, PKN, mDia1, and mDia2, was also capable of immortalizing hMECs. Notably, similar to parental normal cells, Rho-immortalized cells have WT p53 and intact G1 cell cycle arrest on Adriamycin treatment. Rho-immortalized cells were anchorage dependent and were unable to form tumors when implanted in nude mice. Lastly, microarray expression profiling of Rho-immortalized versus parental cells showed altered expression of several genes previously implicated in immortalization and breast cancer progression. Taken together, these results show that RhoA can induce the preneoplastic transformation of hMECs by altering multiple pathways linked to cellular transformation and breast cancer.
Introduction
A large body of evidence implicates Ras-like small G proteins as major players in the regulation of a variety of cellular processes. Rho GTPases cycle between inactive GDP-bound and active GTP-bound states, a transition controlled by guanine nucleotide exchange factor proteins, which convert the GDP-bound to GTP-bound form, and by GTPase-activating proteins, which stimulate the low-intrinsic GTPase activity to convert the active to inactive form (1). It is believed that the multitude of cellular processes regulated by Rho reflects the interaction of the active form with several distinct effector molecules and subsequent activation of these effectors (1-3). For example, Rho effectors such as phosphatidylinositol-4-phosphate 5-kinase, Rho kinase (and related ROCK kinase), formin homology protein p140-Dia, and rhophilin have been linked to the regulation of actin cytoskeleton organization (1, 4-6), and citron kinase seems to regulate cytokinesis (7, 8). Recent evidence suggest a role of Rho effector PKN in cortical actin formation (9) and in G2-M checkpoint regulation (10).
At the cellular level, Rho family small GTPases have emerged as key regulators of cell adhesion, migration, endocytic trafficking, cytokinesis, gene transcription, and cell proliferation through control of the actin cytoskeleton remodeling and other cellular responses to external stimuli (2, 11, 12). The role of Rho G proteins in cell proliferation and oncogenesis is emphasized by the fact that most of their exchange factors were originally identified as oncogenes and by the facilitation of cellular transformation by activated Rho and reversal of various aspects of the transformed phenotype, including invasive behavior, by interrupting Rho function (13-18). Dysfunctional regulation of Rho GTPases has been implicated in certain aspects of cancer development. For instance, overexpression of activated Rho mutants can transform fibroblasts (13). Rho proteins promote cell cycle progression through enhanced cyclin-dependent kinase activity by regulating the levels of cyclin D1, p21WAF1, and p27KIP1 (14). Transcriptional up-regulation of the levels of particular Rho proteins has been described in many types of human cancers, including cancers of the colon, breast, lung, stomach, and pancreas, and was correlated with tumor progression and invasion (15-18). In breast cancer, increased RhoA expression correlated with cancer progression (17, 18), and Rho protein overexpression was shown to contribute to breast cancer cell invasion and metastasis (18). However, the role of Rho proteins in the early steps of transformation of primary human epithelial cells, which are normally programmed to undergo replicative senescence, has not been investigated.
Here, we report that ectopic overexpression of not only a constitutively active RhoA but also the WT RhoA induces the immortalization of primary human mammary epithelial cells (hMEC). Importantly, a point mutant of RhoA, T37A, previously known not to interact with most well-known Rho effectors, such as Rho kinase, PKN, and mDia, also was capable of immortalizing the hMECs. Rho-immortalized hMECs have an intact G1 cell cycle checkpoint, do not exhibit anchorage-independent growth, and do not form tumors in nude mice. Microarray analyses of Rho-immortalized versus parental MECs revealed altered expression of several genes known to be involved in cellular immortalization and breast cancer progression. These results show that ectopic expression of RhoA can induce the preneoplastic transformation of mammary epithelial cells apparently by dysregulating several biochemical pathways linked to cellular transformation and breast cancer.
Materials and Methods
Cell strains and cell culture
Reduction mammoplasty-derived hMECs, 76N and 70N, were grown in the DFCI-1 medium, as described previously (19). RhoA-immortalized cells were grown in DFCI-1 medium supplemented with 100 μg/mL G418 (Sigma).
Plasmid constructs
Rho constructs were subcloned in pLXSN retroviral vector (Clontech) from pTB701 plasmid (kindly provided by Dr. Yoshitaka Ono, Kobe University, Kobe, Japan). RhoA-T19N was PCR amplified from pcDNA-RhoA. T19N (kindly provided by Dr. Arthur Mercurio, University of Massachusetts Medical School, Worcester, MA) and cloned in pLXSN.
Retroviral infection of mammary epithelial cells
Retrovirus-containing culture supernatants were prepared as described previously (20). 76N or 70N cells (5 × 105 per 100-mm dishes) were exposed to retroviral supernatants containing 4 μg/mL polybrene. Stable cell lines were established by selection in G418 (100 μg/mL).
Western blot analysis and antibodies
Cell lysates were quantitated using the bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Inc.). Denatured proteins were resolved on SDS-PAGE gels, transferred to polyvinylidine difluoride membranes (Millipore), and Western blotted using monoclonal antibodies against anti-RhoA (26C4), anti-p53 (DO-1), and anti-p21 (F-5; Santa Cruz Biotechnology, Inc.) and anti-β-actin (AC-15, Abcam).
Glutathione S-transferase pull-down assay
Glutathione S-transferase (GST) fusion proteins were expressed in BL21 bacterial cells and purified with glutathione Sepharose 4B beads (Amersham Biosciences). 293T cells were transfected with myc-tagged Prks-ROCK1 or Prks-ROCK2, flag-tagged Prc-PKN-AL, or mDia1 using calcium phosphate method. The transfectants were lysed in lysis buffer [50 mmol/L Tris-HCl (pH 7.5), 100 mmol/L NaCl, 1 mmol/L DTT, 5 mmol/L MgCl2, 50 mmol/L NaF, 1 mmol/L EDTA, 1 mmol/L Na3VO4, 10% glycerol, 1% NP40, 1 mmol/L phenylmethylsulfonyl fluoride] and spun at 12,000 rpm, and 1 mg each of these supernatants was incubated with 5 μg of GST or various fusion proteins that were loaded with GTP-γ-S in loading buffer [20 mmol/L Tris-HCl (pH 7.5), 1 mmol/L DTT, 10 mmol/L EDTA, 50 mmol/L NaCl, 5% glycerol, 0.1% Triton X-100, 1 mmol/L MgCl2, 100 μmol/L GTP-γ-S] for 4 h at 4°C. Beads were washed and loaded onto 12.5% SDS-PAGE gel. After electrophoresis, the gel was cut into two parts: the upper part that contained ROCK1, ROCK2, PKN, and mDia1 was transferred into polyvinylidene difluoride (PVDF) membrane and probed with anti-myc or anti-flag antibodies and the lower part that contained GST fusion proteins was stained with Coomassie Blue R-250.
