

Abstract
A role for cystatin C (CysC) in the pathogenesis of Alzheimer’s disease (AD) has been suggested by the genetic linkage of a CysC gene (CST3) polymorphism with late-onset AD, the co-localization of CysC with amyloid-β (Aβ) in AD brains, and binding of CysC to soluble Aβ in vitro and in mouse models of AD. This study investigates the binding between Aβ and CysC in the human central nervous system. While CysC binding to soluble Aβ was observed in AD patients and controls, a SDS-resistant CysC/Aβ complex was detected exclusively in brains of neuropathologically normal controls, but not in AD cases. The association of CysC with Aβ in brain from control individuals and in cerebrospinal fluid reveals an interaction of these two polypeptides in their soluble form. The association between Aβ and CysC prevented Aβ accumulation and fibrillogenesis in experimental systems, arguing that CysC plays a protective role in the pathogenesis of AD in humans and explains why decreases in CysC concentration caused by the CST3 polymorphism or by specific presenilin 2 mutations can lead to the development of the disease. Thus, enhancing CysC expression or modulating CysC binding to Aβ have important disease-modifying effects, suggesting a novel therapeutic intervention for AD.
Keywords: Alzheimer’s disease, amyloid-β, amyloid-β protein precursor, cystatin C
INTRODUCTION
Senile plaques, neurofibrillary tangles, and neuronal loss are neuropathological hallmarks of Alzheimer’s disease (AD). Senile plaques comprise an extracellular core of aggregated, fibrillar amyloid-β (Aβ) accompanied by microglial cells, astrocytes, and dystrophic neuronal processes. Although Aβ is the major constituent of amyloid deposits in brain parenchyma and in vascular walls, minor components were identified, such as P-component [1], apolipoprotein E (ApoE) [2], apolipoprotein J [3,4], proteoglycans [5], lysosomal proteinases [6-8], and the proteinases inhibitors, α1-antichymotrypsin, α1-antitrypsin [9], and α2-macroglobulin [10]. Cystatin C (CysC), a cysteine protease inhibitor that is present in all tissues and body fluids examined [11], has also been reported to co-localize with Aβ in parenchymal and vascular amyloid deposits [12-15].
Multiple studies have shown the genetic linkage of the CysC gene (CST3) with late-onset AD (for review [16]). A synergistic risk association between the CST3 polymorphism and APOE ε4 alleles has been reported [17,18]. Furthermore, in vitro studies have shown that CysC binds to Aβ (Aβ1–40 and Aβ1–42) and inhibits fibril formation and oligomerization of Aβ in a concentration-dependent manner [19,20]. Recent observations further confirmed such inhibitory effects in vivo in Aβ-depositing transgenic amyloid-β protein precursor (AβPP) mice overexpressing human CysC. A reduction in Aβ load was observed in the AβPP/CysC double transgenic mice compared to single AβPP transgenic mice [21,22]. Moreover, analysis of human cerebrospinal fluid (CSF) proteins by protein-chip array technology revealed that the combination of five polypeptides, including CysC, two β-2 microglobulin isoforms, a 4.8 kDa VGF polypeptide, and an unknown 7.7 kDa polypeptide could be used for the diagnosis of AD and perhaps the assessment of its severity and progression [23]. Elevated CysC levels were reported in plasma of AD patients compared to healthy donors [24]. These data suggest that CysC, combined with other protein markers, could be a potentially useful CSF/plasma biomarker for the diagnosis of neurological disorders, including AD [23,24].
We have been studying the association of CysC with Aβ in the human central nervous system (CNS) and now report that human CysC binds to Aβ in brain homogenates of both neuropathologically normal controls and AD patients by immunoprecipitation followed by Western blot analysis. This binding is also present in CSF of both AD patients and age-matched non-demented controls, suggesting binding of CysC to a soluble form of Aβ. Strikingly, a band immunoreactive to antibodies to both CysC and Aβ is present in the SDS-extracted membranous fraction of brain homogenates and not in the soluble fraction. This band was found in brains of neuropathologically normal controls, but not in brains of AD patients ranging from early to severe stages of the disease.
MATERIAL AND METHODS
Samples
Postmortem brain tissues from 9 elderly individuals, ranging in age from 50 to 94 years, were examined and diagnosed with neuropathological evidence of various stages of AD according to the guidelines of CERAD [25,26]. We also studied 10 control cases, which were evaluated using the same criteria and found to be normal by neuropathological inspection. Frozen AD and control tissues were obtained from the Harvard Brain Tissue Resource Center at McLean Hospital (Belmont, MA) and Mount Sinai Medical Center (New York, NY). Premortem CSF samples (2 defined AD and 5 age-matched non-demented controls) were obtained via lumbar puncture at “Carlo Besta”, National Neurological Institute, Milan, Italy (Table 1).
Table 1.
