

Abstract
The effect of different total enzyme concentrations on the flux through the bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system (PTS) in vitro was determined by measuring PTS-mediated carbohydrate phosphorylation at different dilutions of cell-free extract of Escherichia coli. The dependence of the flux on the protein concentration was more than linear but less than quadratic. The combined flux–response coefficient of the four enzymes constituting the glucose PTS decreased slightly from values of ≈1.8 with increasing protein concentrations in the assay. Addition of the macromolecular crowding agents polyethylene glycol (PEG) 6000 and PEG 35000 led to a sharper decrease in the combined flux–response coefficient, in one case to values of ≈1. PEG 6000 stimulated the PTS flux at lower protein concentrations and inhibited the flux at higher protein concentrations, with the transition depending on the PEG 6000 concentration. This suggests that macromolecular crowding decreases the dissociation rate constants of enzyme complexes. High concentrations of the microsolute glycerol did not affect the combined flux–response coefficient. The data could be explained with a kinetic model of macromolecular crowding in a two-enzyme group-transfer pathway. Our results suggest that, because of the crowded environment in the cell, the different PTS enzymes form complexes that live long on the time-scale of their turnover. The implications for the metabolic behavior and control properties of the PTS, and for the effect of macromolecular crowding on nonequilibrium processes, are discussed.
Understanding the functioning of the living cell on the basis of processes studied in vitro is a central aim of biochemistry; properties of biomolecules in vitro are extrapolated to the situation in vivo. The concentrations of macromolecules in a biochemical assay are much lower than inside the living cell; total intracellular macromolecule concentrations (RNA and protein) of 340 mg/ml have been measured in exponentially growing Escherichia coli cells, with 70–73% thereof being protein (1). These high concentrations can lead to macromolecular crowding (reviewed in refs. 2 and 3). The ensuing nonideal equilibrium behavior of hemoglobin already was reported by Adair (4). To mimic cellular conditions in a test tube, inert macromolecular crowding agents such as polyethylene glycol (PEG) can be added (2, 3); this then promotes association between macromolecules. For example, addition of PEG can overcome the inhibition of E. coli DNA polymerase I activity by high salt concentrations (5), presumably by effecting the association of DNA and the enzyme, whose interaction was loosened by the high ionic strength.
The effect of high protein concentrations—and of high protein activities caused by macromolecular crowding—on equilibrium phenomena, such as the binding of proteins to DNA, is understood and documented fairly well (6). However, the effects on nonequilibrium processes such as metabolic fluxes and signal transduction have not been investigated experimentally. In vitro, biochemistry often shows reaction rates proportional to the enzyme concentration. Extrapolation to high protein concentration then should be simple indeed. However, for fluxes through signal transduction chains, it was pointed out recently that such simplicity must not be expected (7, 8) because most individual reactions involve two macromolecules rather than one.
This feature of signal transduction chains has been elaborated most extensively for group-transfer pathways, in which a chemical group is transferred along a series of carriers. The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system (PTS; reviewed in ref. 9) is an example of such a group-transfer pathway; the two-component regulatory systems of bacteria have similar properties. When E. coli takes up glucose, a phosphoryl group derived from intracellular phosphoenolpyruvate is transferred along four different PTS proteins [Enzyme I (EI), HPr, IIAGlc, and IICBGlc] to the glucose molecule, yielding intracellular glucose 6-phosphate and pyruvate. On simultaneous equal fractional increases in the concentrations of all glucose PTS enzymes, the flux through the pathway may be expected to vary almost quadratically with enzyme concentration if all of the phosphotransfer reactions are kinetically bimolecular in both directions (i.e., in the case of immediate transfer or a “hit-and-run” mechanism, in which the life-times of the ternary complexes between different PTS enzymes and the bound phosphoryl group are negligible) (7, 8). Such a quadratic dependence contrasts with the linear relationship between enzyme concentrations and rates observed for “ideal” (10) metabolic pathways. Of more importance, it suggests that the fluxes through signal transduction pathways in vivo may be much higher than expected from a simple extrapolation of in vitro results.
