

Abstract
Insulin, a small hormone protein comprising 51 residues in two disulfide-linked polypeptide chains, adopts a predominantly α-helical conformation in its native state. It readily undergoes protein misfolding and aggregates into amyloid fibrils under a variety of conditions. Insulin is a unique model system in which to study protein fibrillization, since its three disulfide bridges are retained in the fibrillar state and thus limit the conformational space available to the polypeptide chains during misfolding and fibrillization. Taking into account this unique conformational restriction, we modeled possible monomeric subunits of the insulin amyloid fibrils using β-solenoid folds, namely, the β-helix and β-roll. Both models agreed with currently available biophysical data. We performed molecular dynamics simulations, which allowed some limited insights into the relative structural stability, suggesting that the β-roll subunit model may be more stable than the β-helix subunit model. We also constructed β-solenoid-based insulin fibril models and conducted fiber diffraction simulation to identify plausible fibril architectures of insulin amyloid. A comparison of simulated fiber diffraction patterns of the fibril models to the experimental insulin x-ray fiber diffraction data suggests that the model fibers composed of six twisted β-roll protofilaments provide the most reasonable fit to available experimental diffraction patterns and previous biophysical studies.
Introduction
Protein misfolding under conditions that destabilize the native state, and subsequent fibrillar aggregation of the aberrant conformers, are involved in the pathogenic processes of many neurodegenerative and systemic diseases, such as Alzheimer's, the prion diseases, and type II diabetes (1,2). Misfolded fibrillar proteins share common structural characteristics even when the native proteins are evolutionarily or structurally unrelated. This observation suggests that the tendency of misfolded proteins to form ordered aggregates, known as amyloid, is a generic property of polypeptide chains (3). Amyloid fibrils are generally unbranched, protease-resistant filaments with dominant β-sheet structures organized in a cross-β fashion, in which the β-strands run perpendicular to the fibril axis (3–5). Electron microscopy (EM), cryo-EM, and atomic force microscopy (AFM) have revealed that the protofilament units are compact β-strand repeats with diameters typically ranging from 15 to 40 Å (5–7). Although amyloid fibrils share common structural properties, their overall morphology can vary depending on the precursor protein, conditions of fibrillization, and the number and arrangement of protofilaments (5,7–9).
Over the last few decades, a great deal of effort has been invested in trying to gain a structural understanding of the mechanisms of protein misfolding and subsequent amyloid fibril formation. For example, recent x-ray crystallography studies of microcrystals formed from short peptides of amyloid-related proteins have revealed a distinctive packing in the β-structure core (10,11). EM and solid-state NMR studies of Aβ1–40 and the fungal HET-s prion protein showed that the misfolded amyloid conformation may adopt an architecture that is structurally unrelated to the native conformation, but similar to β-structures previously observed in nature, namely β-solenoid folds (12–17). The limitations of traditional biophysical methods for studying misfolded proteins have led to the use of molecular modeling to probe plausible structural solutions. Recently, an investigation of the amyloid structure of the mammalian prion protein was reported in which experimental fiber diffraction data was compared to molecular models of prion amyloid (18).
Insulin, a small hormone protein consisting of two polypeptide chains, adopts an α-helical conformation in its native state. The insulin sequence is well conserved among mammalian species, with few variations. Two polypeptide chains are linked by two interchain and one intrachain disulfide bridges (19). Although insulin does not appear to be directly involved with any known human amyloid diseases, native insulin readily converts to an inactive fibrillar form under a wide range of conditions (19). In one clinical study, amyloid-fibril-like deposits containing insulin were found at sites of insulin injection in a diabetic patient (20). An interesting feature of insulin is that its three disulfide bridges are retained in the in vitro and ex vivo fibrillar form (5,19,21). Thus, these disulfide bonds must constrain the possible conformational rearrangements during the α-helix-to-β-sheet transition (21). This conformational constraint makes insulin a unique model system for studying protein misfolding and subsequent amyloid fibrillization.
Native insulin often exists as a zinc-coordinated trimer of dimers under physiological conditions or, alternatively, as a dimer or tetramer in a zinc-free environment (19,21,22). Under conditions of low pH and high temperature, insulin readily dissociates into monomers, and subsequently assembles into fibrils or aggregates (5,19). Insulin fibril formation was first observed in the 1940s, and the nucleation and elongation process was first reported by Waugh (23). In 1972, a fiber diffraction study by Burke et al. showed the cross-β structural pattern of insulin (24). Studies using NMR have demonstrated that the native insulin structure is disrupted before fibrillization occurs (25). Recent mass spectrometry, AFM, hydrogen exchange, and small-angle x-ray scattering (SAXS) studies have shown the formation of small oligomeric species consisting of up to 12 insulin molecules. These species act as elongating units that further assemble into larger irreversible aggregates and ultimately into protofilaments and mature fibrils (26–29). Studies using Fourier transform infrared (FTIR) and circular dichroism (CD) spectroscopy indicate that the initial aggregates retain their predominantly α-helical structure, but that there is a subsequent conversion to a β-sheet structure as the fibrillization process continues (9).
In this study, we modeled a β-rich insulin monomer using either a β-roll or β-helix fold as templates, and assessed the suitability of each of these models as possible subunits of fibrillar insulin. We examined each model based on available biophysical data, and studied the relative stability of the model structures using molecular dynamics (MD) simulations. Fibrillar insulin models were constructed using each of the monomer models, and fiber diffraction patterns were simulated. By comparing the simulated fiber diffraction patterns with those obtained experimentally from insulin fibers, we were able to assess the suitability of each model.