Human brain and CSF samples
| Brain samples for immunoprecipitation | |||
|---|---|---|---|
| Samples | Age | Gender | Source2 |
| AD (n = 3)1 | 86–92 | female (2), male (1) | A, B |
| Control (n = 3) | 53–79 | female (3) | B |
| Brain samples for Western blot analysis | ||||
|---|---|---|---|---|
| Samples | CDR | Age | Gender | Source |
| Early AD (n = 4) | (CDR = 0–0.5) | 50–79 | female (1), male (3) | A, B |
| Moderate AD (n = 3) | (CDR = 1–2) | 80–94 | female (2), male (1) | B |
| Severe AD (n = 2) | (CDR = 5) | 80–94 | female (2) | B |
| Control (n = 2) | (CDR = 0) | 20–30 | male (2) | A, B |
| Control (n = 8) | (CDR = 0) | 50–79 | female (3), male (5) | A, B |
| CSF samples for immunoprecipitation | |||
|---|---|---|---|
| Samples | Age | Gender | Source |
| AD (n = 2) | 47-64 | female (1), male (1) | C |
| Control (n = 5) | 41-69 | female (3), male (2) | C |
Value within parentheses represents number of samples.
Frozen brain tissues were obtained from: (A) the Harvard Brain Tissue Resource Center at McLean Hospital (Belmont, MA), and (B) Mount Sinai Medical Center (New York, NY). (C) premortem CSF samples were obtained from “Carlo Besta”, National Neurological Institute, Milan, Italy.
Brain homogenization
Frozen brain tissue was homogenized in 1:10 weight:volume ratio of ice-cold tissue homogenization buffer (THB) (250 mM sucrose, 20 mM Tris pH 7.4, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 11 μM leupeptin, 7 μM antipain HCl, and 7 μM pepstatin A; all Sigma) [27]. Protein concentration was determined by using Pierce Protein Assay Kit based on the method of Bradford (Pierce, USA).
SDS-extraction of brain homogenates
Brain homogenates in THB buffer were centrifuged at 10,000 × g, 4°C, for 1 hour, providing supernatant (Sup) Sup 1. The resulting pellets were further homogenized in 2% SDS and centrifuged at 10,000 × g, 4°C, for 1 hour, providing Sup 2. The pellet was dissolved in SDS-sample buffer (Invitrogen, LDS buffer), providing Sup 3. The resulting supernatants were used for either Western blotting or immunoprecipitation (see below).
Immunoprecipitation
Monoclonal anti-Aβ antibody 6E10 (Signet) or anti-CysC antibody (Cyst24; Hytest, Finland) coated beads were prepared according to manufacturer’s instructions (Dianyl Biotech, Invitrogen). In brief, 10 μl of suspended rat anti-mouse IgG1 Dynabeads were used per sample. Beads were washed with PBS/0.1% BSA, resuspended with 1.5 μg of anti-Aβ antibody 6E10, and incubated overnight at 4°C with rotation. Coated beads were washed, resuspended with 250 μg brain homogenates in PBS and incubated at 4°C for 2 hours. Washed beads were suspended in sample buffer and the resulting pull-down proteins were applied to 4–12% Bis-Tris gel (Invitrogen) and electro-transferred to nitrocellulose membranes. For immunoprecipitation with anti-CysC antibody (Hytest, Finland), 30 μl of suspended rat anti-mouse IgG1 Dynabeads, 4.5 μg of anti-CysC antibody, and 1 mg brain homogenates were used. A similar immunoprecipitation protocol was carried out in SDS-soluble fraction of brain homogenates, using anti-CysC antibody (R&D Systems, MAB1196) and sheep anti-mouse IgG Dynabeads.
Western blot analysis
Transfer nitrocellulose membranes were incubated with 5% nonfat dry milk TBS buffer for 1 hour at room temperature,followed by incubation with primary antibody overnight at 4°C and secondary antibody for 1 hour. Blots were developed with ECL Super Signal (Pierce). Primary antibodies used are polyclonal anti-CysC antibody (Upstate cell signaling solutions, NY) or monoclonal anti-Aβ antibody, 6E10 (Signet).
RESULTS
CysC binds to soluble Aβ in human brain and CSF
To investigate whether the association between CysC and Aβ observed in vitro [19] and in mouse models [21, 22] occurs in vivo in human CNS, homogenates of human brain tissues obtained from the cortex (Bergman area 8 or 10) of AD patients (n = 3, Table 1) and neuropathologically normal controls (n = 3, Table 1) were used. Western blot analysis with anti-CysC antibody of proteins immunoprecipitated with anti-Aβ antibody revealed binding of human CysC to Aβ in brain homogenates of both AD patients and non-demented controls (Fig. 1A). The converse protocol, in which immunoprecipitation experiments were carried out using anti-CysC antibody followed by Western blotting with anti-Aβ antibody revealed binding of human CysC to Aβ in brain homogenates of AD patients but not non-demented controls (Fig. 1B). Western blot analysis with anti-CysC antibody of proteins immunoprecipitated with anti-CysC antibody revealed precipitation of similar levels of CysC from brain homogenates of both AD patients and neuropathologically normal controls (data not shown). However, Western blot with anti-Aβ antibody of proteins immunoprecipitated with anti-Aβ antibody showed detectable Aβ only in AD brains (Fig. 1C). Demonstration of binding between the two proteins is likely to be limited by the immunoprecipitating capability of the precipitating antibody, the sensitivity of the blotting antibody, and the amount of the proteins in the sample. The concentration of Aβ in the brain of individuals without amyloid deposition is much lower than that of CysC and is not detectable by the anti-Aβ antibody. The data show binding between CysC and Aβ in brain homogenates and the binding in brain tissues that lack amyloid plaques deposition indicates that human CysC binds to a soluble form of Aβ.
