

Abstract
Background
Malignant gliomas remain refractory to treatment despite advances in chemotherapy and surgical techniques. Viral vectors developed to treat gliomas have had low transduction capabilities, limiting their use. Gliomas over-express CD46, CD80, and CD86, all of which bind adenovirus serotype 3.
Methods
To increase the infectivity and replication of oncolytic vectors in malignant brain tumors, we created a conditionally replicating adenovirus, CRAd-Survivin-5/3, which contains a survivin promoter-driving E1A and a chimeric fiber consisting of adenovirus serotype 3 knob.
Results
In vitro, this modified CRAd showed ten- to 100-fold increased cytotoxicity against glioma cells. Ex vivo analysis of primary glioblastoma multiforme samples infected with CRAd-Survivin-5/3 showed an increase in cytotoxicity of 20–30% compared to adenovirus wild-type (AdWT). Innormal human astrocytes and normal brain tissues, CRAd-Survivin-5/3 exhibited 30–40% and 10–15% lower cytotoxicity than AdWT, respectively. In an intracranial xenograft model of glioma, this oncolytic virus increased tumor-free survival and overall lifespan by 50% compared to controls (p < 0.05).
Conclusions
CRAd-Survivin-5/3 represents an attractive alternative to existing vectors and should be tested further in the pre-clinical setting.
Keywords: adenovirus, brain tumor, glioma, survivin, virotherapy
Introduction
Malignant brain tumors, in particular glioblastoma multiforme (GBM), represent one of the most aggressive cancers that is associated with high degree of morbidity and mortality [1,2]. The median survival of GBM patients is approximately 14 months [2]. Despite advances in surgical techniques, radiation therapy and chemotherapy, treatment remains a challenge.
Cancer virotherapy is a relatively novel approach that seeks to target cancer cells over normal healthy tissues. However, efficient delivery methods remain a challenge. Viral vectors that have been used to date include herpes simplex virus, retrovirus, poliovirus measles and adenovirus [3–8] with varying degrees of success. Of these, adenoviral vectors have proven to be safe. Clinical trials using ONYX-015, an oncolytic adenovirus [3], have shown that it is generally well tolerated. However, one of the major challenges in using adenoviral oncolytic vectors comprises the poor infectivity of tumor cells, likely secondary to a paucity of coxsackie and adenovirus receptor on primary tumor cells [9].
Recently, we used modified adenovectors in which the fiber-knob domain was engineered to target receptors that are over-expressed at a higher level in tumors compared to surrounding healthy tissues. We showed that CD46 and CD80/86 are over-expressed on malignant brain tumors and, based on these findings, designed a chimeric adenovector (Ad 5/3) that contains the backbone of Ad 5 fiber with an Ad 3 knob [10–12]. The Ad 5/3 chimeric virus exhibits increased targeting capabilities for cell lines analysed in vitro by at least ten-fold. GBM samples were also analysed for infectivity ex vivo and Ad 5/3 showed a ten-fold increase versus adenovirus wild-type (AdWT). In normal human astrocytes (NHA), Ad 5/3 infection was at least ten-fold lower compared to AdWT [11–13]. These data suggest an improved safety and targeting profile of Ad 5/3 in the setting of malignant glioma.
To further improve the specificity of adenoviral replication in malignant tissues, gliomas were analysed for increased expression of proteins that are absent or expressed at low levels in normal healthy brain tissues. Survivin, a member of the inhibitor of apoptosis family of proteins, is one such gene whose expression is upregulated in tumor cells in up to 79% of GBM cases [11,14–16]. We therefore created a conditionally replicative adenovirus (CRAd) in which the survivin promoter drives E1A gene replication and the fiber contains a modified adenovirus serotype 3 knob (CRAd-Survivin-5/3). We hypothesized that this form of transcriptional and transductional modification would enhance the virotherapy of malignant glioma cells.
In the present study, we characterized the efficacy of this novel dual-specificity conditionally replicating adenovirus. We show that this vector is more effective in sparing normal healthy tissues and, at the same time, achieves better targeting of glioma cells both in vitro and in vivo. Most importantly, we show that, in a murine xenograft model of intracranial glioma, CRAd-Survivin-5/3 significantly prolongs the lifespan of animals with intracranial tumors.