Telomerase assays
Telomerase activity and telomerase length were determined, as described previously (21). Briefly, genomic DNA was isolated from cells using the phenol-chloroform method. Genomic DNA (3–5 μg) was digested with HinfI and RsaI followed by Southern blot analysis using the 32P-labeled TTTAGGG oligonucleotide probe.
DNA damage checkpoint analysis
Cells were treated with 0.5 μg/mL Adriamycin or DMSO for 24 h. For thymidine incorporation, cells were pulsed with [3H]thymidine for 6 h, fixed, and subjected to autoradiography as described previously (21). Labeled nuclei were counted and expressed as % labeled nuclei. Total cell lysates were examined for p53 and p21 protein levels using Western blot analysis.
Anchorage-independent growth in soft agar
A base layer of 0.6% agarose was prepared by diluting a 1.2% sterile stock 1:1 with 2× DMEM or D medium and plating 2 mL per well in six-well plates. The top agarose layer (0.3%; 2 mL) containing 2 × 104 cells was then layered on top of the base layer. The number of colonies was counted after 2 wk; colonies 100 cells or larger were considered positive.
Tumorigenicity assays
Six-week-old female athymic nude (nu/nu) mice (Charles River Laboratories) were injected s.c. close to the fourth mammary gland with 106 cells in 0.2 mL of 1:1 Matrigel (source) and PBS and observed for any tumor growth. Animals were euthanized and necropsies were performed when tumors reached 1 to 1.5 cm in diameter (in case of positive control cell line) or after 6 mo if no tumors were observed. Each cell line was tested in at least five animals. All animal-related procedures were carried out in accordance with the Institutional Animal Care and Use Committee guidelines.
Microarray analyses
RNA was isolated from parental and Rho immortal 76N cells in three independent experiments. RNA quality check, labeling of cRNA, cRNA fragmentation, hybridization of labeled cRNA to GeneChip, and scanning were performed by Microarray Core Facility, Northwestern University. Affymetrix Human Genome U133 Plus 2.0 chips (containing >47,000 transcripts/chip) were used. After hybridization, the chips were scanned by BeneChip Scanner 3000. Statistical analysis of the microarray data was performed by Bioinformatics Core, Northwestern University. Microarray data were collected and achieved in accordance with the MIAME guideline. The annotation of the HG-U133 Plus 2 microarray was updated using the Entrez gene database at the National Center for Biotechnology Information (NCBI). Raw Affymetrix measurements were normalized with a quantile model and quantified with the RMA algorithm using the Bioconductor package. 5’ to 3’ intensity bias and residuals from the RMA model were used for quality assessment of the microarray results. Unsupervised cluster analysis of the samples, genes with fold changes larger than two, was used to confirm the grouping of different phenotypes and experiment replicates. A linear model with Bayesian adjustment (LIMMA) was used to find differentially expressed genes with a statistical confidence of false discovery rate smaller than 0.01. To visualize results, gene expression was clustered using the TreeView program.
Reverse transcription-PCR and quantitative PCR
Total RNA was isolated using Trizol reagent according to the manufacturer’s instructions (Invitrogen). Reverse transcription-PCR (RT-PCR) was performed using SuperScript One-Step RT-PCR kit (Invitrogen). RNA (0.5 μg) was used for each RT-PCR reaction. For quantitative PCR, single-stranded cDNA was produced by reverse transcription using 1 μg RNA in 20 μL reaction (Promega). Quantitative PCR was performed using the SYBR Green reagents on the 7500 Real-Time PCR System (Applied Biosystems).
Results
Overexpression of WT RhoA or activated RhoA-G14V but not RhoA-T19N induces the immortalization of hMECs
The Rho family small GTPases are widely accepted as key regulators of cell adhesion, migration, endocytic trafficking, cytokinesis, gene transcription, and cell proliferation (1, 2, 11, 12). As essentially all of these roles have been assigned based on experiments using immortalized or transformed cell lines that have undergone many genetic alterations, we examined the consequences of RhoA overexpression in primary hMECs. A hMEC strain 76N was infected with retrovirus supernatants generated using the vector, RhoA-WT, RhoA-G14V (constitutively active Rho), or RhoA-T19N (dominant-negative Rho) constructs. Cells were subjected to G418 selection and maintained in G418-containing DFCI-1 medium thereafter. Western blot analysis of lysates after 48 hours of infection showed that all Rho proteins were expressed in transduced cells (Fig. 1A). As expected, 76N cells transduced with vector proliferated initially and then senesced ~20 population doublings (PD; Fig. 1B). Similarly, 76N cells transduced with dominant-negative RhoA-T19N senesced ~20 PDs (Fig. 1B). Both the WT and G14V-expressing cells, however, continued to grow for about a month, followed by about a 2-week “crisis” period where cells stopped growing and eventual emergence of cells that continued to grow with no signs of senescence. These cells have continued to grow beyond 300 PDs without any evidence of senescence, at which time they were frozen. Notably, the G14V-immortalized cells reproducibly expressed much lower levels of RhoA protein compared with the WT-immortalized cells (Fig. 1A). The reason for the lower protein levels is unclear at present; it may reflect the selection of immortal cells expressing relatively low levels of active G14V protein as high levels of active Rho protein are reported to induce apoptosis (22). These experiments were repeated thrice and similar results were obtained. These results show that overexpression of both the WT and constitutively active RhoA proteins leads to immortalization of primary hMECs. Notably, neither the parental cells nor the vector or T19N transduced cells led to immortal derivatives, indicating that the immortalization process is dependent on the expression of active RhoA.
Figure 1.
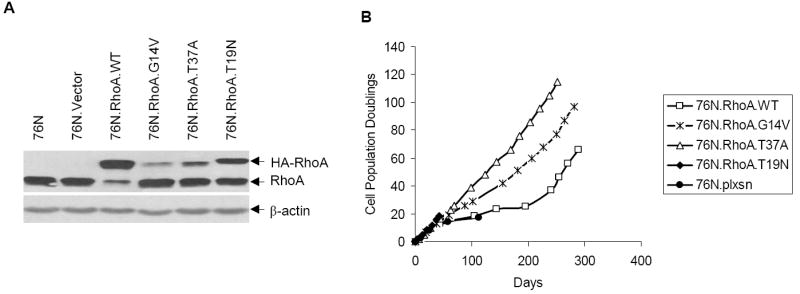
RhoA overexpression induces hMEC immortalization. A, cell lysates from indicated cells were analyzed for RhoA expression using anti-RhoA or β-actin (loading control) antibodies by Western blotting. B, cumulative PDs of cells expressing vector or various Rho mutants.
