

Abstract
We have used magnetic tweezers to study nucleosome assembly on topologically constrained DNA molecules. Assembly was achieved using chicken erythrocyte core histones and histone chaperone protein Nap1 under constant low force. We have observed only partial assembly when the DNA was topologically constrained and much more complete assembly on unconstrained (nicked) DNA tethers. To verify our hypothesis that the lack of full nucleosome assembly on topologically constrained tethers was due to compensatory accumulation of positive supercoiling in the rest of the template, we carried out experiments in which we mechanically relieved the positive supercoiling by rotating the external magnetic field at certain time points of the assembly process. Indeed, such rotation did lead to the same nucleosome saturation level as in the case of nicked tethers. We conclude that levels of positive supercoiling in the range of 0.025–0.051 (most probably in the form of twist) stall the nucleosome assembly process.
Introduction
The eukaryotic genome is organized as chromatin, a complex of DNA and a set of highly positively charged proteins, known as histones (1–3). The basic repeating unit of chromatin structure is the nucleosome, in which ∼147 bps of DNA are wrapped in a left-handed superhelix around an octamer of histones (H2A, H2B, H3, H4) (4–6). Nucleosomes are connected with linker DNA to form a chromatin fiber. The entry and exit portions of nucleosomal DNA are bound by an additional histone protein, linker histone, which stabilizes the nucleosome (7) and contributes significantly to the further folding of chromatin fibers. DNA organized into chromatin is highly compacted and inaccessible to proteins and protein complexes involved in DNA metabolism (transcription, replication, recombination, and repair). Nevertheless, these processes must occur throughout the life of a cell and therefore chromatin structure must be able to dynamically and reversibly change to allow the genetic information in DNA to be processed.
Chromatin assembles in cells during DNA replication and very likely also after passage of RNA polymerase during transcription. In both cases, the DNA is expected to be under tension and also under certain level of torsional stress (8–10). Nuclear DNA is subjected to torsion, because i), DNA is attached to insoluble components of the nuclear matrix in topologically constrained loops (11,12); and ii), the DNA molecule rotates in the catalytic center of immobilized transcriptional machineries (13,14); such rotation positively supercoils DNA in front of the elongating polymerase (10).
The classical biochemical and biophysical methods used in chromatin research are population-average methods that assess properties of the entire population of macromolecules. Recently developed single-molecule methods allow observation of individual molecules at high temporal and spatial resolution (15–21), thus providing molecular parameters important for understanding structural dynamics and molecular heterogeneity. Applying single-molecule techniques to the study of chromatin (22,23) is especially advantageous in view of the complexity of the structure and the enormous heterogeneity in terms of postsynthetic modifications. Studies of chromatin behavior under applied force (24,25) have shown that nucleosomes are disassembled at forces 15–25 pN, depending on the experimental conditions. So far, only a few single molecule studies have addressed questions regarding assembly of chromatin fibers on torsionally unconstrained DNA molecules under tension (26–28). All groups reported that chromatin assembly was significantly slowed down when the DNA template was subjected to forces around 10–12 pN. Topologically constrained DNA, however, mimics more closely the in vivo situation (8,9,11); thus, the study of nucleosome assembly in a topologically constrained system will bring more physiologically relevant answers about the effects of local DNA supercoiling and applied stretching force on formation of nucleosomes.
In this study, we used magnetic tweezers to follow nucleosome assembly in real-time on topologically constrained DNA molecules to more closely approach the physiological reality. We found that higher levels of positive supercoiling are inhibitory to nucleosome formation.
Materials and Methods
Materials
Histone octamers were purified from chicken erythrocytes (Pel Freeze, Rogers, AR) using hydroxyapatite chromatography (29,30) and analyzed on SDS-PAGE (Fig. 1 b). The expression plasmid for yeast nucleosome assembly protein 1 (yNap 1) was a gift from A. Kikuchi (Hiroshima University School of Medicine, Japan). Nucleosome assembly protein 1 (Nap1) was expressed and purified by ion-exchange chromatography following the procedure of Fujii-Nakata et al. (31).
Figure 1.
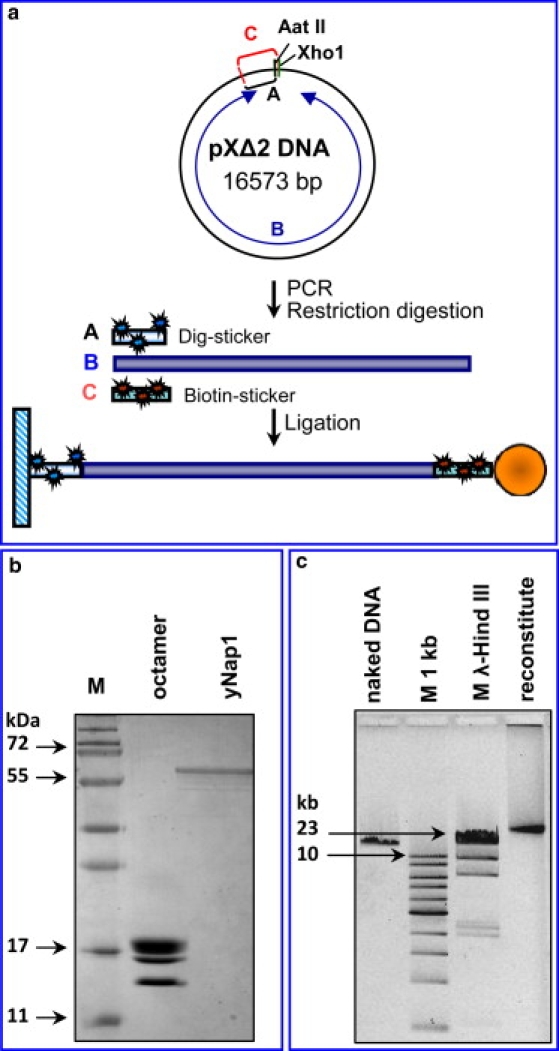
Components used for assembly experiments. (a) Schematic for preparing the DNA construct: the three DNA fragments (dig-sticker, biotin-sticker, and 16.5 kb DNA from pxΔ2 DNA) were prepared separately (see Materials and Methods) and ligated together. (b) Purified histone octamer from chicken erythrocytes and purified recombinant yNap1 were analyzed by 15% SDS-PAGE gel. (c) Analysis of the nucleosome array reconstituted in bulk by 1% agarose gel.