Materials and methods
Cell lines and primary tissue cultures
The human glioma cell lines U87MG and A172 were purchased from the American Type Culture Collection (Manassas, VA, USA). U118MG was provided by Dr Joanne Douglas (University of Alabama, Birmingham, AL, USA). Human glioma cell line No. 10 was obtained from Japan Tissue Bank (Tokyo, Japan). These cells were grown in minimal essential medium with 10% fetal calf serum (FCS), 100 μg/ml penicillin and 100 μg/ml streptomycin. NHA were obtained from Cambrex-Clonetics (East Rutherford, NJ, USA) and were grown in astrocyte growth media supplemented with growth factors as recommended by the manufacturer. HEK293 and 911 cells was gown in Dulbecco's modified Eagle's medium (DMEM) with 10% FCS, 100 μg/ml penicillin and 100 μg/ml streptomycin. GBM and matched normal brain samples were obtained directly from patients undergoing a resection in accordance with a protocol approved by the Institutional Review Board at the University of Chicago. By ‘matched normal brain’, we refer to normal brain tissue (as confirmed by our neuropathologist) that was obtained at the time of tumor resection from an area adjacent to the tumor. The diagnosis of a grade 4 astrocytoma was confirmed by an attending neuropathologist. Glioma and normal brain samples were sliced into 500-μm slices using a tissue slicer (MD4000; KrumDieck, Birmingham, AL, USA). These slices were grown in DMEM containing 10% FCS, 100 μg/ml penicillin and 100 μg/ml streptomycin.
Adenoviral vectors
CRAd-Survivin contains the human survivin promoter, termed CRAd-S, to drive E1 expression. The survivin-controlled E1 expression cassette was placed in the original E1 region of the Ad gene deleting the native E1 promoter as previously described [15]. The AdWT 5 fiber knob region was replaced by Ad type 3 knob [17]. Recombinant adenovirus was created on the basis of homologous recombination in 911 cells between a shuttle vector, pScs/PA/S, which carried the human survivin promoter driving E1 and a pVK700-based wild-type based Ad 5/3 backbone [11]. Replication deficient virus was created by replacing wild-type E1A region with a cassette containing the cytomegalovirus promoter driving the luciferase gene [11]. Replication competent viruses were selected from single plaques on 911 cells, whereas replication deficient virus were selected from 293 cells. They were then expanded in 293 cells and purified by double CsCl gradient ultracentrifugation [18].
Cytotoxicity assay
The toxicity assays performed in the present study were described previously [11]. Briefly, cells were plated on a 24-well plate at a density of 2.5 × 104 cells and were either mock infected or infected with AdWT, Ad 5/3 or CRAd-S-5/3 or replication deficient virus (def 5/3). Cells were infected at 1000, 100, 10 or 1 viral particles (vp)/cell in 0.5 ml of media containing 2% FCS. One hour after infection, media was removed and fresh media was added. Ten days later, the medium was aspirated and the cells were stained with crystal violet. Pictures were taken with a BioRad Chemidoc System (Bio-Rad, Hercules, CA, USA). Toxicity was also measured by lactate dehydrogenase (LDH) release using a CytoTox Cytotoxicity Assay kit (Promega, Madison, WI, USA) in accordance with the manufacturers' instructions.
Quantitative polymerase chain reaction (PCR)
Total DNA was extracted from infected/noninfected cells by DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Gene expression was quantified by quantitative PCR (qPCR) using SYBR GreenER PCR Master Mix (Invitrogen, Carlsbad, CA, USA) and primers recognizing E1A region as described previously [16]. For each experiment, a known amount of AdWT template DNA (106, 105, 104, 103, 102, 101, and 100 copies/μl) was used as a standard curve to quantify the E1A copy numbers of the experimental samples. The total volume of the reaction was 10 μl. DNA amplification was carried out using Opticon 2 system (Bio-Rad). All samples were performed in triplicate.