RhoA-mediated immortalization does not involve Rho effectors, Rho kinase, PKN, and mDia1
The ability of WT and constitutively active RhoA, but not the GDP-binding mutant, to immortalize hMECs suggested that Rho effectors can overcome the senescence checkpoint that limits the life span of normal hMECs. As a large body of literature implicates Rho kinase, PKN, and mDia proteins as major Rho effectors in cell transformation–related phenotypes imparted by active Rho proteins, we wished to examine if RhoA induced hMEC immortalization through these effectors. We used the RhoA-T37A mutant for this purpose as it has been shown in the literature to be incapable of interacting with Rho kinase, PKN, and mDia effectors (23). We first confirmed the reported inability of T37A mutant to interact with specific effector using the well-established pull-down assay using GTP-loaded recombinant GST fusions of Rho proteins (see Materials and Methods). We confirmed that WT and G14V could clearly pull down the Rho kinases ROCK1 and ROCK2 (Fig. 2A and B) as well as PKN (Fig. 2C) and mDia (Fig. 2D); in contrast, T37A failed to pull down these effectors under identical conditions. As expected, the T19N protein, used as a negative control, did not interact with any of the effectors tested (Fig. 2). Next, we used retroviral infection to introduce the T37A protein into hMECs and examined its ability to induce their immortalization. Surprisingly, similar to cells expressing the WT or G14V, cells expressing the T37A mutant continued to grow without any signs of senescence (Fig. 1B). These cells have been cultured for >300 PDs without showing any signs of senescence before cryopreservation. Notably, similar to cells immortalized with G14V, cells immortalized with the T37A mutant also express a substantially lower level of this mutant compared with that in the WT-immortalized cells (Fig. 1A).
Figure 2.
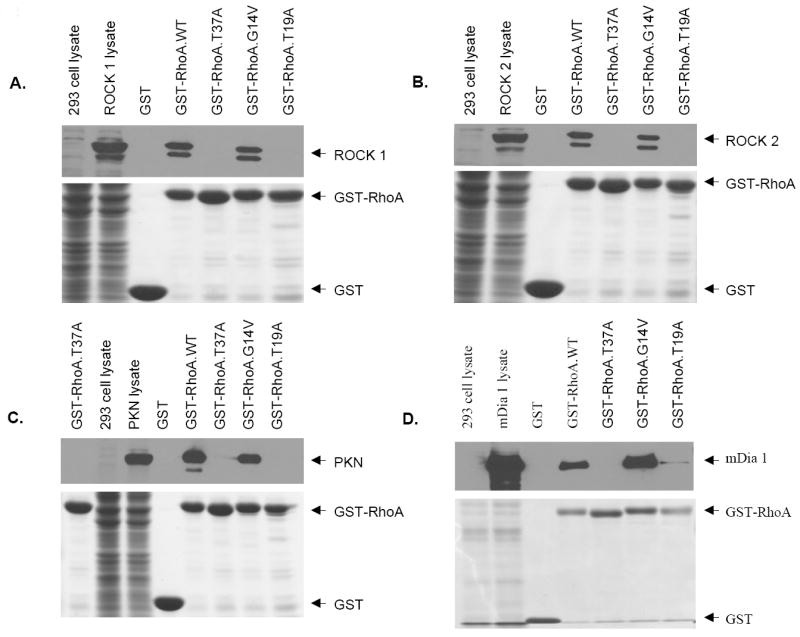
Mutant RhoA-T37A is incapable of interacting with well-known Rho effectors. Various plasmids, Prks-ROCK1, Prks-ROCK2, Prc-PKN-AL, and pFL-mDia1, were transfected into 293T cells, and cell lysates were incubated with GTP-γ-S–loaded GST, or various GST fusion proteins, and loaded into SDS-PAGE gel. After separation of proteins, the gels were cut into two parts: the upper part was transferred to PVDF membrane and probed with anti-myc and anti-flag antibodies to detect myc-tagged ROCK1 or ROCK2 and flag-tagged PKN or mDia1 and the lower part that contains GST or GST fusion proteins was stained with Coomassie Blue R-250.
Taken together, these experiments show that the ability of the ectopically overexpressed RhoA-WT, G14V, and T37A to immortalize hMECs indicates that pathways distinct from the well-known effectors of RhoA can mediate RhoA-dependent immortalization of normal hMECs.
Telomerase activity increases with RhoA-induced immortalization of hMECs
An essentially invariant feature of human cells undergoing immortalization is the induction of telomerase activity (21, 24-27). We therefore assessed the level of telomerase activity in hMECs transduced with WT, G14V, or T37A at different passages using the TRAP assay. As expected, the parental hMECs as well as the vector-transduced cells showed barely detectable levels of telomerase activity (Fig. 3A lanes 2 and 3), whereas the TERT-immortalized 76N cells (positive control) exhibit high telomerase activity (Fig. 3A, lane 1). Notably, telomerase activity increased with increasing PDs in cell lines where immortalization was eventually achieved (Fig. 3A).
Figure 3.
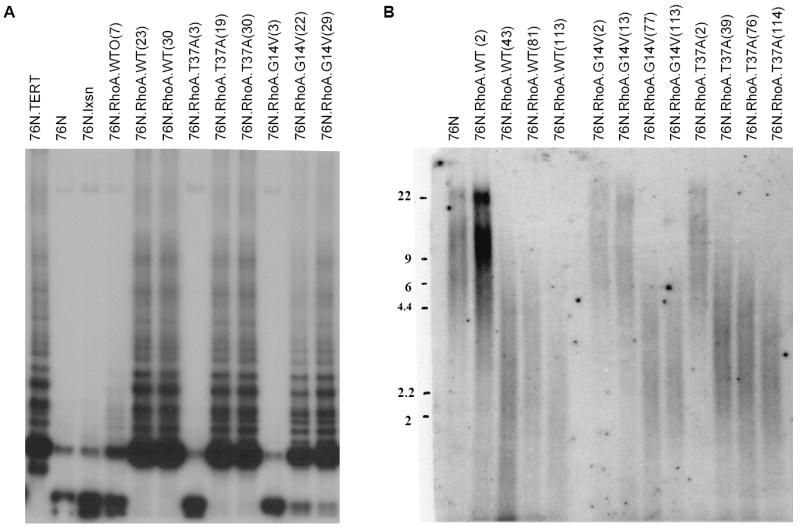
Telomerase activity is induced in immortal cells and the telomere length is maintained. A, telomerase activity at indicated passages (shown in parenthesis) was measured with extracts of 76N.TERT (positive control), 76N, 76N transduced with pLXSN vector (negative control), RhoA-WT, or various Rho mutants. B, the telomere length was determined by digesting genomic DNA from cells. The digested DNA was hybridized with a telomeric probe as described in Materials and Methods.
Induction of telomerase activity is thought to play a key role in negating the telomere attrition associated with replicative senescence by maintaining and/or elongating the telomeres (27). To examine if the induction of telomerase activity during RhoA-induced immortalization contributes toward stabilization and/or elongation of telomeres, we measured telomere length in these cells using the TRF assay. Initially, hMECs transduced with the WT, G14V, or T37A RhoA proteins showed an average telomere length of 6 to 9 kb, similar to that of parental 76N cells; however, with increasing PDs, hMECs immortalized as a result of the overexpression of Rho proteins showed telomeres of ~2.5 kb (Fig. 3B). These cells have maintained the same telomere lengths in subsequent passages (data not shown). These data suggest that telomerase activity in Rho-expressing cells does not result in a net increase in telomere length but seems to maintain telomeres. Collectively, these results are consistent with the idea that ectopic overexpression of RhoA proteins induces the immortalization of hMECs via a telomerase-dependent pathway.