Moreover, in certain metabolic pathways, direct enzyme–enzyme interactions occur, notably when the product of an enzyme is transferred to the subsequent enzyme in the pathway without first equilibrating with the bulk aqueous phase (11–13). When such “metabolite channeling” occurs, the flux also will depend nonlinearly on enzyme concentrations, again complicating the extrapolation from vitrum to vivum. Indeed, metabolite channeling may be hard to demonstrate in vitro, as the degree of channeling itself may decrease when moving from in vivo to in vitro enzyme concentrations (13, 14). In the analysis of channeling, one normally has to consider two competing routes: the “normal” route in which the intermediate is released into the bulk solution and the “channeled” route in which it is transferred directly between the enzymes (see also ref. 14). Group-transfer pathways represent a special case insofar as only the latter route occurs and the transferred group is never released into the bulk phase: channeling is complete. Hence, a group-transfer pathway such as the PTS may serve as a model system for metabolite channeling in which the complication of a varying degree of channeling is absent.
Theoretical analyses have suggested that the dependence of the flux through group-transfer and channeled pathways on the total enzyme concentration may vary between linear and quadratic (and, in fact, beyond either limit) (7, 14). Consequently, a decisive experimental analysis requires a quantifier for this dependence. Such a quantifier is the combined flux–response coefficient (15, 16) of all of the enzymes in the pathway, which is the ratio between the relative increase in flux and the relative increase in total enzyme concentration (at constant relative proportions of all enzymes, and extrapolated to infinitely small changes), and also has been termed enzyme flux-control coefficient (10, 17). For group-transfer pathways and completely channeled metabolic pathways in which boundary substrates are present in excess, the combined flux–response coefficient should =2 at low protein concentrations and dwindle to 1 at high protein concentrations and under conditions of macromolecular crowding (7, 14).
This paper describes experimental evidence for the occurrence of the above phenomenon in vitro by using the PTS as a model system for signal transduction and metabolite channeling. The results suggest that longer-lived complex formation between the PTS enzymes also may occur in vivo. Implications for the control and regulatory properties of the PTS are discussed.
MATERIALS AND METHODS
Preparation of Cell-Free Extracts.
The E. coli strain used in this study, PJ4004, has the genotype F+ asnB32 thi-1 relA1 spoT1 lacUV5 lacY(Am) and is equivalent to LM3118 (18) except for transformation with the plasmid pBR322. Cells were grown at 30°C in minimal Mops medium (19) supplemented with 2.5 μg/ml thiamine, 100 μg/ml ampicillin, and 18.5 mM succinate. Cells were ruptured by passage of a washed and concentrated cell suspension through an Aminco French pressure cell at 1.1 × 108 Pa, and bacterial cell-free extracts were prepared as described (20).
PTS Activity Assay.
Glucose PTS activity in cell-free extracts was determined by measuring the phosphorylation of 10 mM 14C-labeled methyl α-d-glucopyranoside (MeGlc, a nonmetabolizable analogue of glucose) as described (21). Cell-free extract was added at various dilutions; phosphoenolpyruvate was added at a final concentration (in mM) of twice the protein concentration (in mg/ml). Samples were incubated at 37°C for 30 min; we verified that measured fluxes were constant during this time. PEG or glycerol was added to the reaction mixtures as indicated. PEG 6000 (BDH) and PEG 35000 (Merck) were of synthesis grade. Glycerol (87%) of analytical reagent grade was from Merck.
Protein Determinations.
Protein concentrations of cell-free extracts were determined on a Cobas BIO autoanalyzer (Roche, Gipf-Oberfrick, Switzerland) with the bicinchoninic acid assay (22) by using BSA (Sigma) as standard.
Determination of Flux–Response Coefficients.
The combined flux–response coefficient of the four glucose PTS enzymes is defined (15, 16) as
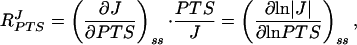 |
1 |
where J is the flux through the pathway at steady state (symbolized by subscript ss), and PTS is the total concentration of the PTS enzymes under conditions in which their relative proportions remain constant. By measuring MeGlc phosphorylation activity for a range of cell-free extract concentrations, the PTS flux could be determined as a function of protein concentration (which was proportional to the total PTS enzyme concentration). The combined flux–response coefficient of the four glucose PTS enzymes was calculated from these data by two independent methods. (i) A third-order polynomial was fitted by least squares regression through the flux vs. protein concentration data in double-logarithmic space. The first derivative of this function yielded the combined flux–response coefficient directly. (ii) The flux vs. protein concentration data were smoothed by fitting a cubic spline in linear space. To obtain the combined flux–response coefficient, the slope was scaled in each point by multiplying with the respective protein concentration and dividing by the flux.