The DNA template consists of three fragments ligated to each other (Fig. 1 a). Two of the fragments, A and C, served as a “stickers” for the attachment of the construct to the surface of the cuvette and the magnetic bead. The constructs were labeled with biotin or digoxigenin on both DNA strands. The attachment of these stickers to the bead and the glass surface leads to a torsionally constrained DNA tether (if the DNA is not nicked (32)). Fragments A and C (1020 and 940 bp, respectively) were obtained by PCR amplification using pXΔ2 DNA as a template (a gift of J. F. Allemand, Laboratoire de Physique Statistique de I'ENS, France, (33)), using the following primers:
Fragment A: PXDXho1(A1), 5′-GCC TGA CTC CCC GTC GTG TA-3′; PXDXho2(A2), 5′-GAG AAG CTC GAG GGG GCG CG-3′. Fragment C: PXDXho1(C1), 5′-GCC TGA CTC CCC GTC GTG TA-3′; pXAat3(C2), 5′-GTT TCT TAG ACG TCA GGT GGC ACT T -3′. Fragment A contains digoxigenin-modified uracil (Roche Applied Science, Indianapolis, IN) in a ratio of 1:6 to nonmodified thymine, fragment C contains biotin-modified uracil (Roche) in a ratio of 1:6 to nonmodified thymine.
Fragment B was obtained by digestion of pXΔ2 DNA with AatII and XhoI (New England BioLabs, Ipswich, MA). The resulting 16.5 kb DNA fragment was purified by gel filtration on Sephacryl S500 or Ultrogel A2.
Surface preparation
The capillaries were cleaned with 0.5 M KOH, treated with 2% APTES (Sigma, St. Louis, MO) in acetone for 15 min and air-dried at room temperature. m-PEG-5000-Succinate (Sigma) was dissolved in fresh 100 mM NaHCO3 buffer containing 0.4 M K2SO4 to a final concentration of 10% (w/v). The PEG solution was then injected in and allowed to react with the capillaries for 15 min, followed by a rinse with water. Finally, the capillaries were incubated with 100 μg/mL antidigoxigenin (Roche) solution in PBS for 30 min and rinsed with water. The antidigoxigenin treated capillaries were stored in water at 4°C (adapted from S. Park, A. Pertsinidis, A. Revyakin, S. Chu, unpublished).
Assembly experiments
The assembly mixture contained a 1:2 (w/w) ratio of chicken histone octamers (10 ng/μL) and yNap1 (20 ng/μL) in 10 mM Tris-HCl, 0.5 mM EDTA, 150 mM NaCl, pH 8.0. The assembly mixture was injected into the capillary and the bead position in (x,y,z) in time was recorded at a rate of 33 frames/s. The flow was stopped when the assembly mixture reached the center of the capillary, where the monitored bead was attached. With the flow on, the hydrodynamic force pushed the bead to the capillary surface. After the flow was stopped, the bead recovered its position in (z) (the distance between the surface and the bead depends on the stretching force applied to the tether (32)). Observing the position of the bead with the flow on and off allowed us to estimate the real DNA extension value for DNA tethered for every particular bead.
Magnetic tweezers
The instrument is set up using established procedures (28,32) with an Olympus IX-71 inverted microscope as the instrument base. A PIFOC (a piezo objective nanopositioner; Physik Instrumente, Germany) allows for a nanometer-resolution objective movement in z dimension and a commercial Olympus nose piece adaptor stage is used to reduce thermal drift. A pair of external magnets is mounted on a platform using a home-built manipulator allowing circular rotation of the magnetic field and movement in all three dimensions above a glass capillary sitting on the stage of the inverted microscope. Capillary tubing is used for solution delivery from a syringe pump at 1 mL/min flow rate.
Data acquisition
Data acquisition and analysis was achieved using home-written C++-based software (provided by Dr. Andrey Revyakin, University of California, Berkeley). We used a PCVision frame grabber and JAI CV-M50 camera from Coreco (1st Vision, Andover, MA) that records 33 frames/s (with a maximal spatial resolution of 720 × 480 pixel/frame). The (x,y,z) coordinates of the magnetic bead were recorded and displayed in real-time. A surface-stuck bead was used as a reference point for drift subtraction during subsequent analysis. Before the assembly experiment, a calibration file was created for each new bead and was used to calculate the z position during assembly. For bead tracking, the bead was initially brought well within the calibration file range using the PIFOC and tracked by video recording up to 150,000 frames.
Results
The experimental setup: ensuring that the DNA tether is topologically constrained
Fig. 1 a shows a schematic of the DNA construct used in the assembly reactions. The electrophoretic analyses of the purified core histones, yNap1 (proteins required for nucleosome assembly), and the nucleosomal array assembled in solution are presented in Fig. 1, b and c. The Nap1-assembled reconstitute (Fig. 1 c, lane 4) migrates more slowly than naked DNA (Fig. 1 c, lane 1), as expected for nucleosomal arrays of this size. We used Nap1 as a nucleosome assembly vehicle because it is known to efficiently assemble nucleosomes in vitro (31,34,35) and because it has been successfully used in similar experiments earlier (28).