Materials and Methods
Molecular modeling of the insulin monomer
The β-roll and β-helix models of C-terminally truncated human insulin (A chain, 1–21; and B chain, 1–22) were built using the iron transporter stabilizer protein SufD (Protein Data Bank (PDB) ID code 1VH4) and the C-terminal domain of N-acetyglucosamine 1-phosphate uridyltransferase GlmU (PDB ID code 1HV9) as templates. For the β-roll model, the structural coordinates from residues 254–274 and 230–249 of 1VH4 were copied and reassembled onto the scaffold so as to follow the selected threading of the insulin A and B chains, respectively (see Results). For the β-helix model, the coordinates from residues 309–323 and 293–312 of 1HV9 were used to build the A and B chains, respectively. β-roll and β-helix polymeric models were constructed by stacking four repeated subunits on top of each other with an intermolecular distance between two neighboring subunits of 4.8 Å. (For details, see Methods in the Supporting Material).
Molecular dynamics simulation of monomeric insulin models
All simulations were performed with the GROMACS software package (30), using the GROMMOS 43a3 force field (31), as described in previous studies (32,33). The model structures used in the simulations were the C-terminally truncated β-roll and β-helix insulin. Models were solvated individually in octahedron boxes filled with water molecules. Sodium ions were used to electroneutralize the system. Solutes, solvent, and counterions were coupled independently to reference temperature baths at 300 K, 345 K, and 375 K, and the pressure was maintained by weakly coupling the system to an external pressure bath at 1 atm. To emulate the acidic, fibrillogenic condition of pH ∼2, two histidine residues, one glutamate residue, and the C-terminus were protonated. Analysis was performed using the built-in programs of the GROMACS software package (30). (For details, see Methods in the Supporting Material).
Molecular modeling and fiber diffraction simulations of the insulin fibril
Molecular models of the fibrils were built by translating the β-roll and β-helix monomeric units along the fibril axis and rotating them around the axis. The degree of rotation varied depending on the helical period of the fibril, which is caused by β-sheet twisting. As a result, two or more protofilaments were used to form a fibril by winding around each other. All insulin fibril models were constructed using InsightII (Accelrys, San Diego, CA) and DISORDER (35), based on structural constraints, such as the helical period, number of protofilaments in the fibril, and the relative positions and orientations of the monomers in the fibril cross section (5,26).
The simulation and analysis of fiber diffraction patterns were conducted using DISORDER (35). For every fibril model, the diffraction patterns, with varying degrees of disorientation between fibrils, were simulated. The value of the disorientation parameter varied from 10–25°. The meridional and equatorial profiles of the simulated and experimental fiber diffraction patterns were then compared by calculating the least-squares residuals (35). Simulated and experimental diffraction patterns were visualized using FIT2D (36). (For details, see Methods in the Supporting Material).
Results
Molecular modeling of the monomeric subunit of the insulin fibril
In modeling the insulin monomeric subunit, we threaded (37) the insulin sequence onto the known β-solenoid folds, namely the parallel β-roll and β-helix, found in the PDB database (Fig. 1 and Fig. S1 in the Supporting Material). In general, β-rolls contain two β-strands per rung with four-residue turns (arcs), which allow the strands to change direction by 180°, whereas the β-helices are composed of three β-strands per rung with three-residue turns (arcs) and a turn angle of ∼120° (15,38). As mentioned above, one of the unique features of insulin is its three disulfide bridges within chains A and B, all of which are retained in the fibrillar state (19,21). Previous mutagenesis and deletion experiments have shown that the C-terminal region of chain B (residues 23–30) is not required for insulin fibrillization (19,39). With these structural constraints in mind, the threading of insulin chains A and B onto a β-roll or β-helix produced relatively simple and unique solutions. Few threading solutions could satisfy the disulfide bridge constraints, structural features of the β-solenoid folds, and the available experimental data, including experiments using insulin single-chain analogs and isomers, fibrillization of isolated chains A and B, and biophysical characterization of insulin fibers using cryo-EM, CD, and FTIR (5,9,15,21,33,40–42).
Figure 1.
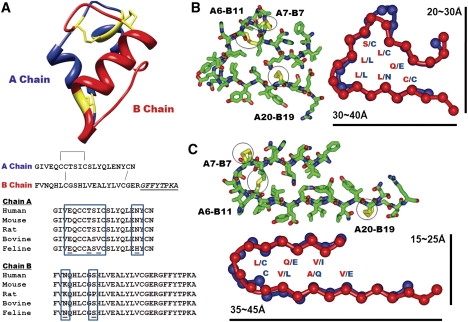
Insulin monomer structure and β-solenoid-based monomeric subunit models for the insulin amyloid fibril. (A) Insulin monomer structure (PDB code 1GUJ), with the sequences of chains A and B. One intrachain and two interchain disulfide bridges are shown in yellow in the structure and by connecting lines in the sequences. The C-terminal region of chain B, underlined and in italic, is not involved in amyloid fibrillization. Multiple sequence alignment of the insulin sequences from five different mammalian species is shown, with sequence variations in the boxes. (B and C) Cross-sectional views of insulin β-solenoid monomeric models, including the C-terminally truncated β-helix (B) and β-roll (C) insulin monomeric subunit models, based on templates from 1HV9 (segment 293–323) and 1VH4 (segment 230–274), respectively. Stick figures are shown with the side chains, with disulfide bridges in yellow. Schematic figures are shown with the amino acid residues facing inside, with approximate dimensions of the structures. Chains A and B are shown in blue and red, respectively.
In the β-roll model, insulin was threaded into two rungs, with each rung comprising one short and one long β-strand connected by tight turns and a disulfide bridge. In the β-helix model, the insulin sequence was also threaded into two rungs of a β-helix with three short β-strands per rung, forming a triangular shape with an opening at one corner. Although chain B of the β-helix model contains typical β-helical turns as observed in nature, chain A contains an unusual four-residue turn with a disulfide bridge, connecting the adjacent β-sheets. Although the types of disulfide bridges introduced into the β-solenoid models are not commonly found in β-solenoid proteins, they are structurally feasible. However, introducing disulfide bridges at the turn regions of the insulin β-helix model resulted in slight distortions to the β-sheets.