Rho-immortalized cells maintain an intact cell cycle checkpoint
We have previously shown that immortalization of hMECs with viral oncogenes, such as human papillomavirus (HPV) E6 or E7, or overexpression of mutant cellular genes, such as mutant p53, causes the abrogation of the DNA damage checkpoint (28-31). In contrast, we have shown that overexpression of another cellular gene, Bmi-1, led to immortalization without abrogating the DNA damage checkpoint (21). To assess the effect of Rho-induced immortalization on DNA damage cell cycle checkpoint, Rho-immortalized cells and normal parental cells as well as the HPV E6–immortalized hMECs (used as positive control) were treated with Adriamycin for 24 hours and assessed for their ability to incorporate [3H]thymidine (an indication of DNA synthesis). As expected, the parental 76N cells failed to incorporate [3H]thymidine after Adriamycin treatment, indicating an intact DNA damage–induced cell cycle arrest. In contrast, the HPV E6–immortalized MECs continued to incorporate [3H]thymidine after Adriamycin treatment, indicating an abrogation of the DNA damage cell cycle checkpoint (Fig. 4A). Importantly, hMECs immortalized by the ectopic expression of each of the RhoA proteins behaved similar to normal parental cells, showing that expression of RhoA does not affect the DNA damage cell cycle checkpoint (Fig. 4A). Consistent with [3H]thymidine incorporation, p53 levels increased dramatically after Adriamycin treatment of 76N as well as RhoA-immortalized cells but not in E6-immortalized cells (Fig. 4B), indicating that p53 expression and function are intact in RhoA-immortalized cells.
Figure 4.
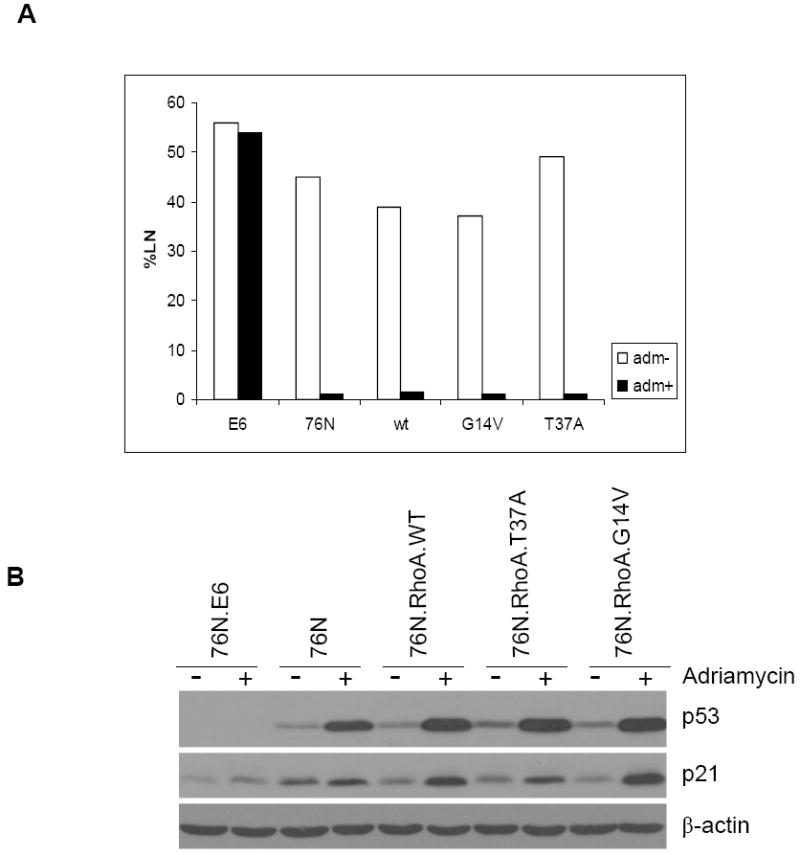
Rho-immortalized cells express normal p53 and maintain intact cell cycle checkpoint. A, 76N (used as positive control), 76N-E6 (used as negative control), and RhoA-immortalized cells were assessed for their ability to synthesize DNA [% labeled nuclei (%LN)] using [3H]thymidine incorporation after Adriamycin treatment. B, immunoblotting of cell lysates with antibodies against p53, p21, or β-actin (as control) after treatment with Adriamycin.
RhoA-induced immortalization is a generalized phenomenon in hMECs
Considering that RhoA expression in one hMEC strain, 76N, reproducibly induced their immortalization, we wished to assess if this is a generalized phenomenon in hMECs. For this purpose, we retrovirally infected an independent hMEC strain 70N with RhoA constructs, as above. Similar to the results obtained with 76N cells, 70N cells expressing RhoA-WT, G14V, or T37A, but not the vector- or T19N-transduced cells, exhibited immortalization (Fig. 5A). We repeated these experiments twice and obtained immortal cells in both cases. 70N cells immortalized with Rho are in continuous passage for >200 PDs with no signs of senescence. Similar to 76N cells, these cells show an intact DNA damage–induced p53 induction response (Fig. 5B).
Figure 5.
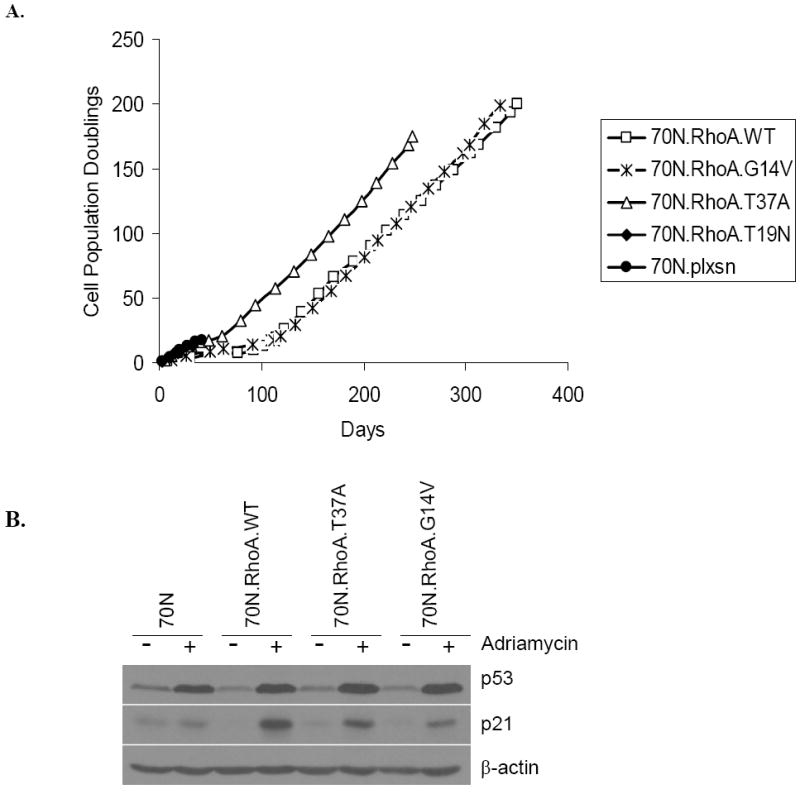
RhoA-induced immortalization is a generalized phenomenon. A, cumulative PDs of 70N cells infected with vector, WT RhoA, constitutively active RhoA (G14V), mutation in effector binding region (T37A), and dominant-negative RhoA (T19N). B, immunoblotting of cell lysates with antibodies against p53, p21, or β-actin (as control) after treatment with Adriamycin.