A schematic of the experimental geometry is presented in Fig. 2 a. We analyzed the bead path on rotation of a single external magnet to find a bead tethered by a single DNA molecule. Beads making symmetrical circles (proving a single DNA tether) were used for the assembly experiments. The length of the tether is a function of the stretching force (32) and can be monitored directly by tracking the z position of the tethered bead or by recording the bead's Brownian motion (x,y position) over time. Under low stretching force, the extension of torsionally constrained DNA is also dependent on the level of torsional stress applied to the molecule by rotating the external magnet pair (32,33). Fig. 2 b shows an example extension versus rotation curve at 0.3 pN obtained by direct real-time data processing (see Materials and Methods). Fig. 2 c presents an example of the Brownian motion data for a relaxed DNA tether and the same molecule after 100 positive (counterclockwise in our setup) rotations under 0.3 pN force. Extensive supercoiling (σ = 0.06) leads to the formation of plectonemes under the low force conditions used, which results in an apparent shortening of the tether and a smaller range of the bead movement in (x,y).
Figure 2.
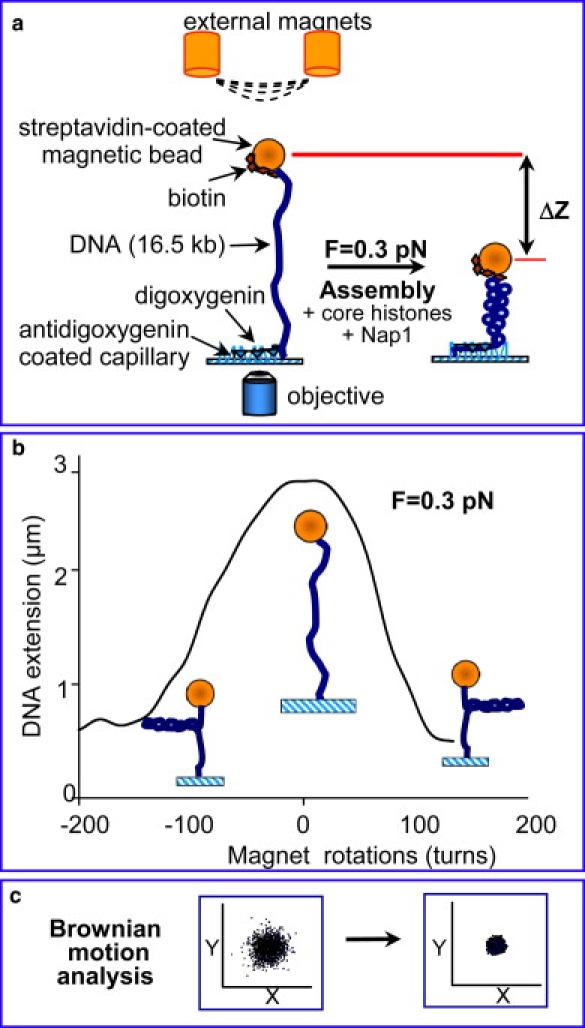
(a) Schematic of the magnetic tweezers setup for studying nucleosome array assembly. A single double-stranded DNA molecule is suspended between a magnetic bead and the surface of the cuvette in a torsionally constrained manner. A mixture of core histones and histone chaperone Nap1 was injected into the cuvette to initiate assembly. The shortening in DNA extension due to formation of nucleosomes was measured by monitoring the movement of the bead in (z), as well as by Brownian motion analysis. (b) Rotation-extension curve on the torsionally constrained DNA at F = 0.3 pN. At low force, rotation of the two external magnets induced (+) or (−) supercoiling in the DNA molecule, which led to plectoneme formation, and thus shortening of the DNA tether. (c) Brownian motion scatter in (x,y) for the naked, relaxed DNA was larger than for the supercoiled DNA or reconstituted nucleosomal array.
After the intactness and the topological state of the DNA tether were verified through extension/rotation curves, DNA length was determined by flow-on/off experiments (turning the pump on/off for a period of several seconds). The buffer flow pushed the attached bead toward the capillary surface and the measured change in extension had to fall within the range expected on the basis of the length of the DNA construct. After locating a qualifying bead and a control bead (attached directly to the capillary surface) in the field of view, assembly solution was rapidly injected into the capillary and bead movements were recorded.
Nucleosome assembly on topologically constrained DNA substrates
When topologically constrained DNA molecules were used as an assembly substrate, the assembly process typically took 45 ± 15 min, gradually slowing down to a complete stop. The change in tether extension varied in the range of 1.45–2.37 μm (Table 1 and Fig. 3 a). This corresponded to a range of 29–47 nucleosomes formed, as calculated from a theoretically estimated shortening of ∼50 nm/nucleosome (24,25,36) (see also Table 1 and Discussion). The DNA has the capacity to accommodate between 83 and 113 nucleosomes, assuming a nucleosome repeat length of 146–200 bp. The lower bound of 146 bp (that is practically the number of bp wrapped around the histone octamer in the crystal structure of the nucleosomal core particle (5)) comes from studies that used Nap1 as the nucleosome reconstitution vehicle (34).
Table 1.