Both insulin models satisfied the sequence and structural features of the β-roll and β-helix folds, such that residues facing the inner core of the structures were mostly similar hydrophobic or small polar residues that stacked on top of each other (15,33,38). In particular, the inner-core residues of the β-helix model were well packed, with similar residues stacking in two chains. The inner-core packing of the β-roll model was less consistent than that of the β-helix model, with glutamine and glutamate residues residing in the core; however, these residues are occasionally found in native β-roll proteins. In addition to structural constraints, the β-solenoid insulin models may also explain the exclusion of the C-terminal region of the B chain from the fibrils. In both models, the C-terminal region (B chain, residues 23–30) did not fit onto the β-solenoid structures due to side-chain constraints. It therefore made logical sense to place this stretch of sequence outside of the β-solenoid core.
The β-solenoid models of the insulin fibrillar subunit also agreed well with studies characterizing the fibrillization of insulin disulfide isomers and isolated chains A and B, and single-chain analogs (21,41,42). For example, insulin structural isomers with alternative pairings of disulfide bridges, which were shown to form amyloid fibrils, were compatible with both the β-roll and β-helix models. These disulfide isomers retained the A20-B19 pairing, whereas the A6-A11 and A7-B7 pairings were alternated with one another. Although these alternative disulfide pairings are not ideally suited to the insulin β-solenoid models shown in Fig. 1, they can be tolerated by using different registers at the turn regions of both models. The cross-seeding experiments using native and isomeric insulin (42) also agreed with compatibility of both the native and isomeric insulin with the β-solenoid models. Furthermore, the β-solenoid models agree with isolated single chain insulin fibrillization experiments. One rung of the β-solenoid structure formed by the isolated single chain of either chain A or chain B can further aggregate into homogeneous protofilaments, with morphologies and dimensions different from those of the wild-type insulin fibrils but retaining the ability to cross-seed with wild-type insulin (21,40,41). Conversely, a single-chain analog in which the C-terminus of chain B was tethered to the N-terminus of chain A was shown to lack fiber-specific α-to-β transition. This can be explained also by the inability of the single-chain analog to adopt a β-solenoid conformation, as such an insulin sequence is not compatible with the sequence and structural requirements of the fold.
A previous cryo-EM study showed that protofibrillar insulin was composed of relatively flat β-sheets with typical sizes ranging from 30–40 Å in diameter (5). Although the β-helix model was relatively symmetrical, with dimensions of 20–30 Å × 30–40 Å, the β-roll model had a long rectangular shape with a width of 15–25 Å and a length of 35–45 Å (Fig. 1, B and C). Although the C-terminal region of chain B is not shown in the figures and was not used to calculate the dimensions, the size estimates of both models were in fair agreement with the approximate dimensions of the insulin protofilament calculated from the cryo-EM experiments. In addition, the β-solenoid structures of insulin fibrillar subunit models can provide the characteristic cross-β pattern and β-signatures, which were consistently observed in fiber diffraction, FTIR, and CD studies of insulin and other fibrillar protein aggregates (9,26,43).
Structural stability of the β-solenoid models of fibrillar insulin subunits
We conducted 40-ns MD simulations using the β-roll and β-helical insulin monomers with intact disulfide bridges under acidic conditions, using a range of temperatures (300–375 K) to emulate the experimental conditions of in vitro insulin fibrillization (see Materials and Methods). Analysis of the positional root mean-square deviations (RMSDs) of the backbone relative to the starting structures indicated that each model reached equilibrium after ∼13 ns. The relative stabilities of the model structures estimated by RMSD revealed much larger differences among the insulin β-helix models than among the β-roll models at the three different temperatures (Fig. 2). The β-helix models showed an increase in RMSD of ∼2 Å when the temperature was increased by 30–45 K. The average RMSDs for Cα, and the interchain backbone-backbone hydrogen bonds, were calculated from three independent trials for both β-solenoid models (Table S1). Although little difference was seen between the β-roll and β-helix models from the interchain hydrogen bonds before and after a 40-ns simulation, the average RMSDs of Cα of the two models consistently showed significantly different stability. This result provides limited evidence that the β-roll model may be relatively more stable than the β-helix model at increased temperatures. For each model, the final structures after a 40-ns MD simulation also revealed that although the β-roll models maintained their initial fold architecture relatively well, the β-helix model displayed an early-stage disruption of the β-helical fold at 345 K and a structural transition to a pseudo-β-roll-like structure at 375 K with two strands per rung with four-residue turns allowing the strands to change direction by ∼180° (Fig. 2 B). We also conducted 100-ns MD simulations of β-roll and β-helix monomer models at 375 K to investigate the structural conversion between these two structures. However, there was no definitive evidence from this particular experiment to suggest that a structural conversion between these two conformations could occur (data not shown).
Figure 2.
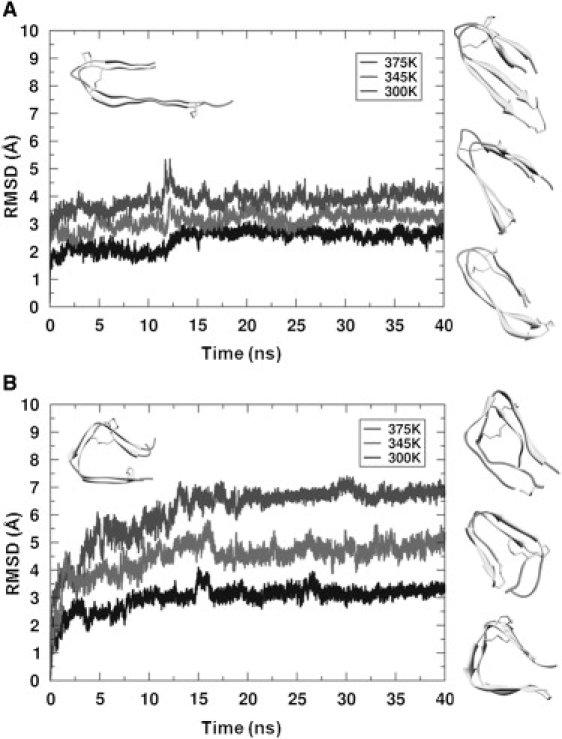
Molecular dynamics simulation of the β-roll and β-helix insulin monomeric models with increasing temperature in acidic conditions. Backbone RMSDs of the β-roll (A) and β-helix (B) models relative to their initial structures (upper left corner) as a function of simulation time at 300 K, 345 K, and 375 K. Final structures are shown at the right in order of increasing temperature (lower to upper).