Numerical Methods.
Simulations and steady-state calculations of the kinetic model were performed on an IBM-compatible personal computer with the metabolic modeling program scamp (23).
RESULTS
Crowding Decreases the PTS Flux–Response Coefficient in Vitro.
Our first aim was to test the prediction (7, 14) that, in group-transfer pathways, the dependence of flux on total enzyme concentration should vary from almost quadratic to almost linear with increasing total enzyme concentration. The flux J through the system varied with the total protein concentration; the dependence was more than linear but less than quadratic (Fig. 1, closed symbols). Addition of 9% PEG 6000 stimulated the PTS flux slightly at lower protein concentrations and inhibited the flux at higher protein concentrations (Fig. 1a, open symbols). A lower assay concentration of PEG 6000 appeared to shift the transition point from stimulation to inhibition of PTS flux to a higher protein concentration; 4.5% PEG 6000 only stimulated the PTS flux in the range of protein concentrations tested (Fig. 1b). PEG 35000, on the other hand, inhibited PTS flux under all conditions tested (Fig. 1c).
Figure 1.

Dependence of PTS flux in vitro on the total protein concentration. The rate of MeGlc phosphorylation by cell-free extracts of E. coli was determined as described in the text. Closed symbols refer to fluxes without extra assay additions. Open symbols refer to fluxes under identical conditions, except that the assay mixture contained, additionally, 9% (m/V) PEG 6000 (a), 4.5% (m/V) PEG 6000 (b), 5% (m/V) PEG 35000 (c), and 1.2 M glycerol (d). Data points reflect means of two independent experiments, except the experiments depicted by the closed symbols in b and d, in which each data point reflects an individual determination. Error bars indicate SEMs. Experiments a–d were performed with different cell-free extracts. The control experiment (closed symbols) was always included as a reference.
Subsequently, the data from Fig. 1 were differentiated in double-logarithmic space to determine the combined flux–response coefficient of the PTS enzymes, RPTSJ (Fig. 2). We used two independent methods: nonlinear least squares regression of a third-order polynomial through the data from Fig. 1 in double-logarithmic space and fitting of a cubic spline through the data in linear space. These approaches were chosen in order to prevent the numerical analysis method from biasing the result. Fig. 2 shows that the two analysis methods agreed satisfactorily; the lines correspond fairly well to the points.
Figure 2.

The effect of PEG 6000, PEG 35000, and glycerol on the combined flux–response coefficient of the PTS enzymes in vitro. The data from Fig. 1 were differentiated and scaled to determine RPTSJ as described in the text. The lines refer to slopes calculated by the first derivative of a third-order polynomial fitted through the flux vs. protein concentration data in double-logarithmic space. The points refer to scaled slopes determined by fitting a cubic spline through the flux vs. protein concentration data in linear space. Solid lines and closed symbols refer to assays without extra additions. Dotted lines and open symbols refer to assays with the following extra additions, as in Fig. 1: 9% (m/V) PEG 6000 (a), 4.5% (m/V) PEG 6000 (b), 5% (m/V) PEG 35000 (c), and 1.2 M glycerol (d).
We first addressed the question of whether, at low protein concentrations, the combined flux–response coefficient for the PTS amounted to 2 rather than to 1, as expected for ideal metabolic pathways. The data for the lower protein concentrations in Fig. 2 all suggest that the answer is indeed close to 2.