Summary of the experimentally measured tether shortening, the estimated number of nucleosomes, the corresponding degree of completion of assembly, and the superhelical density that blocks nucleosome formation
| Experiment number | Tether shortening (μm)∗ | Estimated number of nucleosomes formed† | Completion (%)‡ | Positive superhelical density σ that blocks nucleosome formation§ |
|---|---|---|---|---|
| Partial assembly | ||||
| 1 | 1.9 | 38 | 39 | 0.036 |
| 2 | 2.3 | 46 | 47 | 0.049 |
| 3 | 1.6 | 32 | 33 | 0.028 |
| 4 | 1.45 | 29 | 30 | 0.025 |
| 5 | 2.37 | 47 | 48 | 0.051 |
| 6 | 2.27 | 45 | 46 | 0.047 |
| 7 | 1.6 | 32 | 33 | 0.028 |
| Average | 1.93 ± 0.15 | 39 ± 3 | 39 ± 3 | 0.038 ± 0.004 |
| Full assembly on nicked DNA | ||||
| 1 | 3.52 | 70 | 72 | |
| 2 | 4.48 | 90 | 91 | |
| 3 | 3.21 | 64 | 66 | |
| 4 | 3.90 | 78 | 80 | |
| 5 | 3.85 | 77 | 79 | |
| Average | 3.79 ± 0.21 | 76 ± 4 | 77 ± 4 | |
| Full assembly with magnets rotation | ||||
| 1 | 3.5 | 70 | 71 | |
| 2 | 4.39 | 88 | 90 | |
| Average | 3.94 ± 0.44 | 79 ± 9 | 81 ± 9 | |
Maximal length of the tether is 16,500 bp, which corresponds to ∼5.6 μm (0.34 nm/bp). The maximum length measured experimentally was ∼4.6 μm. The slight difference between the expected and the measured length is due to the low stretching force applied (at low forces, the tether is not maximally extended).
Estimated number of nucleosomes formed is calculated based on the measured shortening and the expected change of DNA tether per nucleosome formed; 147 bp corresponds to ∼50 nm of naked DNA. Taking into account a range of possible nucleosome repeat lengths 146–200 bp, the 16,500 bp can accommodate between 83 and 113 nucleosomes.
We took the average value (98) as the base for estimating the % of completion (number of nucleosomes formed/98).
Superhelical density σ that blocks nucleosome formation is estimated from the number of nucleosomes formed divided by total linking number of the remaining DNA tether (16,500—No. of nucleosomes × 147)/10.4. Values are reported ± SE.
Figure 3.
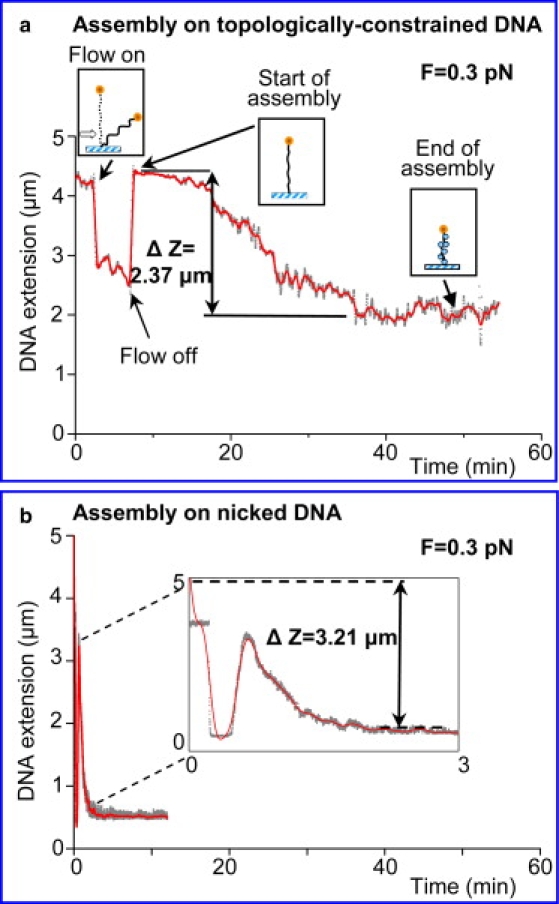
Real-time assembly of nucleosomal arrays followed as decrease in DNA tether length (Δz) as a result of DNA wrapping around the histone octamer. The flow of histone octamers and Nap1 into the cuvette causes apparent shortening of the tether; once the flow was stopped, the bead went back to its original position. The further changes in Δz reflected array assembly. (a) Example assembly curve on torsionally constrained DNA. The Δz (2.37 μm) indicates partial (∼48%) assembly. Even after 50 min the assembly did not go to completion. (b) Example assembly curve on nicked DNA as control. Inset shows more complete assembly (Δz = 3.20 μm; 78% nucleosome saturation) in ∼4 min.
These data indicate that on reaching ∼40% template saturation, the assembly process ceases (complete saturation is estimated based on the length of the DNA tether and a nucleosome repeat length ranging between 146–200 bps). This cessation could be explained by the accumulation of compensatory supercoiling within the DNA as a result of nucleosomal formation. Every nucleosome contains a full negative superhelical wrap of DNA around the histone core, i.e., has a linking number deficit of −1 (37–40); thus, the same level of opposite sense of supercoiling has to accumulate in the rest of the constrained template. In other words, with every nucleosome formed, a compensatory positive helical turn is built into the DNA tether as the magnetic bead is not able to freely rotate and relieve the superhelical stress. Therefore, with ΔLk = −1 per nucleosome formed and assuming that assembly happens only on the B part of the DNA, 16.5 kb in length, the estimated accumulated level of superhelical density σ that precluded further nucleosome formation was between +0.025 and +0.051 (at low stretching force, 0.3 pN, Table 1).
Nucleosome assembly on unconstrained (nicked) DNA substrates
We observed that with the age of the DNA preparation, more unconstrained tethers accumulated. These molecules had the proper length (∼4–4.5 μm), but failed to produce the expected rotation/extension curves (like the one presented in Fig. 2 b), i.e., they did not shorten on rotation of the external magnetic field. When nucleosome assembly was followed on such unconstrained (nicked) molecules, the assembly process happened much faster (within 10 ± 5 min) and to a much fuller extent (77 ± 4%) (Fig. 3 b, and Table 1).