The relative stability of polymeric constructs comprising four repeated units from either the β-roll or β-helix insulin model was also explored using MD simulations to investigate the stability of the β-solenoid insulin oligomers and of the β-solenoid insulin monomers in the oligomeric state. The conformational stability of β-helical polymeric constructs was previously examined to assess their ability to retain their organization and self-assembly status (44,45). We conducted MD simulations at 345 K in acidic conditions for 15 ns. The relative stabilities of the β-roll and β-helix polymeric models were compared using the RMSD of Cα calculated relative to the starting models, and the monomeric subunits located in the middle of polymeric models (second and third subunits) were compared to the initial structures (Fig. S2, A and B). Although the MD simulation of the polymeric models showed improved structural stability relative to the stability of monomeric models in the monomer simulations, the β-roll polymeric model still showed a slightly higher stability than the β-helix polymeric model. The final structures of the second units within the polymeric constructs were closely examined to understand the changes in backbone angle and side-chain packing after 15 ns of MD simulation (Fig. S2 C). Unlike in the single-unit simulation, the hydrophobic cores of these subunits were not exposed to the solvent. In the case of the β-roll subunit, the side-chain packing was relatively well maintained after 15 ns simulation. Most of the structural deviation originated from movements of the C-terminal region of chain B. In contrast, the final structure of the β-helix subunit showed a large degree of structural deviation at the turn region where the two disulfide bonds reside, and this deviation resulted in movements of the backbone and the overall structure of the polymeric construct.
Molecular models of insulin fibrils
We modeled the insulin fibril using both the β-roll and β-helix insulin subunit models as building blocks. Two types of fibrillar molecular models were explored: 1), a twisted-stack arrangement (Fig. 3, A and B); and 2), a helical architecture (Fig. 3 C), based on the previous cryo-EM and SAXS data, respectively. Twenty-three different insulin fibril models were constructed with three different architectural schemes. Of these fibril models, 14 are shown in Fig. 3.
Figure 3.
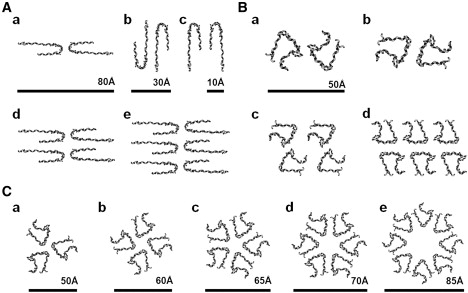
Quaternary structure arrangements to simulate insulin amyloid fibrils. (A) Cross-sectional view of the β-roll-based insulin fibril models with two (a–c), four (d), and six (e) protofilaments. (B) β-helix-based models with two (a and b), four (c), and six (d) protofilaments. Fibril models were constructed based on the measured layer line spacings calculated from the diffraction patterns of insulin amyloid fibrils (8). (C) Trimeric (a), tetrameric (b), pentameric (c), hexameric (d), and octameric (e) models of a β-helix-based protofilament. Models were constructed based on helical periodicity calculated from the insulin oligomeric species observed by SAXS (26).
According to a previous cryo-EM study, two to six protofilaments can be intertwined to make a larger fibril with approximate dimensions of 30 × 40 Å for a single protofilament (5). Since the diameters of the β-solenoid models ranged from ∼20–40 Å, the parallel stacking of monomer subunits along the fibril axis fits the cross-sectional area of a single protofilament. To construct twisted-stack fibril models comprised of two, four, and six protofilaments, the β-solenoid subunits were moved and rotated within the 120 Å × 150 Å × 10 Å lattice (5). Lattice dimensions were based on the approximate dimensions of the six-protofilament fibril cross section. The final fibril models were then constructed by translating these repeating units along the axis of the fibril with helical periods of ∼525 Å, 355 Å, and 426 Å. Helical periods were taken from the previously reported layer line spacing values of two-, four-, and six-protofilament fibrils (5). The final fibril models were distorted crystallite structures that had a long-range twist, with a corresponding helical period.
A previous modeling study based on SAXS data suggested that the helical oligomeric species intertwine into a dense protofilament, and that the two protofilaments twist around each other to form a protofibril (26). Based on the estimated dimensions of insulin oligomeric species observed by SAXS and the tentative model of the insulin fibril proposed in these studies, we constructed five helical models consisting of three, four, five, six, and eight intertwined β-helices. The β-roll subunit model was similarly examined, but it failed to provide reasonable solutions for the insulin fibril (data not shown). In contrast to the proposed model by Vestergaard et al. (26), which has eightfold symmetry formed by eight helical oligomers intertwined to form a protofilament with an average diameter of ∼50 Å, the β-helix-based helical models have diameters ranging from 50 Å for a trimeric model to 85 Å for an octameric model. Considering the dimensions of the insulin protofilament calculated from cryo-EM data (30–40 Å), the only sensible model was the trimeric helical model, in which three β-helix-based protofilaments intertwined with one another. A pair of trimeric β-helical protofilaments twisted around each other to form a protofibril with a diameter of ∼100 Å was consistent with both the cryo-EM (5) and SAXS studies (26).
The fiber diffraction patterns calculated for β-solenoids with parallel and antiparallel β-sheets were different. Fiber structures containing β-solenoids organized in parallel fashion produced a meridional arc at ∼9.6 Å, whereas those with β-solenoids organized in antiparallel fashion produced meridional arcs with a twofold higher periodicity. Since the experimental insulin x-ray diffraction pattern (26) did not show a meridional arc at 19 Å, this allowed us to exclude antiparallel organization of β-solenoid models from consideration.