In vivo tumor formation and treatment
Ten-week-old female nude mice (n = 15 per group; Jackson Laboratories, Bar Harbor, ME, USA) were intracranially (i.c.) injected as per an established protocol approved by the IACUC of the University of Chicago. In brief, mice were anaesthetized with an intraperitoneal injection of ketamine hydrochloride (25 mg/ml)/xylazine (2.5 mg/ml) cocktail. The surgical site was prepared, a midline incision was made, and a 1-mm burr hole centred 2 mm posterior to the coronal suture and 2 mm lateral to the sagittal suture was made. Animals were placed in a stereotactic frame and injected with 3 × 105 cells in a 5-μl volume. Ten days after tumor implantation, 1000 vp per cell of the three different virus, AdWT, Ad 5/3 and CRAd-S-5/3 or sterile phosphate-buffered saline (PBS) (for mock control) were injected into the tumors in a 5-μl volume. Mice were examined daily and those that exhibited signs of disorientation and cachexia were killed.
Statistical analysis
The significant differences between groups were assessed by calculating Student's t-value. p < 0.05 was considered statistically significant. For in vivo results, a Kaplan–Meier survival analysis was plotted and statistical analysis was performed using a Log rank test. Again, p < 0.05 was considered statistically significant.
Results
Enhanced cytotoxicity, infectivity and replication of CRAd-Survivin-5/3 compared to AdWT vector
To assess the in vitro cytotoxic effect of our vector, we plated cells in a 24-well plate and infected them in a serial dilution as described in the materials and methods. Four different viruses were used: CRAd-S-5/3, Ad 5/3, AdWT and replication defective 5/3. In all four cell lines tested, CRAd-S-5/3 showed a ten- to 100-fold increase in toxicity versus AdWT or Ad 5/3 as demonstrated by crystal violet staining (Figure 1A). This killing was the highest in A172 cell line with cytotoxic effects observed at 10 vp/cell, and least in the U118MG cell line with cytotoxic effects observed at 100 vp/cell only. U87MG and No. 10 cell lines exhibited robust cell killing at 100 vp/cell, which declined at 10 vp/cell.
Figure 1.
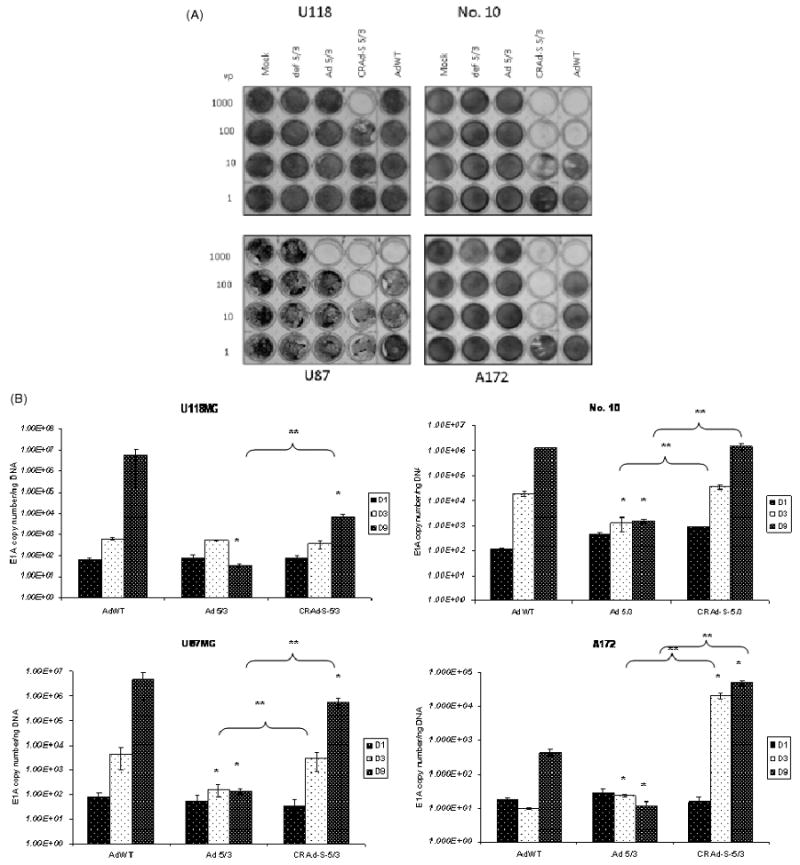
(A) Crystal violet staining of adenovirus infected cells. Cells were infected with adenoviral vectors in a serial dilution. Ten days after infection, cells were stained with crystal violet to assess comparative cytotoxic effect of AdWT, CRAd-Survivin-5/3, Ad 5/3, def 5/3 or mock in transfected GBM cell lines. (B) Replication of adenoviral vectors. U87MG, U118MG, A172 and No. 10 cell lines were infected with 10 vp/cell of the constructs described above and the cells were harvested at days 1, 3 and 9. Total DNA was isolated from these cells and viral replication was measured by qPCR using primers for the Ad 5 E1A gene. Asterisks (*, **) indicate a statistically significant difference (p < 0.05) at specified time points in gene levels relative to AdWT and Ad 5/3, respectively