Fig. 1.

CysC binds to soluble Aβ in vivo, in human brain homogenates, and in CSF. Immunoprecipitation (IP) of proteins from either human brain homogenates or CSF of AD patients or neuropathologically controls (C) with either anti-CysC antibody or anti-Aβ antibody was followed by separation by 4–12% Bis-Tris gradient gel. Immunoprecipitated proteins were Western blotted with either anti-CysC or anti-Aβ antibodies. Neither CysC nor Aβ were detected in the absence of immunoprecipitating antibody or sample (–). Molecular weights are marked on the right and the bands representing CysC and Aβ are marked on the left.
Human CSF samples of AD patients and age-matched non-demented controls (Table 1) were immunoprecipitated using anti-Aβ antibody, and Western blotted with anti-CysC antibody (Fig 1D). The binding between human CysC and Aβ in human CSF further supports the interaction of the proteins in their soluble forms. The specificity of the binding between human CysC and Aβ was demonstrated by control experiments, which included replacement of the immunoprecipitating antibody with normal mouse serum (data not shown) or omitting either primary antibody or brain homogenate (Fig. 1A, C). Neither CysC nor Aβ was observed in these control experiments. Occasionally, a small amount of Aβ bound directly to the beads, but the amount was negligible relative to the amount of protein that bound to the precipitating antibodies, revealing that neither CysC nor Aβ bind the precipitating beads or immunoprecipitate by normal mouse serum.
A SDS-stable band immunoreactive with antibodies to CysC and to Aβ is exclusively present in neuropathologically normal controls
Brain homogenates of AD patients at different stages of the disease (early, moderate, and severe), and neuropathologically normal controls were separated by 4–12% Bis-tris gel electrophoresis and Western blotted with anti-CysC antibody to reveal that all samples contained the monomeric 14 kDa CysC (Fig. 2A). Western blot with anti-Aβ antibody revealed a band of 4 kDa monomeric Aβ in all AD samples, but not controls (Fig. 2B). An additional band of approximately 20 kDa was present in all controls (n = 10, Table 1), but not in moderate or severe AD cases (n = 5, Table 1) following Western blotting with either anti-CysC (Fig. 2A) or anti-Aβ (Fig. 2B) antibodies. The difference in staining intensity of the band by the two antibodies supports the suggestion that the anti-Aβ antibody is a less sensitive blotting antibody compared with the anti-CysC antibody.
Fig. 2.
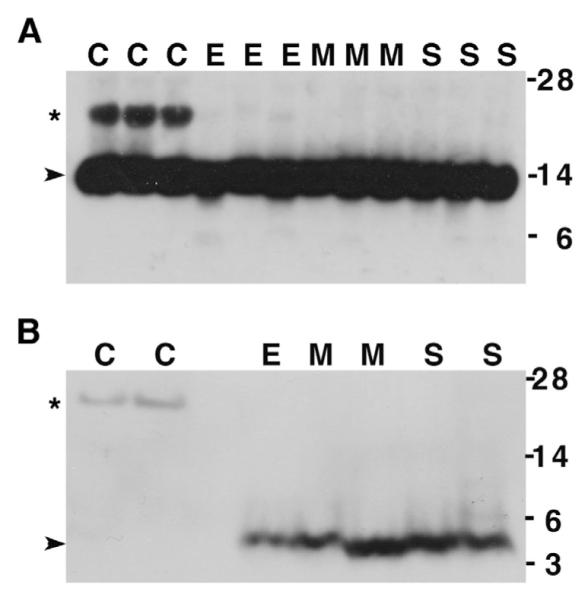
CysC/Aβ complex is present only in brain homogenates of neuropathologically normal controls. Western blot analysis with either anti-CysC antibody (A) or anti-Aβ antibody (B) of brain homogenates separated by 4–12% Bis/tris gel electrophoresis: C = control (CDR = 0); E = early (CDR = 0–0.5); M=moderate (CDR = 1–2); S = severe (CDR = 5) AD patients. The bands of monomeric CysC (14 kDa) and monomeric Aβ (4 kDa) are marked by arrowheads, and the ~20 kDa band is marked by asterisk. Molecular weights are marked on the right.
The detection of a band of the same molecular weight (~20 kDa) by both antibodies suggests that this band contains a complex of CysC with Aβ. The ~20 kDa CysC and Aβ immunoreactive species were detected in brain homogenates of non-affected individuals (controls) but not in brain homogenates of AD patients and was already absent at the earliest stage of the disease in patients with mild cognitive impairment (CDR 0.0–0.5) (Fig. 2A). However, a faint anti-CysC-reactive band of about 20 kDa was occasionally observed in early AD brain homogenates.