RhoA-immortalized cells are anchorage dependent and are unable to form tumors in nude mice
To assess if the immortalization of hMECs initiated by RhoA protein overexpression represents a preneoplastic transformation or a more advanced stage of oncogenic transformation as would be suggested by prior studies of Rho protein overexpression in model cell system (13), we examined their ability to grow in soft agar. Although human tumor cell lines do exhibit anchorage independence for growth, most immortal cells do not exhibit anchorage independence (20). Similar to parental cells, Rho-immortalized cells failed to form colonies in soft agar, whereas Hs578T, a metastatic breast cancer cell line used as a positive control, formed large soft agar colonies (Supplementary Fig. S1). Thus, Rho expression does not confer anchorage independence in hMECs.
To determine whether anchorage-dependent growth of Rho-immortalized cells reflected their incomplete neoplastic transformation, we examined their ability to grow as xenogeneic transplants in nude mice, a trait that correlates well with advanced malignant behavior of human breast cells. For this purpose, we injected 2 × 106 cells mixed with Matrigel into the mammary gland area of nude mouse, as Matrigel has been reported to enhance the tumorigenic potential of human cells (32). As expected, five of five mice injected with MDA-MB-231 cells, a breast tumor cell line known to form tumors in nude mice and used as positive control, formed large tumors. In contrast, none of the RhoA-immortalized cells exhibited any tumor growth (Supplementary Table S1) even when maintained for up to 6 months before euthanasia. Taken together, these experiments clearly show that ectopic overexpression of RhoA induces preneoplastic transformation/immortalization but not full transformation.
Microarray analyses
In view of our results that not only the WT and constitutively active RhoA but also a mutant (T37A) that failed to interact with major oncogenic transformation–relevant effectors could induce the immortalization of primary hMECs, we carried out gene expression profiling analyses to identify the potential pathways that could contribute to RhoA-induced immortalization. Therefore, we compared the gene expression profiles of normal hMECs with those of cells immortalized using RhoA-WT, G14V, or T37A using the Affymetrix Human Genome U133 Plus 2.0 chips with >47,000 transcripts for microarray analysis. The microarray data showed that the expression of ~30 genes was increased, whereas that of a set of ~100 genes was reduced in cells immortalized with RhoA proteins (NCBI Gene Expression Omnibus accession number, GSE 12917; Supplementary Table S2). Based on published links of the candidate genes to cell transformation, we selected a subset of genes, ZNF217, ELF3, S100P, CLCA2, and DAB2, and confirmed altered expression in immortalized cells using RT-PCR, Western blotting, and real-time PCR. Our results show that ZNF217, ELF3, and S100P are overexpressed (Fig. 6A; Supplementary Fig. S2), whereas CLCA2 and DAB2 are down-regulated in RhoA-immortalized hMECs (Fig. 6A and B; Supplementary Fig. S2). Importantly, the altered expression levels of these genes were also observed in several breast cancer cell lines (Fig. 6C and D), implying that these genes may in fact be relevant to Rho-induced immortalization of hMECs and that these genes may be linked to oncogenic transformation in breast cancer.
Figure 6.
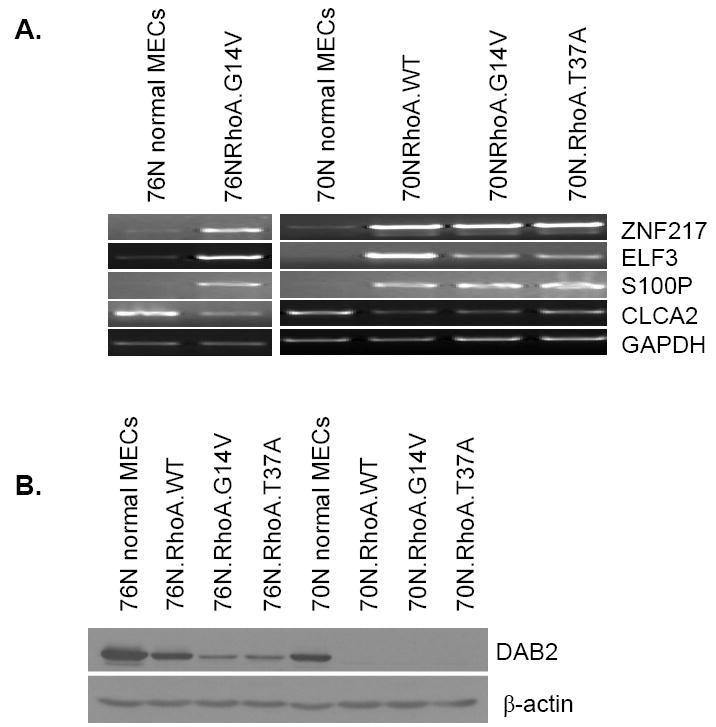
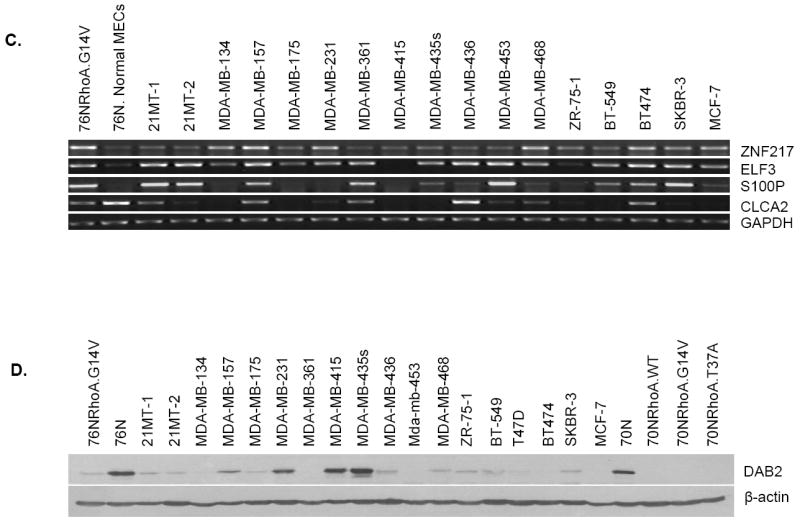
Microarray comparison of Rho-immortalized cells with parental cells identified several differentially expressed genes. Confirmation by RT-PCR and Western blotting. A, RT-PCR analyses showed that ZNF-217, ELF3, and S100P mRNAs were overexpressed in RhoA-immortalized cells, whereas CLCA2 mRNA expression was lower in RhoA-immortalized cells compared with parental 76N or 70N cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a PCR control. B, Western blotting of indicated cell lysates showed that DAB2 protein is decreased in RhoA-immortalized cells compared with parental 76N or 70N cells. β-Actin was used as a loading control. C, RT-PCR analysis of ZNF217, ELF3, S100P, and CLCA2 mRNA expression in breast cancer cell lines. Similar to RhoA-immortalized cells, several breast cancer cell lines showed increased mRNA expression for ZNF217, ELF3, and S100P and decreased mRNA expression for CLCA2 compared with normal 76N cells. D, Western blotting of cell lysates from breast cancer cell lines showed that, similar to Rho-immortalized cells, several breast cancer cell lines express lower levels of DAB2 protein compared with normal 76N and 70N cells.