To confirm that our modified survivin promoter-driven virus replicates better than a virus modified simply at the fiber-knob region (Ad 5/3), we used a qPCR analysis to determine viral replication (Figure 1B). Briefly, cells were infected at 10 vp/cell and harvested in triplicates at 1, 3 and 9 days after infection. In No. 10 cell line, but not U87MG, CRAd-Survivin-5/3 replication was comparable to AdWT at between 105 and 106 viral copies per ng DNA at day 9 after infection. The replication of Ad 5/3 was lower at day 3 compared to CRAd-S-5/3 and AdWT in both No. 10 and U87MG cell lines and was 1000-fold lower in No. 10 cell line and 10,000-fold lower in U87MG (p < 0.05) at day 9 compared to CRAd-S-5/3. In U118MG cell line, CRAd-Survivin-5/3 replication was comparable to AdWT at days 1 and 3 post-infection; however, at day 9, it was lower by approximately 1000-fold (p < 0.05). The A172 cell line had the best replication for CRAd-S-5/3, and was higher than AdWT by 1000-fold at day 3 and by 100-fold at day 9. Ad 5/3 replication was approximately 1000-fold lower in this cell line on both days 3 and 9 compared to CRAd-S-5/3. We also examined the replication of the promoter modified adenovirus having wild-type fiber (AdWT S) and found that it replicates at a sub-optimal level at least 100–10,000-fold lower than CRAd-S-5/3 in all four cell lines analysed (data not shown).
To assess toxicity induced by these viruses in normal tissues in vitro, we used NHA. We infected NHAs with AdWT, Ad 5/3 and CRAd-S-5/3 and quantified E1A levels as a marker of viral replication at days 1 and 3. We also investigated the toxicity induced by these viruses at day 3 after infection in a separate experiment. We found that the replication of CRAd-S-5/3 was ten- to 100-fold lower than AdWT at days 1 and 3 (p < 0.05) (Figure 2A). Ad 5/3 replication was approximately the same as AdWT at day 3. When we analysed toxicity using LDH release as a measure of cell death, AdWT had the highest toxicity of all samples analysed; AdWT toxicity was at least 30–40% higher than CRAd-Survivin-5/3-induced cell death and approximately 60% higher than Ad 5/3 (p < 0.05) (representative data are shown in Figure 2B). The fact that CRAd-S-5/3 had lower cytotoxic effect than AdWT was encouraging and prompted us to investigate primary GBM samples and matched normal brain tissues in an ex vivo manner.
Figure 2.