Our second question concerned the decrease of the combined flux–response coefficient with increasing protein concentrations. Under standard assay conditions, we only observed a slight decrease (Fig. 2, solid lines and closed symbols). It seemed possible that the protein concentrations obtainable in vitro were insufficiently high to afford a substantial decrease in RPTSJ. Therefore, we added macromolecular crowding agents (6) to increase the protein activities: 9% PEG 6000 (Fig. 2a) caused RPTSJ to decrease sharply from 1.8 to values ≈1 whereas a lower concentration (4.5%) of PEG 6000 (Fig. 2b) resulted in a decrease that was less substantial and occurred only at higher protein concentrations (>4.5 mg/ml). The addition of 5% PEG 35000 (Fig. 2c) also resulted in a slight decrease of RPTSJ at high protein concentrations (>4 mg/ml) in comparison to the reference experiment; it would appear that RPTSJ was increased slightly by PEG 35000 addition at the lower protein concentrations. However, the discrepancy between the two fitting methods was greater than in the other cases.
If indeed the decrease in RPTSJ at high protein concentrations in the presence of PEG was caused by macromolecular crowding, a high concentration of an inert small molecule (microsolute) should not generate the same effects because such a molecule should not decrease the access of macromolecules to the solution. To test this, we performed the PTS assay in the presence of 1.2 M glycerol, an uncharged molecule that does not partake in any of the investigated reactions. Although the flux was inhibited with respect to the reference experiment (Fig. 1d), the combined PTS enzyme flux–response coefficient hardly was affected by glycerol (Fig. 2d).
Crowding Effects Reproduced in a Kinetic Model.
To check that a decrease in RPTSJ and a decrease in flux can both be attributed to macromolecular crowding, we constructed a simple kinetic model of a two-enzyme group-transfer pathway, as shown in Fig. 3. The model includes two forms for each enzyme that are not bound to other enzymes or to boundary substrates Xi (S1 and S2 for E1, and S3 and S4 for E2). These Si correspond to the unphosphorylated and phosphorylated enzyme forms in the PTS. The boundary substrates X0 to X3 represent phosphoenolpyruvate, pyruvate, glucose/MeGlc, and glucose 6-phosphate/MeGlc 6-phosphate. The intermediates Q1 to Q3 refer to complexes between the enzymes or between an enzyme and a boundary substrate. We shall use uppercase Roman characters for the name of a species and lowercase italics for its concentration.
Figure 3.

Kinetic model of a two-enzyme group-transfer pathway. Arrows indicate the direction of the flux; all elementary reactions are assumed to be reversible. The rate constants used for the simulation were as follows (ki denotes the rate constant in the forward direction relative to the arrowheads; k−i indicates the rate constant in the reverse direction): k1 = 1, k−1 = 0.5, k2 = 5, k−2 = 2, k3 = 500, k−3 = 250, k4 = 10, k−4 = 20, k5 = 1, k−5 = 0.5, k6 = 5, and k−6 = 1. A series of steady states was calculated in which the concentrations x0 and x2 were varied from 0.5 to 10 and the concentrations x1 and x3 were varied from 0.05 to 1 whereas the total concentrations e1 and e2 were increased from 0.1 to 2 concomitantly. Units are arbitrary.
A series of steady states was calculated in which the total concentrations of the enzymes (e1 and e2) were varied proportionally with the concentrations x0, x1, x2, and x3 (Fig. 4, lines labeled α = 1). RPTSJ decreased from 1.9 to 1.4 (Fig. 4c, lines labeled α = 1). This was the result of a decrease in the flux–response coefficients of the enzymes E1 and E2, as well as of the external metabolites X0 to X3 (data not shown).
Figure 4.

Simulation results of the model described in Fig. 3. Macromolecular crowding agents were assumed to increase the on-rate constants for complex formation (k1, k−2, k3,k−4, k5, and k−6) by a factor α and to decrease the off-rate constants for complex dissociation (k−1, k2, k−3, k4, k−5, and k6) by the same factor α. The other parameters were as per the legend of Fig. 3. For α= 1, crowding effects are assumed to be absent. (a) The flux J. (b) Magnification of the area near the origin in a. (c) The combined flux–response coefficient, for α = 1, 2, and 3.