Simulated versus experimental fiber diffraction patterns of the insulin fibril
Using the β-solenoid insulin fibril models, we simulated fiber diffraction patterns for 1), β-roll and β-helix-based two-, four-, and six-protofilament twisted-stack models; and 2), β-helix-based helical models. The simulated diffraction patterns of the fibril models were calculated and compared with the experimental patterns (Fig. S3, Fig. S4, and Fig. S5). For comparison, the meridional and equatorial arc profiles of the simulated diffraction patterns were compared against those of the experimental diffraction patterns of the insulin amyloid fibril, and the similarity scores were calculated using the least-squares method (Table S2 and Table S3).
In the fiber diffraction simulations of the insulin fibril models, the meridional arc results from the periodic packing of the insulin molecules in the direction along the fiber, whereas the equatorial arc results from periodic β-sheets stacking in the direction across the fiber.
The meridional region of the insulin experimental diffraction pattern is dominated by a strong intensity arc at ∼4.8 Å resolution. This feature corresponds to the distance between adjacent β-strands and indicates that the strands are oriented perpendicular to the fiber axis. This 4.8-Å meridional signal extended all the way into the equatorial region due to its overall strength and to a pronounced disorientation of the amyloid fibrils. A weak, diffuse meridional arc, observed at ∼9.6 Å resolution, is believed to reflect periodic repeats of identical chains along the fiber. The simulated diffraction patterns of all the models showed a strong meridional arc at ∼4.8 Å due to hydrogen-bonded peptide-peptide spacing between the β-sheets. In contrast, the intensity and range of the meridional arc at ∼9.6 Å varied between the models. The diffraction patterns of all the β-roll fibril models consistently showed a relatively weak single peak at ∼9.6 Å. This would be expected from fibril models constructed using two-rung β-solenoid structures as assembling subunits. However, the β-helix fibril models gave diffraction patterns with multiple peaks of various intensity at ∼9.6 Å (Fig. S4). This may reflect some irregularity and distortion in the β-sheets caused by introducing disulfide bridges at the turns of the β-helix subunit model. Although it is difficult to simulate the exact location and intensity of the meridional arcs seen in experimental diffraction patterns, the β-roll fibril models showed a reasonable fit to the meridional arcs (Fig. 4 and Fig. S3).
Figure 4.
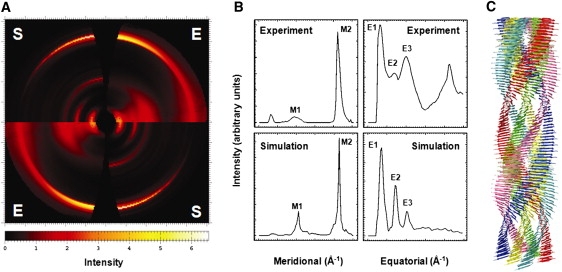
Experimental and simulated diffraction patterns of an insulin amyloid fibril and a six-protofilament β-roll-based insulin fibril model. (A) Quadrant view of the experimental insulin x-ray fiber diffraction pattern (E) (26) and the simulated fiber diffraction patterns of a six-protofilament insulin fibril model using the β-roll monomeric subunit as a building block, constructed by DISORDER (S) (48). The image was prepared by FIT2D (36). (B) Meridional and equatorial arc profiles of the experimental and simulated diffraction patterns, with two meridional arcs, at ∼9.6 Å and ∼4.8 Å (M1 and M2, respectively), and three equatorial arcs, at ∼33 Å, ∼15 Å, and ∼10 Å (E1–E3, respectively). (C) Tentative model of the six-protofilament β-roll-based insulin amyloid fibril. The image was prepared by the UCSF Chimera package (48). The experimental x-ray fiber diffraction pattern of insulin amyloid was collected under the experimental conditions of the SAXS analysis from which the mature fibril consisting of six intertwining protofilaments was evaluated (26).
For the equatorial arcs, a wide range of variations appeared in the number, intensity, and location of arcs among the different fibril models. The experimental diffraction patterns revealed three major equatorial arcs at ∼33 Å, ∼15 Å, and ∼10 Å, which are believed to reflect the stacking distances of β-sheets within the chains and between the subunits along the cross section of the amyloid fibrils (Fig. 4 B). As described above, the 4.8-Å meridional signal was smeared out to such an extent that the diffraction pattern showed a weak signal at 4.8 Å in the equatorial region. Thus, this signal was not considered as one of the equatorial arcs. We focused on identifying fibril models that would generate comparable arcs at the ∼33 Å, ∼15 Å, and ∼10 Å regions by exploring two-, four-, and six-protofilament arrangements of the β-roll and β-helix models, with different arrangements of the subunits and variations in the β-sheet stacking distance (Fig. S3 and Fig. S4). A model fibril composed of six-protofilament β-roll model subunits (Fig. 3 A, e) generated the most comparable triplet of the equatorial arcs (Fig. 4 and Fig. S8). Other fibril models failed to match this pattern of equatorial arcs.
We also simulated and searched the fiber diffraction patterns from the β-helix-based helical models, which were built based on previous SAXS study data, to explore the possibility of alternative arrangements of insulin protofilaments (Fig. S5). However, none of the simulated diffraction patterns of these β-helix fibril models resulted in comparable arcs at the regions discussed above.
Discussion
β-solenoid models satisfy experimental constraints of misfolded insulin
Insulin is a unique system with which to model amyloid fibrils, given the conformational restrictions imposed on the protein by its intrinsic disulfide bridges. These β-solenoid structures provide the cross-β pattern and β-signatures seen in fiber diffraction, FTIR, and CD studies of amyloid fibrils. In addition, previous studies on the fibrillization of insulin single-chain analogs, disulfide isomers, and isolated single chains are consistent with our β-solenoid models. Based on the approximate cross-sectional dimension, both the β-roll and β-helix models were comparable to the insulin fibril, but the β-helix model was a slightly better fit to the estimated diameters of the insulin protofilament.