Discussion
A large number of studies have implicated the crucial role of Rho family GTPases in several cell biological processes linked to oncogenesis: they regulate cell migration through actin cytoskeleton reorganization, participate in transcriptional regulation, and are linked to cell cycle control. Consistent with these functions, Rho proteins have been linked to human cancer (15-18). Rho proteins have been implicated in breast tumor progression: for example, elevated RhoA expression is seen in breast tumors compared with adjacent normal breast tissue, and migration and invasion properties of breast cancer cells were blocked by inhibiting Rho activity (17, 18). In addition, RhoC has been linked to inflammatory breast cancer and overexpression of RhoC in immortalized hMECs induces their transformation (33). Importantly, given the linkage of Rho proteins to integrin receptor signaling and cell migration, essentially all of the previous studies have examined the role of Rho proteins in the context of late events in tumor progression, often with metastatic and invasive behaviors (15-18, 34, 35). In contrast, there have been no studies to date to assess the potential role of Rho proteins in very early events in oncogenic transformation of hMECs.
Here, we have carried out studies to examine the ability of RhoA protein to overcome senescence in normal hMECs. We show using two independent hMEC strains that RhoA overexpression led to their escape from senescence and continuous proliferation. Notably, not only the constitutively active RhoA but also the WT protein overexpression induced the immortalization of normal hMECs. An active Rho GTPase that was needed for immortalization was shown by the inability of a GDP-locked Rho protein to immortalize hMECs. The ability of WT RhoA to immortalize hMECs is significant because activating RhoA mutations are not reported in human cancers but overexpression of WT Rho is a frequent phenomenon in human cancers, including breast cancers. Thus, our results are consistent with the clinical data showing increased RhoA expression with breast tumor progression (17, 18).
Consistent with other models of mammary epithelial cell immortalization, RhoA-immortalized cells exhibit increased telomerase activity and stabilization of telomeres as they overcome the senescence checkpoint. However, we observed increase in telomerase activity in RhoA-immortalized cells after several passages of overexpression of RhoA, suggesting that it may not be a direct effect of RhoA overexpression. Thus, it is difficult to ascertain that increase in telomerase activity is a cause or effect of immortalization.
Unlike other models of deliberate hMEC immortalization, such as the expression of HPV E6 or SV40 large T (19, 20, 28, 31, 36), RhoA-immortalized cells maintained a functional p53 protein and an intact DNA damage cell cycle checkpoint. Thus, in contrast to observations made by us and others that abrogation of p53 function is a crucial event in hMEC immortalization by viral oncogenes, γ-radiation, RhoA-induced immortalization seems to proceed without a requirement to abrogate p53 function. Thus, active RhoA-dependent signals either by themselves or in conjunction with other events that occur in hMECs in culture seem to be sufficient to induce the immortalization of hMECs, without abrogating p53 function. In this regard, it will be of significant interest in the future to explore the role of p16 hypermethylation and loss of expression, and the ensuing loss of Rb function, which characterizes hMEC cultures during their initial selection process in vitro (37) cooperation with Rho to induce immortalization.
In addition to preservation of the p53-dependent G1 cell cycle checkpoint, RhoA-immortalized cells exhibit an inability to grow in an anchorage-independent manner and do not form tumors when implanted in immune-incompetent mice, suggesting that overexpression of RhoA induces a state of preneoplastic transformation of hMECs rather than full transformation. In this regard, the RhoA overexpression model of hMEC immortalization resembles other models that we and others have investigated using viral oncogenes, mutant cellular genes, radiation, or carcinogen treatment; all of these manipulations induce immortalization but not full transformation (19-21, 28-31, 36-38). Thus, the hMEC model described here provides a relatively unique system driven by a breast cancer–relevant cellular gene overexpression with a functional p53 and preneoplastic transformation for biological studies to understand the further genetic alterations that can collaborate with Rho signaling pathways to induce the full transformation of hMECs.
Several downstream effectors have been linked to Rho GTPase functions in normal cells as well as their oncogenic activity measured in rodent fibroblasts. Our initial analyses suggest that the mechanisms by which RhoA overexpression induces the early neoplastic transformation of hMECs are likely to be distinct from traditionally explored pathways. In our studies, we made the unexpected observation that an effector domain mutant of RhoA, T37A, retained the ability to immortalize hMECs. As previously suggested, we found that RhoA-T37A is unable to bind to key effectors of RhoA, Rho kinase (ROCK1 and ROCK2), PKN, and mDia1, which have been linked to RhoA-dependent oncogenic transformation (15-18, 34). These results suggest that hMEC immortalization is unlikely to be through the activation of the well-characterized Rho effectors previously linked to oncogenic transformation.
Our microarray data provide an initial hint about the pathways that might be relevant to RhoA-induced immortalization of hMECs. Our analyses showed ~30 genes whose expression was up-regulated and~ 100 genes whose expression was down-regulated in Rho-immortalized (as well as RhoA-T37A immortalized) cells compared with the normal parental cells (Supplementary Table S2). In our initial work, we used RT-PCR, real-time PCR, and Western blotting to confirm our microarray-based expression changes for a subset of five genes as these are altered in breast cancers. These studies confirmed that RhoA-immortalized cells have a reduced expression of CLCA2 and DAB2, whereas ELF3, S100P, and ZNF217 mRNA expression was up-regulated (Fig. 6A and B; Supplementary Fig. S2). Importantly, several breast cancer cell lines showed that the expression of these genes was altered in the same direction as in RhoA-immortalized hMECs (Fig. 6C and D), consistent with their potential involvement in breast cell transformation.