What was the reason for the decrease in RPTSJ? As e1 and e2 increased, the concentrations of both the free (si) and the complexed (qi) forms of the proteins increased. However, the concentrations of the complexed forms increased more sharply, so that the fraction of complexed enzymes was higher for a total e1 concentration of 2 than for 0.1. At e1 = 0.1, for example, q2/e1 = 0.05 (i.e., 5% of enzyme E1 was complexed with E2) whereas this fraction increased to 0.24 at e1 = 2. For the model system in Fig. 3, the combined flux–response coefficient RPTSJ equals
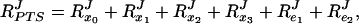 |
2 |
where RJe1 is the flux–response coefficient (15) with respect to the total concentration of E1, etc. The flux–response coefficients in the system obey the following relationship (7):
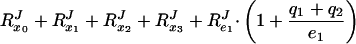 |
3 |
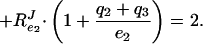 |
If no ternary complexes exist between the different enzymes (i.e., qi = 0), RPTSJ should =2. However, as the fraction of complexed enzymes increases (i.e., qi becomes >0), Eq. 3 shows that RPTSJ will decrease to a value between 1 and 2. In fact, RPTSJ = 2 divided by a correction factor, which lies between 1 and 2 and is a weighted average of the fraction of the enzymes that is, on average, complexed (7).
To model the addition of macromolecular crowding agents to the PTS assay, we first observed that PEG 6000 could either stimulate or inhibit the PTS flux, depending on both the PEG 6000 and the protein concentrations (Fig. 1 a and b). The association between two molecules can, in principle, be increased either by an increase in the apparent association rate constant or by a decrease in the apparent dissociation rate constant. Both changes will lead to an altered equilibrium constant and a higher complex concentration at equilibrium. Minton (6) has argued on thermodynamic grounds that the association of monomers to homopolymers should be stimulated by macromolecular crowding mainly via an enhancement of the association rate constant. However, if macromolecular crowding only increased the association rate constants, the overall effect on the flux only could be stimulatory, as local rates are either unchanged or accelerated. Likewise, if macromolecular crowding only decreased the dissociation rate constants, the overall effect on the flux only could be inhibitory. Because we observed both a stimulation and an inhibition of the flux, depending on the conditions (Fig. 1a and b), we concluded that crowding agents both increase the association rate constants for complex formation and decrease the complex dissociation rate constants. As a first approximation, we assumed the two effects to be equally strong and modulated the rate constants by the same factor (α and 1/α) (Fig. 4).
Increases in α, mimicking the addition of macromolecular crowding agents, inhibited the flux at higher enzyme concentrations (Fig. 4a) and stimulated the flux slightly at lower enzyme concentrations (Fig. 4b). The inhibition for higher enzyme concentrations was stronger for larger values of α. Concomitantly, the combined flux–response coefficient RPTSJ decreased more sharply with enzyme concentration as α was increased (Fig. 4c), approaching a value of 1 for α = 3 and e1 = e2 = 2. Furthermore, the enzyme concentration at which the effect of addition of crowding agent passed from stimulation to inhibition was higher for α = 2 than for α = 3 (Fig. 4b). This was in qualitative agreement with the experimental results for 9% and 4.5% PEG 6000 (Fig. 1a and b).
DISCUSSION
We have studied the implications of varying protein concentrations and macromolecular crowding for nonequilibrium phenomena such as channeled metabolic fluxes and signal transduction. The dependence of the flux through the PTS on total enzyme concentration was determined and quantified in terms of the combined PTS enzyme flux–response coefficient by using both an experimental approach and numerical simulations of a kinetic model of macromolecular crowding. Intracellular conditions were mimicked by adding high concentrations of an inert macromolecule.
Experimentally observed values of the combined response coefficient RPTSJ clearly exceeded 1 (Fig. 2), demonstrating the nonideal dependence of the flux on the total enzyme concentration. One implication of this finding is that such a pathway can exhibit novel control properties: The flux through a group-transfer pathway can respond more sensitively to changes in the concentrations of its constituent enzymes than that through an ideal linear pathway in which the combined flux–response coefficient always =1. When modulating gene expression to manipulate the flux through such pathways, this has stronger effects than in normal ideal (10) pathways. As predicted (8), the summation theorem (24), when written in terms of enzyme concentration-based response coefficients, is not valid for group-transfer pathways or channeled pathways (cf. refs. 7, 8, and 25).