Although the mechanism of amyloid fibrillization remains unclear, it has been suggested that some proteins may undergo a reorganization of their global structures while retaining native structural elements. Other proteins may require substantial unfolding and subsequent conversion of α-helix to β-sheet. In the case of insulin, previous studies suggested that insulin may retain its three disulfide bridges during the conformational rearrangements as it undergoes extensive conversion to β-sheet structure (5). A comparison of the cross-sectional views of the native and β-solenoid insulin model structures showed that a structural transition from the α-helical to the β-solenoid structure may be possible with the disulfide bridge intact. Thus, β-solenoid insulin models may provide a possible explanation for the misfolding and fibrillization events that can occur in a physiological environment.
A multiple-sequence alignment of mammalian insulins showed that some species contain point mutations, such as histidine at A18 for feline and proline at B9 for rat insulin, which may prevent folding to a β-helical structure as they allow folding to a β-roll structure. These two mutations are located in the hydrophobic core of the β-helix model, which cannot accommodate histidine or proline. The same mutations can be tolerated in the β-roll model (Fig. 1). Follow-up experiments on the fibril-forming tendency of insulins from these species or cross-seeding studies with human insulin may provide additional information as to whether the β-roll or β-helix fold is the better modeling solution.
Structural stability of the insulin β-solenoid monomer models
Although research as to whether oligomeric species or mature amyloid fibrils act as toxic agents in the pathogenic process is ongoing, increasing evidence indicates that the monomeric units of misfolded amyloid proteins may act as templating or assembling units for the larger intermediate aggregate species (26,46,47). Some key features of β-roll and β-helix folds that make them good candidates as monomeric structural units of the insulin fibril are the inherent compactness of their folds and the availability of accessible β-faces to initiate and sustain self-assembly. Previous studies of the β-helices have shown that the two-rung unit of the β-helical fold is likely to be the minimal stable unit, and that the packing of the hydrophobic core is the essential determinant of global stability (33). In addition, we have previously shown that native disulfide bridges may help in the stabilization or folding of the β-helix fold (33). Molecular models of β-solenoid insulin indicate that this protein can adopt two full rungs of β-rolls or β-helix folds and accommodate well-packed side chains in its core. However, it is difficult to evaluate the absolute thermodynamic stability of the monomeric unit to evaluate its possible role in the nucleation and elongation steps of amyloid fibrillization. Molecular dynamics simulations can be used to evaluate the relative stability of different insulin monomeric and polymeric models compared to their initial structure under fibrillogenic conditions. Although the actual protein stability may deviate from this relative stability estimation, such simulations may provide some limited insight into how similar models of the same protein would behave under the same fibrillogenic conditions. Based on our simulation of two different β-solenoid insulin models at different temperatures, the β-roll model may be the more stable structure. Although it is difficult to extrapolate these findings to fiber stability, it seems reasonable to speculate that this predicted increase in stability might have an impact on amyloid fiber propagation.
Molecular models of the insulin fibril and a comparison of simulated and experimental fiber diffraction patterns
Although x-ray fiber diffraction studies can provide the characteristic cross-β pattern structure that is a signature of amyloid fibrils, this technique alone has not been very effective in determining the detailed structural arrangement of amyloid fibrils, despite recent improvements in high-resolution fiber diffraction. Diffraction images can provide gross structural information, subject to the quality of the fiber under study. For example, subtle variations in the diffraction image often result from differences between sample preparations. One approach to harnessing the information contained within diffraction images is to start with fibril models and generate theoretical fiber diffraction patterns through computer simulation and compare these patterns to an experimental pattern to validate or refute the models (18). In this way, the most likely fibril structure can be identified from many thousands of possible structural solutions. Although comparing simulated and experimental fiber diffraction patterns does not provide explanations for experimental factors that may affect fiber diffraction, it can be useful for studying the relative difference in diffraction patterns generated by similar fibrillar structures. In this study, only the experimental fiber diffraction data up to 4.4 Å resolution in the meridional direction and 6.7 Å in the radial direction were used, as we could not perform a reasonable background subtraction in the higher-resolution areas. Despite these limitations, and the difficulty of using the entire set of diffraction data, we were still able to generate reasonable models based on the dimensions and helical geometry of the fiber, dimensions of the asymmetric unit, and details of the fold, such as the packing of β-strands in the asymmetric unit. The predictions were made based on ranking of the trial models by calculating least-squares residuals.
Insulin fibril morphology depends on the arrangement and number of protofilaments involved (5). In an attempt to acquire a more detailed understanding of the fibril structure and its arrangement, we used simple monomeric β-solenoid models to build complex fibril models by varying the number of protofilaments and their arrangement. Using a computational method developed to simulate the diffraction pattern of disoriented fibrils, we tested whether the diffraction patterns from the constructed β-solenoid fibril models were comparable with the experimental patterns. A search for the best-fitting model through the enumeration of different models, arrangements, spacings between protofilaments, and degrees of disorientation resulted in the selection of a six-protofilament β-roll fibril model. The final fibril model is consistent with previous studies showing six intertwining protofilaments or three protofibrils formed by two protofilaments each (5,26).