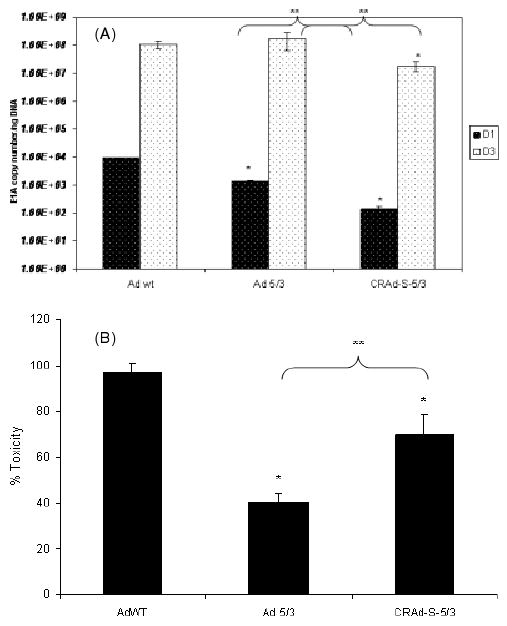
(A) Replication of adenoviral vectors in normal human astrocytes. NHA were infected with AdWT, Ad 5/3 and CRAd-Survivin-5/3 at 10 vp/cell. Cells were harvested at days 1 and 3 post-infection, DNA isolated, and viral replication was measured by qPCR. Asterisks (*, **) indicate a statistically significant difference (p < 0.05) at specified time points in gene levels relative to AdWT and Ad 5/3, respectively. (B) Toxicity to NHA as measured by LDH release assay. After infection by viral vectors mentioned above, cell death was measured using an LDH release assay. Asterisks (*, **) indicate a statistically significant difference (p < 0.05) in toxicity relative to AdWT and Ad 5/3, respectively
Primary GBM samples are targeted by CRAd-Survivin-5/3 ex vivo
Having established CRAd-Survivin-5/3 as a viable adenoviral vector to target gliomas in vitro, we next examined the efficacy of this modified adenovirus to target primary tumor samples ex vivo. Freshly-resected glioma samples from patients undergoing surgery were washed in normal saline solution. The tumors were then cut into 500-μm slices using a tissue-slicer to increase the surface area of the samples for infection with virus. Equal number of slices were infected with CRAd-Survivin-5/3, AdWT and Ad 5/3 viruses, as described above. For each sample, the experiment was performed twice: once to determine toxicity and again to assess the replication of these viruses in primary tissues. Three independent experiments using different tumor samples were performed. In primary brain tumor samples, CRAd-Survivin-5/3 showed a 30% increase in toxicity compared to AdWT and a 10% increase versus Ad 5/3 as measured by LDH release (representative data are shown in Figure 3A). The toxicity assay was performed 3 days after infection and 70% of cells were dead in the CRAd-Survivin-5/3 group, 40% in the AdWT group, and 60% in the Ad 5/3 group. Although the toxicities of Ad 5/3 and CRAd-S-5/3 were not significantly different from each other, they were significantly increased versus AdWT (p < 0.05). Interestingly, when we examined the replication of these viruses in the same samples, we found that the replication of CRAd-Survivin-5/3 was initially higher by approximately five-fold versus AdWT (day 1) but progressively decreased and, by day 3, was approximately ten-fold lower than AdWT (Figure 3B). On both days 1 and 3, values among the different groups were significantly different (p < 0.05). This effect was observed in all the samples that were analysed.
Figure 3.
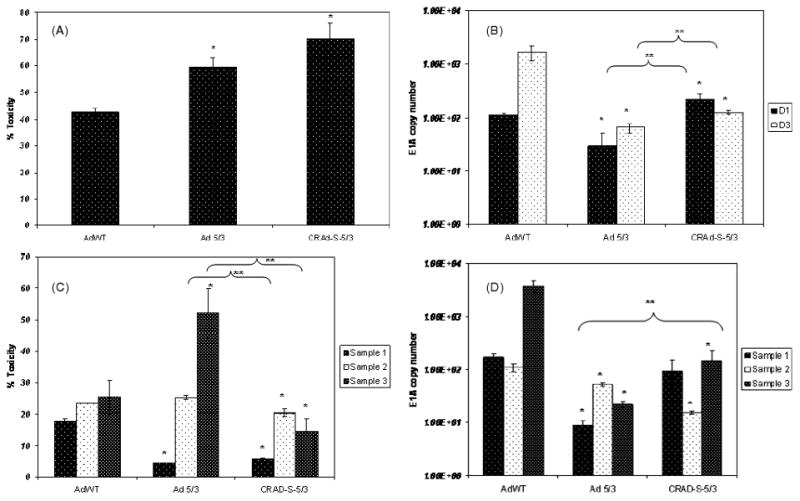
Ex vivo analysis of primary patient samples. (A) Toxicity and (B) replication of viral vectors AdWT, Ad 5/3 and CRAd-Survivin-5/3 in primary GBM samples. Infection was carried out as described earlier and tissue slices were analysed for LDH release to measure toxicity or with qPCR for replication. A representative example is shown. (C) Toxicity and (D) replication of viral vectors in matched normal brain tissues. Asterisks (*, **) indicate a statistically significant difference (p < 0.05) at specified time points in toxicity/gene levels relative to AdWT and Ad 5/3, respectively
In patient-matched, normal brain samples, the cytotoxicity of CRAd-Survivin-5/3 was 10–15% lower than that induced by AdWT at day 3 after infection in all three samples analysed (p < 0.05) (Figure 3C). We also investigated the replication of these viruses in normal brain slices, and found that the replication of CRAd-Survivin-5/3 was also decreased in these samples by ten- to 50-fold (Figure 3D). Only sample 1 was not statistically lower than AdWT replication (p < 0.05). Of note, Ad 5/3 had a higher toxicity than AdWT in sample 3, although it had lower replication as assessed by E1A copy numbers. Taken together, these results demonstrate that CRAd-Survivin-5/3 is an effective oncolytic virus in GBM samples and exhibits an enhanced safety profile in normal tissue versus AdWT.