Sequential centrifugation of brain homogenates was used to identify the fraction that contains the ~20 kDa band. Western blot analysis with anti-CysC antibody of the soluble or insoluble fractions of brain homogenates revealed the presence of the 14 kDa monomeric CysC in all fractions. Following centrifugation of the homogenates at 10,000 × g for an hour, the supernatant (Sup 1 in Fig. 3) was removed and the pellets were solubilized in 2% SDS, and then centrifuged again to remove insoluble material (Sup 3 in Fig. 3) from the SDS-soluble material (Sup 2 in Fig. 3). The band of ~20 kDa was found only in the SDS-soluble fractions of precipitated material (Sup 2 in Fig. 3) from non-demented control individuals and not from AD patients. The size of the ~20 kDa band is distinct from the 28 kDa dimer formed by high concentration of CysC, as demonstrated for human urinary CysC (Fig. 3).
Fig. 3.

Western blot analysis with anti-CysC antibody reveals SDS-stable CysC/Aβ complex in the cellular fraction of brain homogenates of neuropathologically normal controls. Brain homogenates from neuropathologically normal controls (C) and AD patients were centrifuged (soluble fraction, Sup 1). The pellet was extracted with 2% SDS and centrifuged (Sup 2). The pellet of the SDS extraction was dissolved in SDS-sample buffer (Sup 3). Arrowheads mark CysC monomer (14 kDa) and dimer (28 kDa) observed in urinary human CysC (uCysC) and the ~20 kDa complex is marked by an asterisk. Molecular weight markers are on the right.
The presence of the SDS-stable ~20 kDa band in the SDS-soluble protein fractions (Sup 2) from brain homogenate of control individuals was confirmed by Western blot analysis with anti-CysC antibody of proteins immunoprecipitated with anti-CysC (Fig. 4). That this band represents a complex between CysC and Aβ was confirmed by Western blot analysis with anti-CysC antibody of proteins precipitated by anti-Aβ antibodies (Fig. 4). A band of approximately 20 kDa was apparent in only the SDS-soluble fraction from controls, not in that from AD patients (Fig. 4).
Fig. 4.
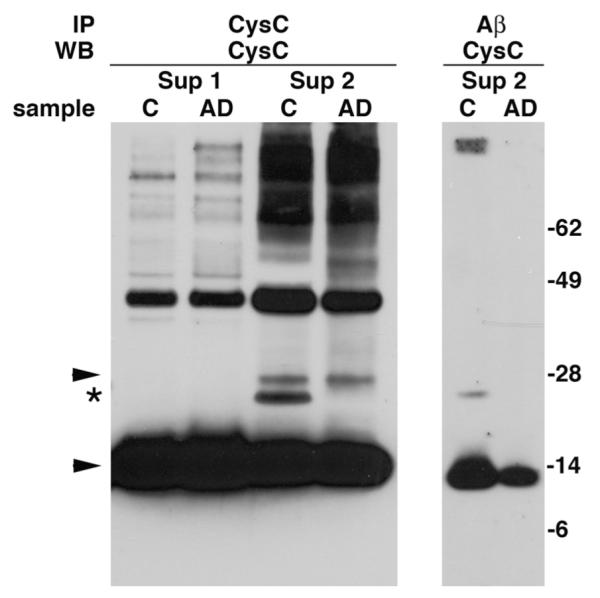
SDS-stable CysC/Aβ complex immunoprecipitated from the cellular fraction of brain homogenates of neuropathological normal controls. Soluble fractions (Sup 1) and SDS-soluble cellular fractions (Sup 2) of brain homogenates from neuropathological normal controls (C) and AD patients were immunoprecipitated with either anti-CysC or anti-Aβ antibodies and Western blotted with anti-CysC antibody. Arrowheads mark CysC monomer (14 kDa) and dimer (28 kDa) and the ~20 kDa complex is marked by an asterisk. Molecular weight markers are on the right.
DISCUSSION
Immunohistochemical studies revealed that CysC co-localizes with Aβ predominantly in amyloid-laden vascular walls and in senile plaque cores of amyloid in brains of patients with AD, Down’s syndrome, hereditary cerebral hemorrhage with amyloidosis, Dutch type, and cerebral infarction [12-15,28], and in non-demented aged individuals [15]. CysC also co-localizes with Aβ deposits in the brain of aged rhesus and squirrel monkeys [29] as well as dogs [30]. In vitro observations have demonstrated a specific, concentration dependent, high affinity binding between CysC and Aβ [19]. Based on these findings it was suggested that the presence of CysC within amyloid plaques is a residue of its association with soluble Aβ.