Although the comparison of fiber diffraction patterns showed that the six-protofilament β-roll insulin fibril model agreed well with the experimental data, there were two major discrepancies. First, a model could not be identified that would generate a meridional arc at ∼33 Å and also showed a disoriented meridional signal at ∼4.8 Å that extended to the equatorial region (Fig. 4 B). However, we believe that these peculiar signatures may have been caused by misorientation of the amyloid filaments within the dried fibril which could not be simulated by the program DISORDER. Second, there was a large difference in intensity between the equatorial arcs. As shown in Fig. 4 A, the experimental diffraction pattern showed relatively stronger and wider peaks, especially at ∼10 Å, than did any of our simulated patterns. Although differences between the actual insulin and the fibril model are major concerns, these may have resulted from disorientation of the amyloid fibrils due to heterogeneities arising in the fibrils. The physical nature of amyloid fibril disorientation is still poorly understood and therefore difficult to account for in computational simulations. Also, the flexible C-terminus of the B chain, which was not included in this modeling study, may affect the diffraction signal. As an attempt to explore the possible contribution of conformational heterogeneity within the fibrils to disorientation of the amyloid fibrils and their diffraction patterns, we conducted fiber diffraction simulations for six-protofilament, twisted-stack fibril models, based on the final structures of the β-solenoid insulin monomer units from the 40-ns MD simulations, as well as hybrid fibril models, comprising both β-roll and β-helix monomer units (Fig. S6 and Fig. S7). Although diffraction patterns built from the final structures of the MD simulations did not show any significant improvements, the hybrid fibril models showed better fits for the meridional and equatorial profiles (Table S2). Further studies are necessary to probe how conformational heterogeneity can be applied in the search for a better-fitting fibril model, and to understand the nature of the disorientation of amyloid fibrils. These efforts will no doubt include the generation of higher-resolution fiber diffraction patterns with increasing resolution of the meridional and equatorial arcs. Even with improved patterns, fiber diffraction simulations and analysis can only be effective when the information we gather from molecular models is reasonably accurate in terms of the dimensions and basic architecture of the amyloid fibrils. However, despite the recognized limitations, we are satisfied that method of fiber diffraction simulation described in this article can be an effective tool to study fiber structure where techniques that require higher resolution (e.g., x-ray crystallography) are not applicable.
Conclusions
Solid-state NMR, fiber diffraction, EM, and AFM studies of Aβ1–40 and the fungal HET-s prion protein have shed light on structural properties of amyloid fibrils. Amyloid fibrils share a structural architecture resembling that of β-solenoid folds such as β-rolls and β-helices. However, studies suggest, there is also morphological diversity among the different amyloid fibrils, which may reflect structural diversity of the monomeric structural unit. To further understand the detailed structural arrangements of the amyloid fibril, we used several computational tools to model and investigate the insulin monomeric subunit and fibril. We have shown here that the β-solenoid is an architecture likely to be adopted by insulin in the fibrillogenic state, although further studies are necessary to examine the discrepancies between our theoretical models and experimental data.
Supporting Material
Three tables, eight figures, and methods are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01526-4.
Supporting Material
Acknowledgments
We thank Dr. Alexander Borovinskiy for valuable discussions and assistance with DISORDER, Dr. Minna Groenning for providing us with the x-ray fiber diffraction pattern of insulin, and Sarit Helman, M.P.H., for critically reading the manuscript.
This work was supported by National Institutes of Health grants AG21601.
References
- 1.Dobson C.M. Protein folding and its links with human disease. Biochem. Soc. Symp. 2001;68:1–26. [PubMed] [Google Scholar]
- 2.Prusiner S.B., Scott M.R., DeArmond S.J., Cohen F.E. Prion protein biology. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 3.Serpell L.C., Benson M., Liepnieks J.J., Fraser P.E. Structural analyses of fibrinogen amyloid fibrils. Amyloid. 2007;14:199–203. doi: 10.1080/13506120701461111. [DOI] [PubMed] [Google Scholar]
- 4.Serpell L.C., Smith J.M. Direct visualisation of the β-sheet structure of synthetic Alzheimer's amyloid. J. Mol. Biol. 2000;299:225–231. doi: 10.1006/jmbi.2000.3650. [DOI] [PubMed] [Google Scholar]
- 5.Jimenez J.L., Nettleton E.J., Bouchard M., Robinson C.V., Dobson C.M. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2002;99:9196–9201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khurana R., Ionescu-Zanetti C., Pope M., Li J., Nielson L. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys. J. 2003;85:1135–1144. doi: 10.1016/S0006-3495(03)74550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jimenez J.L., Guijarro J.I., Orlova E., Zurdo J., Dobson C.M. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldsbury C.S., Cooper G.J., Goldie K.N., Muller S.A., Saafi E.L. Polymorphic fibrillar assembly of human amylin. J. Struct. Biol. 1997;119:17–27. doi: 10.1006/jsbi.1997.3858. [DOI] [PubMed] [Google Scholar]
- 9.Bouchard M., Zurdo J., Nettleton E.J., Dobson C.M., Robinson C.V. Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 2000;9:1960–1967. doi: 10.1110/ps.9.10.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson R., Sawaya M.R., Balbirnie M., Madsen A.O., Riekel C. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sawaya M.R., Sambashivan S., Nelson R., Ivanova M.I., Sievers S.A. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 12.Luhrs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wasmer C., Lange A., Van Melckebeke H., Siemer A.B., Riek R. Amyloid fibrils of the HET-s(218–289) prion form a β solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 14.Sachse C., Fandrich M., Grigorieff N. Paired β-sheet structure of an Aβ(1–40) amyloid fibril revealed by electron microscopy. Proc. Natl. Acad. Sci. USA. 2008;105:7462–7466. doi: 10.1073/pnas.0712290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kajava A.V., Steven A.C. β-rolls, β-helices, and other β-solenoid proteins. Adv. Protein Chem. 2006;73:55–96. doi: 10.1016/S0065-3233(06)73003-0. [DOI] [PubMed] [Google Scholar]