The combined flux–response coefficient need not be constant for one and the same pathway; in fact, the experimental results (Fig. 2) show that RPTSJ can vary with experimental conditions. In particular, it should decrease at high enzyme concentrations. Because the intracellular protein concentration is ≈40 times higher than the highest protein concentration used in the assay in vitro (6.5 mg/ml) (1), RPTSJ may well be much closer to 1 than to 2 in the cell. This suggestion is supported by the observation that addition of PEG 6000 to the in vitro assay to mimic intracellular conditions caused RPTSJ to decrease sharply to values ≈1 (Fig. 2a). Furthermore, Van der Vlag et al. (26) have measured a value of 0.8 for the sum of the enzyme flux–response coefficients of the glucose PTS in Salmonella typhimurium in vivo: i.e., less than the expected value between 1 and 2 (7). The flux–response coefficients of phosphoenolpyruvate, pyruvate, MeGlc, or MeGlc 6-phosphate were not determined, but a flux–response coefficient of 0.7 was assigned to the MeGlc/MeGlc 6-phosphate couple on the grounds that it equaled RIICBJ, and a value of 1.5 (i.e., 2 × 0.7 + 0.1) was deduced for RPTSJ when including the boundary metabolites in the sum (26). After the present studies, it might seem that −0.5 (i.e., the difference between 1.5 and 2) of the flux control should be caused by the complexes between the PTS components. This, however, would not be quite an accurate conclusion: If complexation between IIAGlc and IICBGlc were in part responsible for the decrease of the total control below 2, then the flux control residing in the MeGlc/MeGlc 6-phosphate couple should be <0.7. Indeed, from the dependence of the PTS uptake flux in vivo on the MeGlc concentration, we expect RIICBJ to amount to 0.2 only; the apparent KM of IICBGlc for MeGlc uptake in S. typhimurium is 170 μM (27), and the uptake assays (26) were performed at 500 μM MeGlc. Accordingly, we expect that RPTSJ in vivo is much closer to 1 than to 1.4 and hence that complex formation between the PTS components caused by macromolecular crowding inside the cell bereaves the PTS proteins of half their potential flux control. More detailed experimentation will have to tell.
If the combined flux–response coefficient of a group-transfer pathway is <2, this points to the existence of ternary complexes between the pathway components (Eq. 3; see also ref. 7). The closer the coefficient is to 2, the more the pathway resembles a perfect dynamic channel where the transfer of the group is immediate on hit-and-run collisions between the enzymes and between enzymes and boundary substrates; the closer it is to 1, the longer is the lifetime of the ternary complexes (14) and the higher are their relative concentrations. Determining RPTSJ can therefore be a good indicator of the nature of the channeling process: i.e., whether ternary complexes between the complexes persist or not. Intracellular values for RPTSJ of ≈1 would indeed suggest that a significant proportion of the PTS enzymes may exist in the complexed state in the cell.
The agreement between model and experiment suggests that the observed results may be caused by complex formation enhanced by macromolecular crowding. The general dependence of the flux on protein concentration, the decrease in RPTSJ for higher enzyme concentrations, the stimulatory effect of PEG 6000 at low enzyme concentrations, and its inhibitory effect at higher enzyme concentrations all could be reproduced by a simplified kinetic model of only two group-transfer enzymes relying on simple kinetic assumptions. The established theories on macromolecular crowding (2, 3, 6, 13) focused on equilibrium aspects and therefore did not consider details of the association and dissociation rate constants but instead considered only their ratio (i.e., the equilibrium constant). Our results show that, for the model to match experimental data, both rate constants have to be affected by macromolecular crowding, and in opposite directions. The effect of macromolecular crowding on the dissociation rate constant is counterintuitive to us but may help explaining the basis of gel-shift assays of DNA–protein complexes.
Mao et al. (28) reported the isolation, purification and reconstitution of a fusion protein comprising the four glucose PTS enzymes of E. coli, in which the different enzymes were joined by flexible linkers. In agreement with our results, the phosphorylation activity of an equimolar mixture of the individual isolated enzymes varied more than linearly but less than quadratically with the total enzyme concentration (figure 2A in ref. 28). In addition, the specific phosphotransferase activity of the fusion protein was 3–4× higher than that of the equimolar mixture of the isolated enzymes (28). The proximity of the different active sites in the fusion protein evidently stimulated the phosphotransfer reaction. This is in agreement with our result that PTS activity could be stimulated by PEG under some conditions. Crowding may lead to a closer proximity of the active sites by promoting complex formation between the PTS enzymes.