Co-localization of CysC with Aβ deposits was also found in brains of transgenic mice overexpressing human AβPP [15,31]. We have recently shown binding between CysC and Aβ in brains and plasma of CysC overexpressing transgenic mice [21,22]. Crossbreeding AβPP transgenic mice with CysC overexpressing transgenic mice resulted in inhibition of Aβ deposition in the brains of double transgenic mice compared to AβPP single transgenic mice [21,22]. This finding is in agreement with in vitro studies showing that CysC inhibits Aβ fibril formation [19]. Furthermore, in vitro studies have shown that CysC inhibits the formation of high molecular weight Aβ oligomeric assemblies ([20] and our unpublished data). Senile plaques are one of the typical neuropathological features in the cortex and hippocampus of AD patients and are mainly composed of Aβ peptide in its fibrillar form. Aβ fibril formation is preceded by multiple conformational changes of intermediate Aβ species including trimer, pentamer, and higher molecular weight complexes. The intermediate Aβ species were found to be neurotoxic at lower concentrations than fibrillar Aβ, and their levels in the brain correlate better with the severity of cognitive impairment than does the density of plaque deposits (for review [32]).
Here we demonstrate that human CysC binds to soluble Aβ in human brain and CSF. While the monomeric form of Aβ is absent and a stable CysC/Aβ complex is found in brains of non-demented individuals that do not have Aβ deposits, only the monomeric form of Aβ is found in AD brains. This suggests that the heightened tendency of Aβ to bind itself in AD brains competes with its binding to CysC. A similar competition between the binding of variant CysC to itself and its binding to Aβ was previously described. An L68Q substitution in CysC in patients with hereditary cerebral hemorrhage with amyloidosis, Icelandic type (HCHWA-I) [33], causes CysC amyloid deposition in the CNS vasculature, resulting in cerebral hemorrhagic strokes [34]. In vitro studies have shown that while CysC forms concentration-dependentdimers, the HCHWA-I variant dimerizes at lower concentrations than the wild type protein [35]. Western blot analysis of brain homogenates prepared from transgenic mice expressing the L68Q variant of human CysC revealed a 14 kDa monomeric CysC and a CysC 28 kDa dimer [21, 22]. Overexpression of AβPP in transgenic mice overexpressing the L68Q variant CysC resulted in lower levels of the dimeric form of CysC compared to those in mice expressing only CysC [21,22]. Consistent with the greater tendency of HCHWA-I CysC to dimerize than the wild type protein, the dimer was not observed in the brain of mice expressing the human wild type gene. The data suggest that self-aggregation of each polypeptide competes with the binding of one to the other.
Moreover, the CysC/Aβ complex was identified in centrifugation fractions that contain cellular membranes and intracellular compartments such as nuclei, large membrane fragments, and lysosomes. In vitro studies have shown that the binding of CysC to Aβ is concentration dependent [19]. High concentrations of both CysC and Aβ in intracellular vesicular compartments would support their binding. Immunohistochemical analysis of brain sections of AD patients and non-demented individuals revealed that the anti-CysC antiserum labeled neurons, astrocytes, and cells within the walls of large leptomeningeal vessels [15]. In particular, strong punctate immunoreactivity within the cytoplasm and neuronal processes was primarily limited to pyramidal neurons in cortical layers III and IV [15,36]. Using an antibody specific to the carboxyl-terminus of Aβ42, punctate intracellular immunoreactivity was observed in the same neuronal subpopulation strongly stained for CysC [15]. This suggests that Aβ accumulates in a specific population of pyramidal neurons in the brain, the same cell type in which CysC is highly expressed. The accumulation of both polypeptides in neuronal vesicular compartments would support inter-molecular binding.
While a L68Q variant form of CysC forms amyloid deposits in the CNS of HCHWA-I patients [34], the wild type protein is non-amyloidogenic. Both forms of the protein bind Aβ and inhibit Aβ aggregation and amyloid deposition [19-22]. CysC is one of several proteins that are potentially amyloidogenic,but have direct physical interaction with Aβ when soluble. It was reported that gelsolin and transthyretin bind Aβ and inhibit Aβ fibril formation [37-40]. Moreover, cellular prion protein was recently identified as a high-affinity cell-surface receptor for soluble Aβ oligomers on neurons and the interaction does not require the infectious prion conformation [41]. It remains to be determined whether the primary structure of amyloidogenic proteins modulates their interaction with each other, resulting in a defense mechanism against amyloidosis. We propose that CysC is a carrier of soluble Aβ and plays a role in inhibiting the aggregation of Aβ. Modulation of CysC concentration or the association between CysC and Aβ in the human CNS would prevent amyloid deposition by a mechanism similar to that in mouse models. Multiple studies have shown changes in CysC serum concentrations in a variety of conditions, including aging (for review [42]). Changes in CysC concentrations were also observed in the brain in response to different types of injury, such as ischemia or induction of epilepsy (for review [16]). Altered CysC trafficking and a reduction in its secretion are caused by two presenilin 2 mutations (PS2 M239I and T122R), linked to familial AD [43]. Moreover, the CST3 gene G73A polymorphism results in an amino acid change from Ala to Thr at the −2 position for signal peptidase cleavage, altering the hydrophobicity profile of the signal sequence [44]. This results in a less efficient cleavage of the signal peptide and thus a reduced secretion of CysC [45-47]. The decreased CysC secretion associated with the CST3 polymorphism suggests a mechanism for the high-risk of late-onset sporadic AD related to this polymorphism [16]. The inhibition of Aβ aggregation caused by binding of CysC to Aβ suggests a mechanism by which a reduced CysC brain concentration is associated with AD. Therefore, we hypothesize that endogenous CysC is a carrier of soluble Aβ in body fluids such as CSF and blood, as well as in the neuropil, where it plays an ongoing role in inhibiting the association of Aβ into insoluble plaques. Modulation of CysC levels and/or binding to Aβ may have important disease-modifying effect and provide an alternative entry point for novel therapeutic intervention for AD.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Neurological Disorders and Stroke (NS42029), National Institute on Aging (AG017617), The American Heart Association (0040102N) and the Alzheimer’s Association (IIRG-07-59699).