CRAd-Survivin-5/3 virotherapy extends survival in an in vivo model of intracranial tumor
To further establish that CRAd-Survivin-5/3 is an effective adenoviral therapy against GBM, we investigated the ability of this virus to improve survival in an in vivo model. Nude (athymic) mice were i.c. implanted with 3 × 105 U87MG cells. This cell line is established as a model for i.c. tumor and, in our in vitro experiments exhibited approximately ten-fold higher cell killing than AdWT. Seven days after implantation of the cells, a few representative mice were killed to demonstrate tumor growth inside the brain. The remaining mice were then divided into four groups: (i) a mock group receiving an i.c. injection of 5 μl of PBS; (ii) AdWT; (iii) Ad 5/3; and (iv) CRAd-Survivin-5/3. The last three groups received an i.c. injection of 100 vp/cell of the respective virus. Mice were monitored daily and killed when they appeared disoriented and cachectic. Planned sacrifices were made at days 1, 3, 7 and 10 to assess viral replication. Late-stage viral replication was also investigated using a tumor sample from mice dying after 35 days. Mice in the mock group survived the shortest time span from tumor implantation, with deaths at regular intervals from 22 days onward and a median survival of 28.5 days. However, the group that received the virus survived significantly longer. The median survival rates for AdWT-, Ad 5/3- and CRAd-S-5/3-injected groups were 35, 36 and 43.5 days, respectively (p < 0.05) (Figure 4A). At day 45, all mice from the mock-injected group either died or had to be killed to ease pain and suffering. At this point, 30% of AdWT injected group and 20% of the Ad 5/3-injected group survived; however, 50% of the CRAd-S-5/3-injected group still remained symptom free (p < 0.05).
Figure 4.
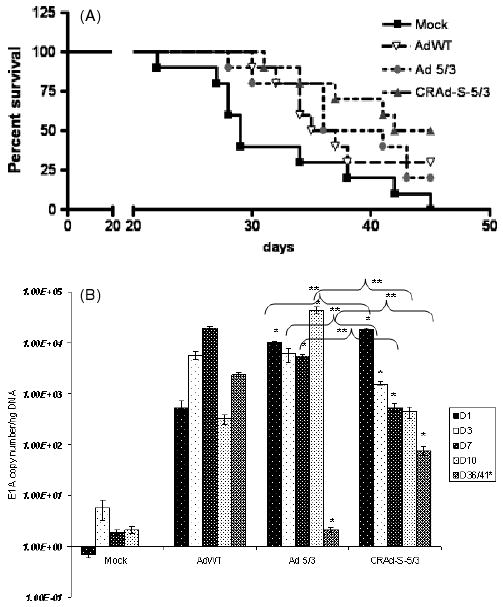
In vivo analysis of CRAd-Survivin-5/3. (A) Survival graph showing different groups days after virus injection. (B) Replication of AdWT, Ad 5/3 and CRAd-Survivin-5/3 in mouse brain tumors. Animals were killed at days shown and total DNA was isolated from the brains. Replication of adenovirus was measured by qPCR. Asterisks (*, **) indicate a statistically significant difference (p < 0.05)at specified time points in gene levels relative to AdWT and Ad 5/3, respectively
In mice that were deliberately killed at time points indicated above, we isolated DNA from the brain containing tumor and determined E1A copy numbers. Figure 4B shows that, at day 1, CRAd-Survivin-5/3 had the highest level of E1A copies at 104 viral copies per ng tissue DNA. This is more than 100-fold higher than AdWT. From day 3 onwards, however, this level fell off to 103 at day 3 and 1.5 × 102 at days 7 and 10. This observation was similar to that observed in primary GBM samples and is most likely a result of a decrease in i.c. tumor size and cell death. Interestingly, at day 36, AdWT and CRAd-Survivin-5/3 were still actively replicating at 104 and 102, respectively, whereas, at a comparable time point (day 41), Ad 5/3 showed no actively replicating virus inside the mouse brain. Overall, our studies performed in vivo demonstrated an increased cytotoxicity leading to a longer survival of animals in an i.c. model of glioma.