Although a high concentration of glycerol in the PTS assay inhibited the flux (Fig. 1d), the combined enzyme flux–response coefficient essentially was unaffected (Fig. 2d). This suggests that the degree of complexation of the PTS enzymes was similar in the presence and absence of glycerol. The observed inhibition of the flux points to some change in enzyme activities or to a reduction in diffusion rates. Indeed, high microsolute concentrations also are known to affect macromolecule equilibria (reviewed in ref. 29), but the near-identical values for RPTSJ after glycerol addition, compared with those in the absence of glycerol, suggest that the decrease in RPTSJ observed after PEG addition (and hence the increased complex formation between the PTS enzymes) can be attributed specifically to macromolecular crowding. Irrespective of more specific inhibitory effects, increasing the protein concentration or adding PEG always led to a decrease in RPTSJ, as expected from enhanced macromolecular crowding.
The inhibitory effect observed with PEG 35000, over the entire protein concentration range tested, points to a different mechanism of action from that of PEG 6000. PEG 35000 addition resulted in a sharper decrease of RPTSJ with increasing protein concentration (Fig. 2c). Because the molecular weight of PEG 35000 is between those of the different PTS enzymes whereas PEG 6000 is smaller than all of them [subunit molecular weights of EI, HPr, IIAGlc, and IICBGlc from E. coli are 63489, 9109, 18099, and 50645, respectively (30–32)], this is not expected on the basis of equilibrium macromolecular crowding (2). Perhaps the association and dissociation rate constants (and hence, the net PTS flux) are influenced differently by PEG 35000 than by PEG 6000.
Further studies along these lines with the PTS and other signal transduction pathways may reveal drastic differences between in vivo function and in vitro assay and may shed light on the mechanisms underlying this behavior. For example, a role has been proposed for macromolecular crowding in compensating changes induced by osmotic stress and decreased free cytoplasmic water content (33). Of even greater importance, such studies perhaps may reveal how in vitro studies can be formatted to mimic the crowded conditions inside the cell.
Acknowledgments
We thank Drs. J. van der Vlag and K. van Dam for helpful discussions and the following organizations for financial support: the South African Foundation for Research Development, the Harry Crossley Foundation, and The Netherlands Organization for Scientific Research.
ABBREVIATIONS
- PTS
phosphoenolpyruvate:carbohydrate phosphotransferase system
- MeGlc
methyl α-d-glucopyranoside
- PEG
polyethylene glycol
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Zimmerman S B, Trach S O. J Mol Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-v. [DOI] [PubMed] [Google Scholar]
- 2.Zimmerman S B, Minton A P. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- 3.Garner M M, Burg M B. Am J Physiol. 1994;266:C877–C892. doi: 10.1152/ajpcell.1994.266.4.C877. [DOI] [PubMed] [Google Scholar]
- 4.Adair G S. Proc R Soc Lond Ser A. 1928;120:573–603. [Google Scholar]
- 5.Zimmerman S B, Harrison B. Proc Natl Acad Sci USA. 1987;84:1871–1875. doi: 10.1073/pnas.84.7.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minton A P. Mol Cell Biochem. 1983;55:119–140. doi: 10.1007/BF00673707. [DOI] [PubMed] [Google Scholar]
- 7.Kholodenko B N, Westerhoff H V. Biochim Biophys Acta. 1995;1229:256–274. [Google Scholar]