Footnotes
Authors’ disclosures available online (http://www.jalz.com/disclosures/view.php?id=25).
REFERENCES
- [1].Coria F, Castano E, Prelli F, Larrondo-Lillo M, van Duinen S, Shelanski ML, Frangione B. Isolation and characterization of amyloid P component from AD and other types of cerebral amyloidosis. Lab Invest. 1988;58:454–458. [PubMed] [Google Scholar]
- [2].Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- [3].McGeer PL, Kawamata T, Walker DG. Distribution of clusterin in Alzheimer brain tissue. Brain Res. 1992;579:337–341. doi: 10.1016/0006-8993(92)90071-g. [DOI] [PubMed] [Google Scholar]
- [4].Choi-Miura NH, Ihara Y, Fukuchi K, Takeda M, Nakano Y, Tobe T, Tomita M. SP-40,40 is a constituent of Alzheimer’s amyloid. Acta Neuropathol. 1992;83:260–264. doi: 10.1007/BF00296787. [DOI] [PubMed] [Google Scholar]
- [5].Snow AD, Willmer J, Kisilevsky R. Sulfated glycosaminoglycans: a common constituent of all amyloids? Lab Invest. 1987;56:120–123. [PubMed] [Google Scholar]
- [6].Bernstein HG, Kirschke H, Wiederanders B, Schmidt D, Rinne A. Antigenic expression of cathepsin B in aged human brain. Brain Res Bull. 1990;24:543–549. doi: 10.1016/0361-9230(90)90157-u. [DOI] [PubMed] [Google Scholar]
- [7].Nakamura Y, Takeda M, Suzuki H, Hattori H, Tada K, Hariguchi S, Hashimoto S, Nishimura T. Abnormal distribution of cathepsins in the brain of patients with Alzheimer’s disease. Neurosci Lett. 1991;130:195–198. doi: 10.1016/0304-3940(91)90395-a. [DOI] [PubMed] [Google Scholar]
- [8].Nixon RA, Cataldo AM, Paskevich PA, Hamilton DJ, Wheelock TR, Kanaley-Andrews L. The lysosomal system in neurons. Involvement at multiple stages of Alzheimer’s disease pathogenesis. Ann N Y Acad Sci. 1992;674:65–88. doi: 10.1111/j.1749-6632.1992.tb27478.x. [DOI] [PubMed] [Google Scholar]
- [9].Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor α1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell. 1988;52:487–501. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- [10].Van Gool D, De Strooper B, Van Leuven F, Triau E, Dom R. α2-Macroglobulin expression in neuritic-type plaques in patients with Alzheimer’s disease. Neurobiol Aging. 1993;14:233–237. doi: 10.1016/0197-4580(93)90006-w. [DOI] [PubMed] [Google Scholar]
- [11].Barrett AJ. The cystatins: a diverse superfamily of cysteine peptidase inhibitors. Biomed Biochim Acta. 1986;45:1363–1374. [PubMed] [Google Scholar]
- [12].Maruyama K, Ikeda S, Ishihara T, Allsop D, Yanagisawa N. Immunohistochemical characterization of cerebrovascular amyloid in 46 autopsied cases using antibodies to β protein and cystatin C. Stroke. 1990;21:397–403. doi: 10.1161/01.str.21.3.397. [DOI] [PubMed] [Google Scholar]
- [13].Vinters HV, Nishimura GS, Secor DL, Pardridge WM. Immunoreactive A4 and γ-trace peptide colocalization in amyloidotic arteriolar lesions in brains of patients with Alzheimer’s disease. Am J Pathol. 1990;137:233–240. [PMC free article] [PubMed] [Google Scholar]
- [14].Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T. Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci. 1993;116:135–141. doi: 10.1016/0022-510x(93)90317-r. [DOI] [PubMed] [Google Scholar]
- [15].Levy E, Sastre M, Kumar A, Gallo G, Piccardo P, Ghetti B, Tagliavini F. Codeposition of cystatin C with amyloid-β protein in the brain of Alzheimer’s disease patients. J Neuropathol Exp Neurol. 2001;60:94–104. doi: 10.1093/jnen/60.1.94. [DOI] [PubMed] [Google Scholar]
- [16].Levy E, Jaskolski M, Grubb A. The role of cystatin C in cerebral amyloid angiopathy and stroke: cell biology and animal models. Brain Pathol. 2006;16:60–70. doi: 10.1111/j.1750-3639.2006.tb00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Beyer K, Lao JI, Gomez M, Riutort N, Latorre P, Mate JL, Ariza A. Alzheimer’s disease and the cystatin C gene polymorphism: an association study. Neurosci Lett. 2001;315:17–20. doi: 10.1016/s0304-3940(01)02307-2. [DOI] [PubMed] [Google Scholar]