Rotation of the external magnets to relieve the accumulating positive supercoiling in topologically constrained molecules leads to much more complete assembly
The comparison between the behavior of topologically constrained DNA templates and unconstrained (nicked) molecules hints at the possibility that accumulation of positive supercoiling in the remaining free (nonnucleosomal) DNA may be the main cause for the incomplete assembly on the constrained templates. We have therefore carried out further experiments on torsionally constrained DNA molecules, in which we mechanically relieved the accumulating positive supercoiling at certain time points (after some assembly had already occurred) by rotating the external magnetic field (clockwise in our experimental setup). We chose to relieve the stress at 20 min after the start of assembly based on the experiments with torsionally constrained DNA, a time point where the process slowed down considerably (see Fig. 3 a) and the observed tether shortening exceeded ∼2 μm. As can be seen from the curves in Fig. 4, rotation of the external magnets did lead to more complete assembly, comparable to that observed on nicked DNA molecules (see Table 1). The whole assembly processes took longer (>60 min) than on unconstrained DNA molecules, but at the end a more complete assembly was achieved. In one case we observed a minor (∼0.1 μm) tether extension (jump) at the point when the magnets were rotated. All other cases of external magnet rotation yielded no observable extension of the tether, which points out that the magnet rotation did not result in relieving any considerable amount of writhe (plectonemes).
Figure 4.
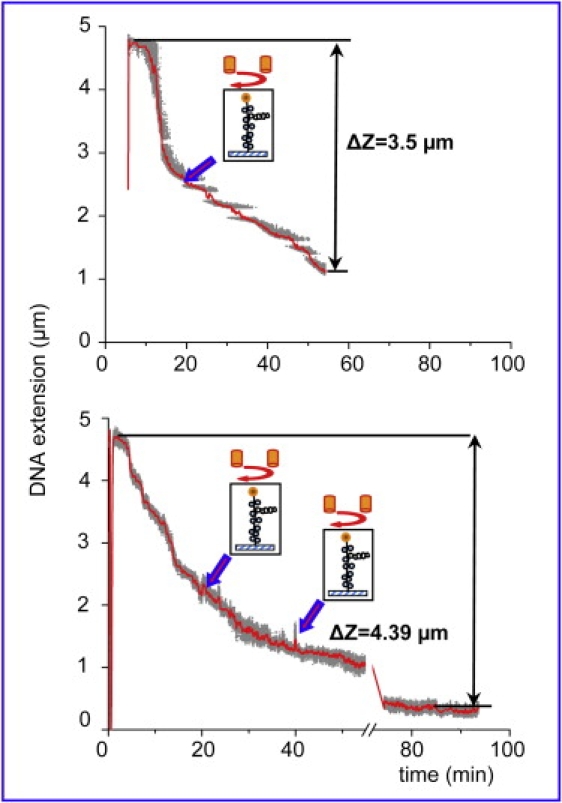
Assembly curves on torsionally constrained DNA after intermittent relief of the accumulating supercoiling stress by rotating the external magnetic field. (Top curve) The external magnets were rotated once after 20 min of assembly (52 clockwise rotations that neutralize the positive supercoiling accumulating in the DNA template). The mechanically induced relaxation led to further assembly (Δz = 3.5 μm). (Bottom curve) The magnets were rotated twice: at 20 min (52 negative turns) and at 40 min (20 negative turns). More complete assembly (Δz = 4.39 μm) was achieved in ∼93 min.
Discussion
Previous literature reports on single-molecule chromatin assembly used DNA tethers that were free to swivel around the ends, i.e., dissipated the torsional stress that is expected to accumulate as a result of nucleosome formation. We used topologically constrained tethers to study the dependence of nucleosome assembly on torsion (a situation that is more physiologically relevant, see Introduction).
We have been successful in following assembly of individual nucleosomal arrays in real-time in the presence of Nap1. The constrained DNA template exhibits different behavior from unconstrained DNA during the nucleosome assembly process as it shortens significantly less (to ∼40% of the original DNA length; Fig. 3 a) than the nicked DNA (∼77%; Fig. 3 b). We carried out control experiments to show that Nap1 by itself does not contribute to the tether shortening (data now shown).
To estimate how many nucleosomes were formed, we made a relatively simple assumption about the tether length change per nucleosome formed, as explained in Table 1. A total of 147 bp of DNA contained in the nucleosome corresponds to 50 nm in length. One could argue that when a nucleosome forms, 147 bp (∼50 nm) of naked DNA is replaced with a nucleosome of ∼10 nm in diameter. It is however, very difficult, if not impossible, to theoretically take this into account because the effect of replacing naked DNA with a nucleosome of certain dimensions on the overall length of the tether would depend on the particular geometry of the fiber: the overall trajectory, the nucleosome-to-nucleosome orientation, etc. This is impossible to know (or model) at present. Therefore, we lean toward the value ∼50 nm per nucleosome. Our estimate is supported by actual experimental results in Bancaud et al. (36), who directly compared the length of preassembled defined nucleosomal arrays with that of the respective naked DNA template (obtained by stripping the histones from the fiber directly in the flow cell of their MT setup). Finally, in a separate set of experiments carried out on a shorter (8 kb) piece of DNA containing tandemly repeated nucleosome positioning sequence, we were able to observe somewhat discreet steps of ∼50 nm (Fig. S1 in the Supporting Material).
To test whether the reason for the partial assembly is indeed the buildup of positive supercoiling (that is expected to counteract the negative wrapping of the DNA in the nucleosome particle), we have mechanically relieved the positive supercoiling by rotating the external magnets (clockwise in our setup) at specific time points of the assembly. We have seen resumption of assembly, to a much more complete nucleosome saturation. Thus, chromatin assembly on torsionally constrained DNA is highly sensitive to DNA positive supercoiling. Nucleosome formation stops at “stalling” superhelical density of +0.038.