- 16.Jenkins J., Pickersgill R. The architecture of parallel β-helices and related folds. Prog. Biophys. Mol. Biol. 2001;77:111–175. doi: 10.1016/s0079-6107(01)00013-x. [DOI] [PubMed] [Google Scholar]
- 17.Petkova A.T., Ishii Y., Balbach J.J., Antzutkin O.N., Leapman R.D. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wille H., Bian W., McDonald M., Kendall A., Colby D.W. Natural and synthetic prion structure form x-ray fiber diffraction. Proc. Natl. Acad. Sci. USA. 2009 doi: 10.1073/pnas.0909006106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brange J., Andersen L., Laursen E.D., Meyn G., Rasmussen E. Toward understanding insulin fibrillation. J. Pharm. Sci. 1997;86:517–525. doi: 10.1021/js960297s. [DOI] [PubMed] [Google Scholar]
- 20.Dische F.E., Wernstedt C., Westermark G.T., Westermark P., Pepys M.B. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158–161. doi: 10.1007/BF00276849. [DOI] [PubMed] [Google Scholar]
- 21.Devlin G.L., Knowles T.P., Squires A., McCammon M.G., Gras S.L. The component polypeptide chains of bovine insulin nucleate or inhibit aggregation of the parent protein in a conformation-dependent manner. J. Mol. Biol. 2006;360:497–509. doi: 10.1016/j.jmb.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Bryant C., Spencer D.B., Miller A., Bakaysa D.L., McCune K.S. Acid stabilization of insulin. Biochemistry. 1993;32:8075–8082. doi: 10.1021/bi00083a004. [DOI] [PubMed] [Google Scholar]
- 23.Waugh D.F., Thompson R.E., Weimer R.J. Assay of insulin in vitro by fibril elongation and precipitation. J. Biol. Chem. 1950;185:85–95. [PubMed] [Google Scholar]
- 24.Burke M.J., Rougvie M.A. Cross- protein structures. I. Insulin fibrils. Biochemistry. 1972;11:2435–2439. doi: 10.1021/bi00763a008. [DOI] [PubMed] [Google Scholar]
- 25.Hua Q.X., Weiss M.A. Mechanism of insulin fibrillation: the structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J. Biol. Chem. 2004;279:21449–21460. doi: 10.1074/jbc.M314141200. [DOI] [PubMed] [Google Scholar]
- 26.Vestergaard B., Groenning M., Roessle M., Kastrup J.S., van de Weert M. A helical structural nucleus is the primary elongating unit of insulin amyloid fibrils. PLoS Biol. 2007;5:e134. doi: 10.1371/journal.pbio.0050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nettleton E.J., Tito P., Sunde M., Bouchard M., Dobson C.M. Characterization of the oligomeric states of insulin in self-assembly and amyloid fibril formation by mass spectrometry. Biophys. J. 2000;79:1053–1065. doi: 10.1016/S0006-3495(00)76359-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmad A., Uversky V.N., Hong D., Fink A.L. Early events in the fibrillation of monomeric insulin. J. Biol. Chem. 2005;280:42669–42675. doi: 10.1074/jbc.M504298200. [DOI] [PubMed] [Google Scholar]
- 29.Jansen R., Dzwolak W., Winter R. Amyloidogenic self-assembly of insulin aggregates probed by high resolution atomic force microscopy. Biophys. J. 2005;88:1344–1353. doi: 10.1529/biophysj.104.048843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 31.Daura X., Mark A.E., Van Gunsteren W.F. Parametrization of aliphatic CHn united atoms of GROMOS96 force field. J. Comput. Chem. 1998;19:535–547. [Google Scholar]
- 32.Langedijk J.P., Fuentes G., Boshuizen R., Bonvin A.M. Two-rung model of a left-handed β-helix for prions explains species barrier and strain variation in transmissible spongiform encephalopathies. J. Mol. Biol. 2006;360:907–920. doi: 10.1016/j.jmb.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 33.Choi J.H., Govaerts C., May B.C., Cohen F.E. Analysis of the sequence and structural features of the left-handed β-helical fold. Proteins. 2008;73:150–160. doi: 10.1002/prot.22051. [DOI] [PubMed] [Google Scholar]
- 34.Reference deleted in proof.
- 35.Borovinskiy, A. 2006. DISORDER (http://fibernet.vanderbilt.edu/software/disorder).
- 36.Hammersley A. ESRF; Grenoble, France: 1997. FIT2D: An Introduction and Overview. [Google Scholar]
- 37.Bowie J.U., Luthy R., Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 38.Hennetin J., Jullian B., Steven A.C., Kajava A.V. Standard conformations of β-arches in β-solenoid proteins. J. Mol. Biol. 2006;358:1094–1105. doi: 10.1016/j.jmb.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 39.Brange J., Dodson G.G., Edwards D.J., Holden P.H., Whittingham J.L. A model of insulin fibrils derived from the x-ray crystal structure of a monomeric insulin (despentapeptide insulin) Proteins. 1997;27:507–516. [PubMed] [Google Scholar]
- 40.Hong D.P., Fink A.L. Independent heterologous fibrillation of insulin and its B-chain peptide. Biochemistry. 2005;44:16701–16709. doi: 10.1021/bi051658y. [DOI] [PubMed] [Google Scholar]
- 41.Hong D.P., Ahmad A., Fink A.L. Fibrillation of human insulin A and B chains. Biochemistry. 2006;45:9342–9353. doi: 10.1021/bi0604936. [DOI] [PubMed] [Google Scholar]
- 42.Huang K., Maiti N.C., Phillips N.B., Carey P.R., Weiss M.A. Structure-specific effects of protein topology on cross-β assembly: studies of insulin fibrillation. Biochemistry. 2006;45:10278–10293. doi: 10.1021/bi060879g. [DOI] [PubMed] [Google Scholar]
- 43.Groenning M., Norrman M., Flink J.M., van de Weert M., Bukrinsky J.T. Binding mode of thioflavin T in insulin amyloid fibrils. J. Struct. Biol. 2007;159:483–497. doi: 10.1016/j.jsb.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Haspel N., Zanuy D., Aleman C., Wolfson H., Nussinov R. De novo tubular nanostructure design based on self-assembly of β-helical protein motifs. Structure. 2006;14:1137–1148. doi: 10.1016/j.str.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 45.Zheng J., Zanuy D., Haspel N., Tsai C.J., Aleman C. Nanostructure design using protein building blocks enhanced by conformationally constrained synthetic residues. Biochemistry. 2007;46:1205–1218. doi: 10.1021/bi061674a. [DOI] [PubMed] [Google Scholar]
- 46.Collins S.R., Douglass A., Vale R.D., Weissman J.S. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004;2:e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyama B.H., Kelly M.J., Gross J.D., Weissman J.S. The structural basis of yeast prion strain variants. Nature. 2007;449:233–237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 48.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.