- [18].Cathcart HM, Huang R, Lanham IS, Corder EH, Poduslo SE. Cystatin C as a risk factor for Alzheimer disease. Neurology. 2005;64:755–757. doi: 10.1212/01.WNL.0000151980.42337.44. [DOI] [PubMed] [Google Scholar]
- [19].Sastre M, Calero M, Pawlik M, Mathews PM, Kumar A, Danilov V, Schmidt SD, Nixon RA, Frangione B, Levy E. Binding of cystatin C to Alzheimer’s amyloid β inhibits amyloid fibril formation. Neurobiol Aging. 2004;25:1033–1043. doi: 10.1016/j.neurobiolaging.2003.11.006. [DOI] [PubMed] [Google Scholar]
- [20].Selenica ML, Wang X, Ostergaard-Pedersen L, Westlind-Danielsson A, Grubb A. Cystatin C reduces the in vitro formation of soluble Aβ1-42 oligomers and protofibrils. Scand J Clin Lab Invest. 2007;67:179–190. doi: 10.1080/00365510601009738. [DOI] [PubMed] [Google Scholar]
- [21].Kaeser SA, Herzig MC, Coomaraswamy J, Kilger E, Selenica ML, Winkler DT, Staufenbiel M, Levy E, Grubb A, Jucker M. Cystatin C modulates cerebral β-amyloidosis. Nat Genet. 2007;39:1437–1439. doi: 10.1038/ng.2007.23. [DOI] [PubMed] [Google Scholar]
- [22].Mi W, Pawlik M, Sastre M, Jung SS, Radvinsky DS, Klein AM, Sommer J, Schmidt SD, Nixon RA, Mathews PM, Levy E. Cystatin C inhibits amyloid-β deposition in Alzheimer’s disease mouse models. Nat Genet. 2007;39:1440–1442. doi: 10.1038/ng.2007.29. [DOI] [PubMed] [Google Scholar]
- [23].Carrette O, Demalte I, Scherl A, Yalkinoglu O, Corthals G, Burkhard P, Hochstrasser DF, Sanchez JC. A panel of cerebrospinal fluid potential biomarkers for the diagnosis of Alzheimer’s disease. Proteomics. 2003;3:1486–1494. doi: 10.1002/pmic.200300470. [DOI] [PubMed] [Google Scholar]
- [24].Straface E, Matarrese P, Gambardella L, Vona R, Sgadari A, Silveri MC, Malorni W. Oxidative imbalance and cathepsin D changes as peripheral blood biomarkers of Alzheimer disease: a pilot study. FEBS Lett. 2005;579:2759–2766. doi: 10.1016/j.febslet.2005.03.094. [DOI] [PubMed] [Google Scholar]
- [25].Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- [26].Mirra SS, Hart MN, Terry RD. Making the diagnosis of Alzheimer’s disease. A primer for practicing pathologists. Arch Pathol Lab Med. 1993;117:132–144. [PubMed] [Google Scholar]
- [27].Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-β quantitation. Methods Mol Biol. 2005;299:267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- [28].Haan J, Maat-Schieman MLC, van Duinen SG, Jensson O, Thorsteinsson L, Roos RAC. Co-localization of β/A4 and cystatin C in cortical blood vessels in Dutch, but not in Icelandic hereditary cerebral hemorrhage with amyloidosis. Acta Neurol Scand. 1994;89:367–371. doi: 10.1111/j.1600-0404.1994.tb02648.x. [DOI] [PubMed] [Google Scholar]
- [29].Wei L, Walker LC, Levy E. Cystatin C: Icelandic-like mutation in an animal model of cerebrovascular β amyloidosis. Stroke. 1996;27:2080–2085. doi: 10.1161/01.str.27.11.2080. [DOI] [PubMed] [Google Scholar]
- [30].Uchida K, Kuroki K, Yoshino T, Yamaguchi R, Tateyama S. Immunohistochemical study of constituents other than β-protein in canine senile plaques and cerebral amyloid angiopathy. Acta Neuropathol (Berl) 1997;93:277–284. doi: 10.1007/s004010050615. [DOI] [PubMed] [Google Scholar]
- [31].Steinhoff T, Moritz E, Wollmer MA, Mohajeri MH, Kins S, Nitsch RM. Increased cystatin C in astrocytes of transgenic mice expressing the K670N-M671L mutation of the amyloid precursor protein and deposition in brain amyloid plaques. Neurobiol Dis. 2001;8:647–654. doi: 10.1006/nbdi.2001.0412. [DOI] [PubMed] [Google Scholar]