The creation of supercoiling in the DNA/chromatin template is a reality during the translocation of the transcription bubble along the DNA double helix (10). Because the eukaryotic RNA polymerases are immobile (8,41), the transcription bubble does not rotate, but only translocates with respect to the fixed ends of the DNA (42) (reviewed in Lavelle (9)). The rotating DNA creates positive superhelical torsion ahead of the advancing polymerase and negative supercoiling in the wake of the enzyme. The biological significance of supercoiling in processes, such as transcription through nucleosomes, has become especially prominent after the observation that core RNA polymerase II activity was inhibited on nucleosomal templates. This was in contrast to the holoenzyme, which was not markedly repressed. This difference was attributed to the presence of topoisomerase IIα in the holoenzyme (41). These in vitro results are in agreement with the well-documented role of topoisomerase in eukaryotic gene transcription in vivo (43,44). The in vitro work also showed that chromatin prevented the diffusion of superhelical tension off the free ends of the linear template. None of these phenomena are understood at the mechanistic level.
Recent magnetic tweezers studies (36,45) indicated that nucleosomal arrays can absorb a significant amount of positive stress, without nucleosome disassembly. This behavior was explained by conformational transitions in nucleosomes: due to thermal fluctuations the entry/exit linkers alternate their relative orientation, passing from the canonical “negatively” crossed configuration (as observed in crystals) to a “positively” crossed state, possibly going through an “open” state (in which the linkers do not cross at all). Applying positive torsional stress shifts the equilibrium toward the positively crossed state. When the torsional stress reaches a critical value, the nucleosomes undergo a structural transition, through a chiral transition of the DNA superhelix (going from negatively to positively wrapped), while preserving the whole histone octamer (for further in-depth discussion, see Zlatanova et al. (46)). In such a structural transition, the linking number per nucleosome changes from −1 to +1, so that every nucleosome absorbs up to two turns when subjected to high levels of positive torsional stress; in other words, the L-octasome to R-octasome transition acts as a topological buffer. Thus, already existing “canonical” nucleosomes may convert into R-octasomes under positive torsion. Chromatin containing positively supercoiled DNA was shown to possess conformationally altered nucleosomes in vivo (47); however, the exact structure of these altered nucleosomes remains to be determined (46).
But can nucleosomes form on positively supercoiled DNA to start with? Some studies reported classical nucleosomes on positively supercoiled DNA (48,49), whereas others found that nucleosomes failed to form on positively supercoiled DNA and that H2A/H2B and H3/H4 had different affinities for DNA of different signs of supercoiling (50).
Our results seem to conform with the idea that positive supercoiling is inhibitory to nucleosome formation. ∼40% template saturation achieved on torsionally constrained DNA would result in σ ∼ 0.038. This level of superhelical density seems higher than the point of DNA buckling instability on naked DNA in the low-force regime (the transition to plectoneme formation occurs at σ = 0.01–0.015; see Fig. 2 b). On the other hand, the model described in Bancaud et al. (36) postulates that nucleosomal arrays can act as a topological buffer and absorb more torsion than naked DNA. Because our substrate consists of a mix of DNA and nucleosomes at each time point of assembly, it is difficult to make a reasonable prediction of when the twist/writhe transition would occur. The situation gets more complicated in view of the observation of Bancaud et al. (36) that on semisaturated fibers the extra positive (or negative) stress leads to length-versus-rotation slope of ∼25 nm/turn and this slope seems to depend on the saturation level. The authors also state that plectonemes cannot form on the DNA spacers flanking their nucleosomal array because the torque required for reaching the buckling transition is lower for nucleosomal fiber (3 pN nm rad−1). Our data are not easily comparable because we observe the process of assembly (demonstrated as tether shortening) under progressively changing conditions within the fiber. Although we cannot completely exclude the possibility of plectoneme formation in our experiments on topologically constrained templates (that would definitely shift our superhelical density estimate to lower values), we do not see strong evidence for their existence. Thus, we do not observe any tether extension after the magnet rotations are introduced, which points out that until that time no plectonemes had been formed. Additionally, Bancaud et al. (36) observed a progressive tether shortening of their nucleosome fibers after introducing extra positive rotations, supposedly due to formation of plectonemes within the fiber. Our DNA in constrained tether assembly experiments does not reach possible minimal tether length, thus supporting the idea that no plectoneme formation occurs in our assembly experiments.
A possible contributor to the tether shortening might be nucleosome-nucleosome interactions, that might be expected to occur at the physiological ionic strength we work at. The strength of these interactions remains a matter of debate (51), and the degree to which they occur depends on the nucleosome saturation levels in the arrays. In our topologically constrained DNA experiments, assembly stalls long before saturation and therefore we do not expect major contribution of nucleosome-nucleosome interaction to the observed shortening. Considering that an error in the estimated number of nucleosomes would play a role, our superhelical density estimate should be considered an upper limit.
Thus, we favor the possibility that it is the accumulation of torsional stress (change in the twist of the DNA) that changes the conformation of the DNA in such a way that it becomes refractory to proper interactions with histones to form nucleosomes. This interpretation may also explain earlier results (48,49) that showed nucleosome formation on positively supercoiled DNA plasmids in which the DNA was not torsionally stressed.
Supporting Material
A figure is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01515-X.
Supporting Material
Acknowledgments
The authors thank Dr. A. Revyakin (University of California, Berkeley) for providing data acquisition software, the unpublished surface modification protocol, technical advice, and critical reading of the manuscript, C. Geisler for help with supercoiled DNA preparation, and C. Seebart for help with Nap1 purification.
This work was supported by the National Science Foundation (grant 0504239).