Prior studies have shown that ELF3/ESE1, an ETS family transcription factor, is up-regulated in a subset of breast tumors as well as during tumorigenic progression of MCF-12A hMEC line (39, 40). Similarly, several studies have implicated S100P in cellular immortalization (26, 41) and overexpression of S100P contributes to tumorigenesis as it promotes tumor growth, invasion, and cell survival (42). ZNF217 is frequently amplified in breast cancer (43), and its overexpression has been shown to induce mammary epithelial cell immortalization (38). CLCA2 (chloride channel, calcium activated, family member 2) is reportedly lost during tumor progression in human breast cancer; CLCA2 was found to be expressed in normal breast epithelium but not in breast cancer (44). Another study showed that expression of CLCA2 in CLCA2-negative MDA-MB-231 and MDA-MB-435 cells reduced the Matrigel invasion in vitro and metastatic tumor formation of MDA-MB-231 cells in nude mice (45). DAB2 (disabled 2) or DOC-2 (differentially expressed in ovarian carcinoma 2), originally isolated as a potential tumor suppressor gene from human ovarian carcinoma, is involved in modulating multiple signaling pathways and protein trafficking (46). Decreased expression of DOC-2/DAB2 has been observed in several cancers, including prostate, mammary, colon, and choriocarcinoma (46, 47). DOC-2/hDab-2 expression in breast cancer cells resulted in sensitivity to suspension-induced cell death (anoikis; ref. 48). Significantly, our analyses of Oncomine database7 showed that S100P overexpression in breast cancers is correlated with high tumor grade in two breast cancer data sets, and its expression is higher in invasive breast cancers compared with breast ductal carcinoma in situ (Supplementary Fig. S3). Similarly, DAB2 expression is down-regulated in breast cancers in one data set and its down-regulation is correlated with lymphocytic infiltration and tumor grade in another two data sets (Supplementary Fig. S4). Thus, future studies to perturb the expression of these candidate genes in RhoA-immortalized hMEC system as well as analyses of how their expression is controlled by Rho-dependent signaling pathways should add significantly to our understanding of early oncogenic transformation of hMECs with direct relevance to human breast cancer.
In conclusion, the present study shows that RhoA, implicated in breast cancer oncogenesis by clinical studies and well known as a critical gatekeeper of receptor signals into multiple cell biological pathways, can induce the immortalization of hMECs. Notably, mammary epithelial cell immortalization by an effector domain mutant of RhoA that is incapable of interacting with well-characterized Rho effectors previously implicated in oncogenic transformation strongly suggests that RhoA-induced early transformation of hMECs proceeds to novel pathways. The system described here should prove suitable for future analyses to uncover the nature of these pathways and to link them to oncogenic pathways in breast cancer.
Supplementary Material
Acknowledgments
Grant support: Department of Defense grants W81XWH-05-1-0231 and W81XWH-07-1-0351 and NIH grants CA94143 and CA96844 (V. Band) and NIH grants CA87986, CA105489, CA99900, CA116552, and CA99163 (H. Band). V. Band acknowledges the support of the Duckworth family through the Duckworth Family Chair for Breast Cancer Research. H. Band acknowledges the support from the Jean Ruggles-Romoser Chair for Cancer Research.
Footnotes
The work presented here was initiated while the principal investigators (LL, NP and VB) were at the New England Medical Center, Tufts University, Boston, MA and subsequently at Evanston Northwestern Healthcare Research Institute, Department of Medicine, Feinberg School of Medicine, Northwestern University, Evanston, IL.
References
- 1.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Pt 2):241–55. [PMC free article] [PubMed] [Google Scholar]
- 2.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 3.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz M. Rho signalling at a glance. J Cell Sci. 2004;117:5457–8. doi: 10.1242/jcs.01582. [DOI] [PubMed] [Google Scholar]
- 5.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 6.Peck JW, Oberst M, Bouker KB, Bowden E, Burbelo PD. The RhoA-binding protein, Rhophilin-2, regulates actin cytoskeleton organization. J Biol Chem. 2002;277:43924–32. doi: 10.1074/jbc.M203569200. [DOI] [PubMed] [Google Scholar]
- 7.Madaule P, Eda M, Watanabe N, et al. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature. 1998;394:491–4. doi: 10.1038/28873. [DOI] [PubMed] [Google Scholar]
- 8.Piekny A, Werner M, Glotzer M. Cytokinesis: welcome to the Rho zone. Trends Cell Biol. 2005;15:651–8. doi: 10.1016/j.tcb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Lim MA, Yang L, Zheng Y, Wu H, Dong LQ, Liu F. Roles of PDK-1 and PKN in regulating cell migration and cortical actin formation of PTEN-knockout cells. Oncogene. 2004;23:9348–58. doi: 10.1038/sj.onc.1208147. [DOI] [PubMed] [Google Scholar]
- 10.Su C, Deaton RA, Iglewsky MA, Valencia TG, Grant SR. PKN activation via transforming growth factor-β1 (TGF-β1) receptor signaling delays G2/M phase transition in vascular smooth muscle cells. Cell Cycle. 2007;6:739–49. doi: 10.4161/cc.6.6.3985. [DOI] [PubMed] [Google Scholar]
- 11.Fukata M, Nakagawa M, Kaibuchi K. Roles of Rho-family GTPases in cell polarization and directional migration. Curr Opin Cell Biol. 2003;15:590–7. doi: 10.1016/s0955-0674(03)00097-8. [DOI] [PubMed] [Google Scholar]
- 12.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Debidda M, Wang L, Zang H, Poli V, Zheng Y. A role of STAT3 in Rho GTPase-regulated cell migration and proliferation. J Biol Chem. 2005;280:17275–85. doi: 10.1074/jbc.M413187200. [DOI] [PubMed] [Google Scholar]
- 14.Welsh CF. Rho GTPases as key transducers of proliferative signals in G1 cell cycle regulation. Breast Cancer Res Treat. 2004;84:33–42. doi: 10.1023/B:BREA.0000018425.31633.07. [DOI] [PubMed] [Google Scholar]
- 15.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 16.Ridley AJ. Rho proteins and cancer. Breast Cancer Res Treat. 2004;84:13–9. doi: 10.1023/B:BREA.0000018423.47497.c6. [DOI] [PubMed] [Google Scholar]
- 17.Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer. 2002;87:635–44. doi: 10.1038/sj.bjc.6600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burbelo P, Wellstein A, Pestell RG. Altered Rho GTPase signaling pathways in breast cancer cells. Breast Cancer Res Treat. 2004;84:43–8. doi: 10.1023/B:BREA.0000018422.02237.f9. [DOI] [PubMed] [Google Scholar]