- [32].Walsh DM, Selkoe DJ. Aβ oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- [33].Levy E, Lopez-Otin C, Ghiso J, Geltner D, Frangione B. Stroke in Icelandic patients with hereditary amyloid angiopathy is related to a mutation in the cystatin C gene, an inhibitor of cysteine proteases. J Exp Med. 1989;169:1771–1778. doi: 10.1084/jem.169.5.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gudmundsson G, Hallgrimsson J, Jonasson TA, Bjarnason O. Hereditary cerebral haemorrhage with amyloidosis. Brain. 1972;95:387–404. doi: 10.1093/brain/95.2.387. [DOI] [PubMed] [Google Scholar]
- [35].Wei L, Berman Y, Castano EM, Cadene M, Beavis RC, Devi L, Levy E. Instability of the amyloidogenic cystatin C variant of hereditary cerebral hemorrhage with amyloidosis, Icelandic type. J Biol Chem. 1998;273:11806–11814. doi: 10.1074/jbc.273.19.11806. [DOI] [PubMed] [Google Scholar]
- [36].Deng A, Irizarry MC, Nitsch RM, Growdon JH, Rebeck GW. Elevation of cystatin C in susceptible neurons in Alzheimer’s disease. Am J Pathol. 2001;159:1061–1068. doi: 10.1016/S0002-9440(10)61781-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chauhan VP, Ray I, Chauhan A, Wisniewski HM. Binding of gelsolin, a secretory protein, to amyloid β-protein. Biochem Biophys Res Commun. 1999;258:241–246. doi: 10.1006/bbrc.1999.0623. [DOI] [PubMed] [Google Scholar]
- [38].Ray I, Chauhan A, Wegiel J, Chauhan VP. Gelsolin inhibits the fibrillization of amyloid β-protein, and also defibrillizes its preformed fibrils. Brain Res. 2000;853:344–351. doi: 10.1016/s0006-8993(99)02315-x. [DOI] [PubMed] [Google Scholar]
- [39].Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK, Zagorski MG, Talafous J, Eisenberg M, Saunders AM, Roses AD, Goldgaber D. Transthyretin sequesters amyloid β protein and prevents amyloid formation. Proc Natl Acad Sci U S A. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc Natl Acad Sci U S A. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Madero M, Sarnak MJ, Stevens LA. Serum cystatin C as a marker of glomerular filtration rate. Curr Opin Nephrol Hypertens. 2006;15:610–616. doi: 10.1097/01.mnh.0000247505.71915.05. [DOI] [PubMed] [Google Scholar]
- [43].Ghidoni R, Benussi L, Paterlini A, Missale C, Usardi A, Rossi R, Barbiero L, Spano P, Binetti G. Presenilin 2 mutations alter cystatin C trafficking in mouse primary neurons. Neurobiol Aging. 2007;28:371–376. doi: 10.1016/j.neurobiolaging.2006.01.007. [DOI] [PubMed] [Google Scholar]
- [44].Finckh U, von Der Kammer H, Velden J, Michel T, Andresen B, Deng A, Zhang J, Muller-Thomsen T, Zuchowski K, Menzer G, Mann U, Papassotiropoulos A, Heun R, Zurdel J, Holst F, Benussi L, Stoppe G, Reiss J, Miserez AR, Staehelin HB, Rebeck GW, Hyman BT, Binetti G, Hock C, Growdon JH, Nitsch RM. Genetic association of a cystatin C gene polymorphism with late-onset Alzheimer disease. Arch Neurol. 2000;57:1579–1583. doi: 10.1001/archneur.57.11.1579. [DOI] [PubMed] [Google Scholar]
- [45].Benussi L, Ghidoni R, Steinhoff T, Alberici A, Villa A, Mazzoli F, Nicosia F, Barbiero L, Broglio L, Feudatari E, Signorini S, Finckh U, Nitsch RM, Binetti G. Alzheimer disease-associated cystatin C variant undergoes impaired secretion. Neurobiol Dis. 2003;13:15–21. doi: 10.1016/s0969-9961(03)00012-3. [DOI] [PubMed] [Google Scholar]
- [46].Paraoan L, Ratnayaka A, Spiller DG, Hiscott P, White MR, Grierson I. Unexpected intracellular localization of the AMD-associated cystatin C variant. Traffic. 2004;5:884–895. doi: 10.1111/j.1600-0854.2004.00230.x. [DOI] [PubMed] [Google Scholar]
- [47].Noto D, Cefalu AB, Barbagallo CM, Pace A, Rizzo M, Marino G, Caldarella R, Castello A, Pernice V, Notarbartolo A, Averna MR. Cystatin C levels are decreased in acute myocardial infarction: effect of cystatin C G73A gene polymorphism on plasma levels. Int J Cardiol. 2005;101:213–217. doi: 10.1016/j.ijcard.2004.03.018. [DOI] [PubMed] [Google Scholar]