References
- 1.van Holde K.E. Springer-Verlag; New York: 1989. Chromatin. [Google Scholar]
- 2.Widom J. Structure, dynamics, and function of chromatin in vitro. Annu. Rev. Biophys. Biomol. Struct. 1998;27:285–327. doi: 10.1146/annurev.biophys.27.1.285. [DOI] [PubMed] [Google Scholar]
- 3.Zlatanova J., Leuba S.H. Elsevier; Amsterdam, New York: 2004. Chromatin Structure and Dynamics: State-of-the-Art. [Google Scholar]
- 4.Arents G., Moudrianakis E.N. Topography of the histone octamer surface: repeating structural motifs utilized in the docking of nucleosomal DNA. Proc. Natl. Acad. Sci. USA. 1993;90:10489–10493. doi: 10.1073/pnas.90.22.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 6.van Holde K., Zlatanova J. The nucleosome core particle: does it have structural and physiologic relevance? Bioessays. 1999;21:776–780. doi: 10.1002/(SICI)1521-1878(199909)21:9<776::AID-BIES9>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 7.Zlatanova J., Seebart C., Tomschik M. The linker-protein network: control of nucleosomal DNA accessibility. Trends Biochem. Sci. 2008;33:247–253. doi: 10.1016/j.tibs.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Cook P.R. The organization of replication and transcription. Science. 1999;284:1790–1795. doi: 10.1126/science.284.5421.1790. [DOI] [PubMed] [Google Scholar]
- 9.Lavelle C. Forces and torques in the nucleus: chromatin under mechanical constraints. Biochem. Cell Biol. 2009;87:307–322. doi: 10.1139/O08-123. [DOI] [PubMed] [Google Scholar]
- 10.Liu L.F., Wang J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berezney R. Regulating the mammalian genome: the role of nuclear architecture. Adv. Enzyme Regul. 2002;42:39–52. doi: 10.1016/s0065-2571(01)00041-3. [DOI] [PubMed] [Google Scholar]
- 12.Zlatanova J.S., van Holde K.E. Chromatin loops and transcriptional regulation. Crit. Rev. Eukaryot. Gene Expr. 1992;2:211–224. [PubMed] [Google Scholar]
- 13.Harada Y., Ohara O., Takatsuki A., Itoh H., Shimamoto N. Direct observation of DNA rotation during transcription by Escherichia coli RNA polymerase. Nature. 2001;409:113–115. doi: 10.1038/35051126. [DOI] [PubMed] [Google Scholar]
- 14.Pomerantz R.T., Ramjit R., Gueroui Z., Place C., Anikin M. A tightly regulated molecular motor based upon T7 RNA polymerase. Nano Lett. 2005;5:1698–1703. doi: 10.1021/nl0509125. [DOI] [PubMed] [Google Scholar]
- 15.March issue devoted to reviews on single-molecule approaches. Science. 1999;283 [Google Scholar]
- 16.Bustamante C., Bryant Z., Smith S.B. Ten years of tension: single-molecule DNA mechanics. Nature. 2003;421:423–427. doi: 10.1038/nature01405. [DOI] [PubMed] [Google Scholar]
- 17.Greenleaf W.J., Woodside M.T., Block S.M. High-resolution, single-molecule measurements of biomolecular motion. Annu. Rev. Biophys. Biomol. Struct. 2007;36:171–190. doi: 10.1146/annurev.biophys.36.101106.101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joo C., Balci H., Ishitsuka Y., Buranachai C., Ha T. Advances in single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- 19.Leuba S.H., Zlatanova J. Pergamon; Amsterdam, New York: 2001. Biology at the Single Molecule Level. [Google Scholar]
- 20.van Holde K.E. Biochemistry at the single-molecule level: minireview series. J. Biol. Chem. 1999;274:14515. doi: 10.1074/jbc.274.21.14515. [DOI] [PubMed] [Google Scholar]
- 21.Zlatanova J., van Holde K. Single-molecule biology: what is it and how does it work? Mol. Cell. 2006;24:317–329. doi: 10.1016/j.molcel.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 22.Zlatanova J., Leuba S.H. Chromatin fibers, one-at-a-time. J. Mol. Biol. 2003;331:1–19. doi: 10.1016/s0022-2836(03)00691-0. [DOI] [PubMed] [Google Scholar]
- 23.Zlatanova J., Leuba S.H. Magnetic tweezers: a sensitive tool to study DNA and chromatin at the single-molecule level. Biochem. Cell Biol. 2003;81:151–159. doi: 10.1139/o03-048. [DOI] [PubMed] [Google Scholar]
- 24.Bennink M.L., Leuba S.H., Leno G.H., Zlatanova J., de Grooth B.G. Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nat. Struct. Biol. 2001;8:606–610. doi: 10.1038/89646. [DOI] [PubMed] [Google Scholar]
- 25.Brower-Toland B.D., Smith C.L., Yeh R.C., Lis J.T., Peterson C.L. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl. Acad. Sci. USA. 2002;99:1960–1965. doi: 10.1073/pnas.022638399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennink M.L., Pope L.H., Leuba S.H., de Grooth B.G., Greve J. Single chromatin fibre assembly using optical tweezers. Single Mol. 2001;2:91–97. [Google Scholar]
- 27.Ladoux B., Quivy J.P., Doyle P., du Roure O., Almouzni G. Fast kinetics of chromatin assembly revealed by single-molecule videomicroscopy and scanning force microscopy. Proc. Natl. Acad. Sci. USA. 2000;97:14251–14256. doi: 10.1073/pnas.250471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leuba S.H., Karymov M.A., Tomschik M., Ramjit R., Smith P. Assembly of single chromatin fibers depends on the tension in the DNA molecule: magnetic tweezers study. Proc. Natl. Acad. Sci. USA. 2003;100:495–500. doi: 10.1073/pnas.0136890100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomschik M., Karymov M.A., Zlatanova J., Leuba S.H. The archaeal histone-fold protein HMf organizes DNA into bona fide chromatin fibers. Structure. 2001;9:1201–1211. doi: 10.1016/s0969-2126(01)00682-7. [DOI] [PubMed] [Google Scholar]