Discussion
We characterized the efficacy of a novel conditionally replicating adenovirus that has the fiber domain of Ad 5 and the knob from Ad 3. The E1A gene of this virus is driven by a tumor-specific promoter, survivin. The complexity of this adenoviral vector confers specificity in targeting as well as in its replication once inside a tumor cell.
Using glioma cell lines, we compared the virolytic activities of CRAd-S-5/3 with AdWT and Ad 5/3. Our data shows the enhanced transduction of CRAd-Survivin-5/3 compared to the wild-type adenovirus. We also compared the replication of these viruses to examine the effect of the survivin promoter-driven viral replication leading to cell killing. Our data suggests that, in vitro, the dual modification in CRAd-Survivin-5/3 enhances viral tropism to glioma cells that have higher levels of CD46 receptors. In addition, survivin-driven E1A gene expression increases the replication of this virus and the combination of both is a potent tool towards specificity in tumor targeting.
One salient feature of the present study is that the success of virotherapy depends on the selectivity of the agent. In our case, this selectivity is achieved by the use of tumor selective promoter (survivin), as well as transductional modification of the adenoviral fiber (with Ad 3 knob). We have previously shown the inherent variability of both survivin [11,15] and Ad 3 receptor expression [12,13] in human gliomas. It is precisely such differences in the expression of survivin and/or CD46/CD80/CD86 that explain the differences in the cytotoxic effect expressed by the virus in different cell lines. This finding is important because it points the way towards individualized virotherapies based on the molecular profile of the tumor.
In NHA, the toxicity and replication of CRAd-S-5/3 was significantly (p < 0.05) lower than AdWT. This was expected because our virus was designed to be relatively ineffective against normal tissues. We also used normal brain tissues obtained from patients to examine the toxicity of our virus ex vivo. Overall, our virus showed lowered toxicity and lower replication than AdWT.
In vivo, we implanted nude mice with intracranial tumors and injected these tumors with CRAd-Survivin-5/3, AdWT, Ad 5/3 virus, and a mock group. In our study, although the mock injected group had a half-life of 28.5 days, those that were infected with CRAd-Survivin-5/3 survived significantly longer, with a half-life of 43.5 days. By contrast, AdWT and Ad 5/3 had median survival of 35 and 36 days, respectively. Although we did not observe continued higher replication of CRAd-Survivin-5/3, we believe that this might be the result of an decrease in i.c. tumor size associated with cell lysis. This is also supported by the number of survivors in the CRAd-S-5/3 group (50%) that are symptom free after 45 days.
Oncolytic adenoviruses are being increasingly used in studies both in vitro and in vivo for many different cancers [19,20]. However, their potential in gliomas is often understated. Because of their tendency to infiltrate the brain parenchyma, gliomas are often not completely resectable and tend to recur frequently. CRAds present a valuable tool for killing these ‘rogue’ cells, particularly if more efficient methods of delivery can be devised. However, just as a single drug cannot comprise a cure for all patients, different CRAd vectors need to be tested in pre-clinical settings to achieve a combinatorial effect for treating gliomas. Our vector, CRAd-S-5/3, is one such candidate that needs to be investigated further.