- 19.Band V, Zajchowski D, Kulesa V, Sager R. Human papilloma virus DNAs immortalize normal human mammary epithelial cells and reduce their growth factor requirements. Proc Natl Acad Sci U S A. 1990;87:463–7. doi: 10.1073/pnas.87.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Band V. In vitro models of early neoplastic transformation of human mammary epithelial cells. Methods Mol Biol. 2003;223:237–48. doi: 10.1385/1-59259-329-1:237. [DOI] [PubMed] [Google Scholar]
- 21.Dimri GP, Martinez JL, Jacobs JJ, et al. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002;62:4736–45. [PubMed] [Google Scholar]
- 22.Del Re DP, Miyamoto S, Brown JH. RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J Biol Chem. 2007;282:8069–78. doi: 10.1074/jbc.M604298200. [DOI] [PubMed] [Google Scholar]
- 23.Su W, Chardin P, Yamazaki M, Kanaho Y, Du G. RhoA-mediated phospholipase D1 signaling is not required for the formation of stress fibers and focal adhesions. Cell Signal. 2006;18:469–78. doi: 10.1016/j.cellsig.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 24.Stampfer MR, Yaswen P. Culture models of human mammary epithelial cell transformation. J Mammary Gland Biol Neoplasia. 2000;5:365–78. doi: 10.1023/a:1009525827514. [DOI] [PubMed] [Google Scholar]
- 25.Yaswen P, Stampfer MR. Epigenetic changes accompanying human mammary epithelial cell immortalization. J Mammary Gland Biol Neoplasia. 2001;6:223–34. doi: 10.1023/a:1011364925259. [DOI] [PubMed] [Google Scholar]
- 26.Gudjonsson T, Villadsen R, Ronnov-Jessen L, Petersen OW. Immortalization protocols used in cell culture models of human breast morphogenesis. Cell Mol Life Sci. 2004;61:2523–34. doi: 10.1007/s00018-004-4167-z. [DOI] [PubMed] [Google Scholar]
- 27.Counter CM, Hahn WC, Wei W, et al. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci U S A. 1998;95:14723–8. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wazer DE, Liu XL, Chu Q, Gao Q, Band V. Immortalization of distinct human mammary epithelial cell types by human papilloma virus 16 E6 or E7. Proc Natl Acad Sci U S A. 1995;92:3687–91. doi: 10.1073/pnas.92.9.3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao Q, Hauser SH, Liu XL, Wazer DE, Madoc-Jones H, Band V. Mutant p53-induced immortalization of primary human mammary epithelial cells. Cancer Res. 1996;56:3129–33. [PubMed] [Google Scholar]
- 30.Cao Y, Gao Q, Wazer DE, Band V. Abrogation of wild-type p53-mediated transactivation is insufficient for mutant p53-induced immortalization of normal human mammary epithelial cells. Cancer Res. 1997;57:5584–9. [PubMed] [Google Scholar]
- 31.Liu Y, Chen JJ, Gao Q, et al. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J Virol. 1999;73:7297–307. doi: 10.1128/jvi.73.9.7297-7307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullen P, Ritchie A, Langdon SP, Miller WR. Effect of Matrigel on the tumorigenicity of human breast and ovarian carcinoma cell lines. Int J Cancer. 1996;67:816–20. doi: 10.1002/(SICI)1097-0215(19960917)67:6<816::AID-IJC10>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 33.Kleer CG, Zhang Y, Pan Q, et al. WISP3 and RhoC guanosine triphosphatase cooperate in the development of inflammatory breast cancer. Breast Cancer Res. 2004;6:R110–5. doi: 10.1186/bcr755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamai T, Tsujii T, Arai K, et al. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res. 2003;9:2632–41. [PubMed] [Google Scholar]
- 35.Kamai T, Kawakami S, Koga F, et al. RhoA is associated with invasion and lymph node metastasis in upper urinary tract cancer. BJU Int. 2003;91:234–8. doi: 10.1046/j.1464-410x.2003.03063.x. [DOI] [PubMed] [Google Scholar]
- 36.Toouli CD, Huschtscha LI, Neumann AA, et al. Comparison of human mammary epithelial cells immortalized by simian virus 40 T-antigen or by the telomerase catalytic subunit. Oncogene. 2002;21:128–39. doi: 10.1038/sj.onc.1205014. [DOI] [PubMed] [Google Scholar]
- 37.Wong DJ, Foster SA, Galloway DA, Reid BJ. Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest. Mol Cell Biol. 1999;19:5642–51. doi: 10.1128/mcb.19.8.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nonet GH, Stampfer MR, Chin K, Gray JW, Collins CC, Yaswen P. The ZNF217 gene amplified in breast cancers promotes immortalization of human mammary epithelial cells. Cancer Res. 2001;61:1250–4. [PubMed] [Google Scholar]
- 39.Chang CH, Scott GK, Kuo WL, et al. ESX: a structurally unique Ets overexpressed early during human breast tumorigenesis. Oncogene. 1997;14:1617–22. doi: 10.1038/sj.onc.1200978. [DOI] [PubMed] [Google Scholar]
- 40.Prescott JD, Koto KS, Singh M, Gutierrez-Hartmann A. The ETS transcription factor ESE-1 transforms MCF-12A human mammary epithelial cells via a novel cytoplasmic mechanism. Mol Cell Biol. 2004;24:5548–64. doi: 10.1128/MCB.24.12.5548-5564.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guerreiro Da Silva ID, Hu YF, Russo IH, et al. S100P calcium-binding protein overexpression is associated with immortalization of human breast epithelial cells in vitro and early stages of breast cancer development in vivo. Int J Oncol. 2000;16:231–40. [PubMed] [Google Scholar]
- 42.Wang G, Platt-Higgins A, Carroll J, et al. Induction of metastasis by S100P in a rat mammary model and its association with poor survival of breast cancer patients. Cancer Res. 2006;66:1199–207. doi: 10.1158/0008-5472.CAN-05-2605. [DOI] [PubMed] [Google Scholar]
- 43.Collins C, Rommens JM, Kowbel D, et al. Positional cloning of ZNF217 and NABC1: genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc Natl Acad Sci U S A. 1998;95:8703–8. doi: 10.1073/pnas.95.15.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Cowell JK, Sossey-Alaoui K. CLCA2 tumour suppressor gene in 1p31 is epigenetically regulated in breast cancer. Oncogene. 2004;23:1474–80. doi: 10.1038/sj.onc.1207249. [DOI] [PubMed] [Google Scholar]
- 45.Gruber AD, Pauli BU. Tumorigenicity of human breast cancer is associated with loss of the Ca2+-activated chloride channel CLCA2. Cancer Res. 1999;59:5488–91. [PubMed] [Google Scholar]
- 46.Sheng Z, Sun W, Simth E, Cohen C, Sheng Z, Xu XX. Restoration of positioning control following disabled-2 expression in ovarian and breast tumor cells. Oncogene. 2000;10:4847–54. doi: 10.1038/sj.onc.1203853. [DOI] [PubMed] [Google Scholar]
- 47.Bagadi SA, Prasad CP, Srivastava A, Prashad R, Gupta SD, Ralhan R. Frequent loss of Dab2 protein and infrequent promoter hypermethylation in breast cancer. Breast Cancer Res Treat. 2007;104:277–86. doi: 10.1007/s10549-006-9422-6. [DOI] [PubMed] [Google Scholar]
- 48.Wang SC, Makino K, Xia W, et al. DOC-2/hDab-2 inhibits ILK activity and induces anoikis in breast cancer cells through an Akt-independent pathway. Oncogene. 2001;20:6960–4. doi: 10.1038/sj.onc.1204873. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.