- 30.von Holt C., Brandt W.F., Greyling H.J., Lindsey G.G., Retief J.D. Isolation and characterization of histones. Methods Enzymol. 1989;170:431–523. doi: 10.1016/0076-6879(89)70061-6. [DOI] [PubMed] [Google Scholar]
- 31.Fujii-Nakata T., Ishimi Y., Okuda A., Kikuchi A. Functional analysis of nucleosome assembly protein, NAP-1. The negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J. Biol. Chem. 1992;267:20980–20986. [PubMed] [Google Scholar]
- 32.Strick T.R., Allemand J.F., Bensimon D., Bensimon A., Croquette V. The elasticity of a single supercoiled DNA molecule. Science. 1996;271:1835–1837. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 33.Allemand J.F., Bensimon D., Lavery R., Croquette V. Stretched and overwound DNA forms a Pauling-like structure with exposed bases. Proc. Natl. Acad. Sci. USA. 1998;95:14152–14157. doi: 10.1073/pnas.95.24.14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McQuibban G.A., Commisso-Cappelli C.N., Lewis P.N. Assembly, remodeling, and histone binding capabilities of yeast nucleosome assembly protein 1. J. Biol. Chem. 1998;273:6582–6590. doi: 10.1074/jbc.273.11.6582. [DOI] [PubMed] [Google Scholar]
- 35.Zlatanova J., Seebart C., Tomschik M. Nap1: taking a closer look at a juggler protein of extraordinary skills. FASEB J. 2007;21:1294–1310. doi: 10.1096/fj.06-7199rev. [DOI] [PubMed] [Google Scholar]
- 36.Bancaud A., Conde e Silva N., Barbi M., Wagner G., Allemand J.F. Structural plasticity of single chromatin fibers revealed by torsional manipulation. Nat. Struct. Mol. Biol. 2006;13:444–450. doi: 10.1038/nsmb1087. [DOI] [PubMed] [Google Scholar]
- 37.Crick F.H. Linking numbers and nucleosomes. Proc. Natl. Acad. Sci. USA. 1976;73:2639–2643. doi: 10.1073/pnas.73.8.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Germond J.E., Hirt B., Oudet P., Gross-Bellark M., Chambon P. Folding of the DNA double helix in chromatin-like structures from simian virus 40. Proc. Natl. Acad. Sci. USA. 1975;72:1843–1847. doi: 10.1073/pnas.72.5.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keller W. Determination of the number of superhelical turns in simian virus 40 DNA by gel electrophoresis. Proc. Natl. Acad. Sci. USA. 1975;72:4876–4880. doi: 10.1073/pnas.72.12.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prunell A. A topological approach to nucleosome structure and dynamics: the linking number paradox and other issues. Biophys. J. 1998;74:2531–2544. doi: 10.1016/S0006-3495(98)77961-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mondal N., Parvin J.D. DNA topoisomerase IIα is required for RNA polymerase II transcription on chromatin templates. Nature. 2001;413:435–438. doi: 10.1038/35096590. [DOI] [PubMed] [Google Scholar]
- 42.Mondal N., Parvin J.D. Transcription from the perspective of the DNA: twists and bumps in the road. Crit. Rev. Eukaryot. Gene Expr. 2003;13:1–8. doi: 10.1615/critreveukaryotgeneexpr.v13.i1.10. [DOI] [PubMed] [Google Scholar]
- 43.Collins I., Weber A., Levens D. Transcriptional consequences of topoisomerase inhibition. Mol. Cell. Biol. 2001;21:8437–8451. doi: 10.1128/MCB.21.24.8437-8451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kouzine F., Sanford S., Elisha-Feil Z., Levens D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat. Struct. Mol. Biol. 2008;15:146–154. doi: 10.1038/nsmb.1372. [DOI] [PubMed] [Google Scholar]
- 45.Bancaud A., Wagner G., Conde E.S.N., Lavelle C., Wong H. Nucleosome chiral transition under positive torsional stress in single chromatin fibers. Mol. Cell. 2007;27:135–147. doi: 10.1016/j.molcel.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 46.Zlatanova J., Bishop T.C., Victor J.M., Jackson V., van Holde K. The nucleosome family: dynamic and growing. Structure. 2009;17:160–171. doi: 10.1016/j.str.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 47.Lee M.S., Garrard W.T. Positive DNA supercoiling generates a chromatin conformation characteristic of highly active genes. Proc. Natl. Acad. Sci. USA. 1991;88:9675–9679. doi: 10.1073/pnas.88.21.9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark D.J., Felsenfeld G. Formation of nucleosomes on positively supercoiled DNA. EMBO J. 1991;10:387–395. doi: 10.1002/j.1460-2075.1991.tb07960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clark D.J., Ghirlando R., Felsenfeld G., Eisenberg H. Effect of positive supercoiling on DNA compaction by nucleosome cores. J. Mol. Biol. 1993;234:297–301. doi: 10.1006/jmbi.1993.1585. [DOI] [PubMed] [Google Scholar]
- 50.Jackson V. What happens to nucleosomes during transcription? In: Zlatanova J., editor. Chromatin Structure and Dynamics: State-of-the-Art. Elsevier; Amsterdam: 2004. pp. 467–491. [Google Scholar]
- 51.Kruithof M., Chien F.T., Routh A., Logie C., Rhodes D. Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nat. Struct. Mol. Biol. 2009;16:534–540. doi: 10.1038/nsmb.1590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.