- 8.van Dam K, van der Vlag J, Kholodenko B N, Westerhoff H V. Eur J Biochem. 1993;212:791–799. doi: 10.1111/j.1432-1033.1993.tb17720.x. [DOI] [PubMed] [Google Scholar]
- 9.Postma P W, Lengeler J W, Jacobson G R. Microbiol Rev. 1993;57:543–594. doi: 10.1128/mr.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kholodenko B N, Molenaar D, Schuster S, Heinrich R, Westerhoff H V. Biophys Chem. 1995;56:215–226. doi: 10.1016/0301-4622(95)00039-z. [DOI] [PubMed] [Google Scholar]
- 11.Srere P A. Annu Rev Biochem. 1987;56:89–124. doi: 10.1146/annurev.bi.56.070187.000513. [DOI] [PubMed] [Google Scholar]
- 12.Srivastava D K, Bernhard S A. Annu Rev Biophys Biophys Chem. 1987;16:175–204. doi: 10.1146/annurev.bb.16.060187.001135. [DOI] [PubMed] [Google Scholar]
- 13.Agius L, Sherratt H S A, editors. Channelling in Intermediary Metabolism. London: Portland Press; 1997. [Google Scholar]
- 14.Kholodenko B N, Cascante M, Westerhoff H V. Mol Cell Biochem. 1995;143:151–168. doi: 10.1007/BF01816949. [DOI] [PubMed] [Google Scholar]
- 15.Fell D A. Biochem J. 1992;286:313–330. doi: 10.1042/bj2860313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohwer J M. Ph.D. thesis. Amsterdam: Univ. of Amsterdam; 1997. [Google Scholar]
- 17.Kholodenko B N, Westerhoff H V. Trends Biochem Sci. 1995;20:52–54. doi: 10.1016/s0968-0004(00)88955-0. [DOI] [PubMed] [Google Scholar]
- 18.Jensen P R, Michelsen O. J Bacteriol. 1992;174:7635–7641. doi: 10.1128/jb.174.23.7635-7641.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neidhardt F C, Bloch P L, Smith D F. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Postma P W. J Bacteriol. 1977;129:630–639. doi: 10.1128/jb.129.2.630-639.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roseman S, Meadow N D, Kukuruzinska M A. Methods Enzymol. 1982;90:417–423. doi: 10.1016/s0076-6879(82)90165-3. [DOI] [PubMed] [Google Scholar]
- 22.Smith P K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk D C. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 23.Sauro H M. Comput Appl Biosci. 1993;9:441–450. doi: 10.1093/bioinformatics/9.4.441. [DOI] [PubMed] [Google Scholar]
- 24.Kacser H, Burns J A, Fell D A. Biochem Soc Trans. 1995;23:341–366. doi: 10.1042/bst0230341. [DOI] [PubMed] [Google Scholar]
- 25.Brand M D, Vallis B P S, Kesseler A. Eur J Biochem. 1994;226:819–829. doi: 10.1111/j.1432-1033.1994.00819.x. [DOI] [PubMed] [Google Scholar]
- 26.van der Vlag J, van’t Hof R, van Dam K, Postma P W. Eur J Biochem. 1995;230:170–182. doi: 10.1111/j.1432-1033.1995.0170i.x. [DOI] [PubMed] [Google Scholar]
- 27.Stock J B, Waygood E B, Meadow N D, Postma P W, Roseman S. J Biol Chem. 1982;257:14543–14552. [PubMed] [Google Scholar]
- 28.Mao Q, Schunk T, Gerber B, Erni B. J Biol Chem. 1995;270:18295–18300. doi: 10.1074/jbc.270.31.18295. [DOI] [PubMed] [Google Scholar]
- 29.Timasheff S N. Annu Rev Biophys Biomol Struct. 1993;22:67–97. doi: 10.1146/annurev.bb.22.060193.000435. [DOI] [PubMed] [Google Scholar]
- 30.Saffen D W, Presper K A, Doering T L, Roseman S. J Biol Chem. 1987;262:16241–16253. [PubMed] [Google Scholar]
- 31.Nelson S O, Schuitema A R J, Benne R, van der Ploeg L H T, Plijter J J, Aan F, Postma P W. EMBO J. 1984;3:1587–1593. doi: 10.1002/j.1460-2075.1984.tb02015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erni B, Zanolari B. J Biol Chem. 1986;261:16398–16403. [PubMed] [Google Scholar]
- 33.Record M T, Jr, Courtenay E S, Cayley S, Guttman H J. Trends Biochem Sci. 1998;23:190–194. doi: 10.1016/s0968-0004(98)01207-9. [DOI] [PubMed] [Google Scholar]