Acknowledgments
This work was supported by the National Cancer Institute (R01-CA122930), the National Institute of Neurological Disorders and Stroke (K08-NS046430), the Alliance for Cancer Gene Therapy Young Investigator Award, and the American Cancer Society (RSG-07-276-01-MGO).
References
- 1.Sathornsumetee S, Rich JN. Designer therapies for glioblastoma multiforme. Ann NY Acad Sci. 2008;1142:108–132. doi: 10.1196/annals.1444.009. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Chiocca EA, Abbed KM, Tatter S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958–966. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 4.Yazaki T, Manz HJ, Rabkin SD, et al. Treatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1. Cancer Res. 1995;55:4752–4756. [PubMed] [Google Scholar]
- 5.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 6.Temme A, Herzig E, Weigle B, et al. Inhibition of malignant glioma cell growth by a survivin mutant retrovirus. Hum Gene Ther. 2005;16:209–222. doi: 10.1089/hum.2005.16.209. [DOI] [PubMed] [Google Scholar]
- 7.Allen C, Paraskevakou G, Iankov I, et al. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Mol Ther. 2008;16:1556–1564. doi: 10.1038/mt.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selznick LA, Shamji MF, Fecci P, et al. Molecular strategies for the treatment of malignant glioma – genes, viruses, and vaccines. Neurosurg Rev. 2008;31:141–155. doi: 10.1007/s10143-008-0121-0. discussion 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesniak MS. Gene therapy for malignant glioma. Expert Rev Neurother. 2006;6:479–488. doi: 10.1586/14737175.6.4.479. [DOI] [PubMed] [Google Scholar]
- 10.Zheng S, Ulasov IV, Han Y, et al. Fiber-knob modifications enhance adenoviral tropism and gene transfer in malignant glioma. J Gene Med. 2007;9:151–160. doi: 10.1002/jgm.1008. [DOI] [PubMed] [Google Scholar]
- 11.Ulasov IV, Rivera AA, Sonabend AM, et al. Comparative evaluation of survivin, midkine and CXCR4 promoters for transcriptional targeting of glioma gene therapy. Cancer Biol Ther. 2007;6:679–685. doi: 10.4161/cbt.6.5.3957. [DOI] [PubMed] [Google Scholar]
- 12.Ulasov IV, Tyler MA, Zheng S, et al. CD46 represents a target for adenoviral gene therapy of malignant glioma. Hum Gene Ther. 2006;17:556–564. doi: 10.1089/hum.2006.17.556. [DOI] [PubMed] [Google Scholar]
- 13.Ulasov IV, Rivera AA, Han Y, et al. Targeting adenovirus to CD80 and CD86 receptors increases gene transfer efficiency to malignant glioma cells. J Neurosurg. 2007;107:617–627. doi: 10.3171/JNS-07/09/0617. [DOI] [PubMed] [Google Scholar]
- 14.Das A, Tan WL, Teo J, et al. Expression of survivin in primary glioblastomas. J Cancer Res Clin Oncol. 2002;128:302–306. doi: 10.1007/s00432-002-0343-4. [DOI] [PubMed] [Google Scholar]
- 15.Van Houdt WJ, Haviv YS, Lu B, et al. The human survivin promoter: a novel transcriptional targeting strategy for treatment of glioma. J Neurosurg. 2006;104:583–592. doi: 10.3171/jns.2006.104.4.583. [DOI] [PubMed] [Google Scholar]
- 16.Nandi S, Ulasov IV, Tyler MA, et al. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008;68:5778–5784. doi: 10.1158/0008-5472.CAN-07-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tekant Y, Davydova J, Ramirez PJ, et al. Oncolytic adenoviral therapy in gallbladder carcinoma. Surgery. 2005;137:527–535. doi: 10.1016/j.surg.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 18.Graham FL, Smiley J, Russell WC, et al. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Gao L, Yeagy B, et al. Virus combinations and chemotherapy for the treatment of human cancers. Curr Opin Mol Ther. 2008;10:371–379. [PubMed] [Google Scholar]
- 20.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]