

Abstract
Single nucleotide polymorphisms (SNPs) in the KLK3 gene on chromosome 19q13.33 are associated with serum prostate-specific antigen (PSA) levels. Recent genome wide association studies of prostate cancer have yielded conflicting results for association of the same SNPs with prostate cancer risk. Since the KLK3 gene encodes the PSA protein that forms the basis for a widely used screening test for prostate cancer, it is critical to fully characterize genetic variation in this region and assess its relationship with the risk of prostate cancer. We have conducted a next-generation sequence analysis in 78 individuals of European ancestry to characterize common (minor allele frequency, MAF >1%) genetic variation in a 56 kb region on chromosome 19q13.33 centered on the KLK3 gene (chr19:56,019,829–56,076,043 bps). We identified 555 polymorphic loci in the process including 116 novel SNPs and 182 novel insertion/deletion polymorphisms (indels). Based on tagging analysis, 144 loci are necessary to tag the region at an r 2 threshold of 0.8 and MAF of 1% or higher, while 86 loci are required to tag the region at an r 2 threshold of 0.8 and MAF >5%. Our sequence data augments coverage by 35 and 78% as compared to variants in dbSNP and HapMap, respectively. We observed six non-synonymous amino acid or frame shift changes in the KLK3 gene and three changes in each of the neighboring genes, KLK15 and KLK2. Our study has generated a detailed map of common genetic variation in the genomic region surrounding the KLK3 gene, which should be useful for fine-mapping the association signal as well as determining the contribution of this locus to prostate cancer risk and/or regulation of PSA expression.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-009-0751-5) contains supplementary material, which is available to authorized users.
Introduction
Prostate cancer is the most commonly diagnosed non-cutaneous cancer in men in the US (Jemal et al. 2008). A widely used test for prostate cancer screening is based on measuring prostate-specific antigen (PSA) protein levels in serum. However, elevated PSA levels can also be caused by nonmalignant conditions such as benign prostatic hyperplasia and prostatitis (Punglia et al. 2006). Although the PSA test has led to the diagnosis of earlier stage prostate cancers, the specificity and sensitivity of the test is not optimal for clinical applications (Punglia et al. 2003; Thompson et al. 2004). Consequently, large randomized screening trials are currently ongoing to assess the benefits of the PSA test for prostate cancer screening and mortality rates. Although interim results have been published (Andriole et al. 2009; Schroder et al. 2009) the benefits, if any, of the PSA test as a diagnostic screening tool for prostate cancer are still not clear.
A recent genome wide association study (GWAS) of prostate cancer reported that several single nucleotide polymorphisms (SNPs) on chromosome 19q13.33 were associated with an increased risk of prostate cancer (Eeles et al. 2008b). The most significant SNP, rs2735839, is located 600 bp downstream of the KLK3 gene, which encodes kallikrein 3 (hK3), also known as PSA. A notable feature of this study was that control individuals were selected to have low PSA levels (<0.5 ng/ml). In a separate GWAS of prostate cancer conducted within the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, the findings on chromosome 19 were not confirmed for prostate cancer susceptibility (Thomas et al. 2008). Further analysis of the results from the PLCO study indicated an association between rs2735839 and PSA levels; interestingly, when the controls were restricted to those with very low PSA levels, a strong association was observed with prostate cancer risk (Ahn et al. 2008). In the first study that reported the chromosome 19 association with prostate cancer, the authors reported evidence for association of rs2735839 and prostate cancer in additional studies unselected for PSA levels but the level of significance was substantially less (Eeles et al. 2008a) than in the discovery set with the low PSA controls (Eeles et al. 2008b). Additional studies have also shown association between rs2735839 and PSA levels in control individuals and with less aggressive prostate cancer, but selection biases for elevated PSA may have played a role (Kader et al. 2009; Xu et al. 2008). Thus, there is evidence that the locus on chromosome 19q13.33 is associated with PSA levels and possibly also prostate cancer.
To conduct follow-up studies in this region of chromosome 19q33.13, we generated a map of common SNPs and insertion/deletion polymorphisms (indels) through a deep sequencing analysis of a 56 kb region flanking rs2735839 (chr19:56,019,829–56,076,043 bps; NCBI Build 36.3) in 78 unrelated individuals of European ancestry using a novel solution-based sequence capture method, combined with the Roche-454 platform (Rothberg and Leamon 2008). This region chosen for targeted resequencing is centered on the KLK3 gene but also includes the neighboring genes KLK15 (centromeric) and KLK2 (telomeric). We identified 555 polymorphic loci including 116 novel SNPs and 182 novel indels. Eleven coding variants were identified in the KLK3 gene, including five that result in non-synonymous amino acid changes and one that causes a frameshift in the protein. Four coding variants were identified in the neighboring KLK15 gene and five in the KLK2 gene. This catalog of common genetic variation establishes a foundation for comprehensively tagging the region in individuals of European ancestry for fine-mapping studies.
Materials and methods
Samples
DNA samples were from 41 prostate cancer cases and 33 controls individuals (total 74) from the National Cancer Institute’s (NCI) Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial (Gohagan et al. 2000). They were drawn from samples analyzed in the initial scan of the Cancer Genetics Markers of Susceptibility (CGEMS) Initiative (http://cgems.cancer.gov) (Thomas et al. 2008; Yeager et al. 2007). Approximately half of the samples from prostate cancer cases were selected on the basis of high PSA levels (>4 ng/ml, n = 21) and the other half with low PSA levels (<4 ng/ml, n = 20). In a similar manner, approximately half of the control samples came from individuals with low normal PSA levels (<0.5 ng/ml, n = 17) while the other half were within the high normal range (2.5–3.9 ng/ml, n = 16).
DNA samples from 18 individuals in HapMap CEU pedigrees were sequenced: 2 three generation CEPH families (14 individuals from families #1350 and #1444) and two sets of parents (4 individuals from families #1334 and #1340). Finally, two trios from YRI pedigrees were included (6 individuals from families #5 and #16).
Region sequenced
We sequenced a 56 kb genomic region on chromosome 19 (56,019,829–56,076,043 bps, NCBI Build 36.3) selected based on the pattern of linkage disequilibrium (LD) around rs2735839 in the HapMap CEU samples using Haploview (Barrett et al. 2005). The region was selected to include the most significant SNP associated with prostate cancer and PSA levels, rs2735839, and the neighboring region based on the genetic map (Ahn et al. 2008; Eeles et al. 2008b). The boundaries were extended to include adjacent blocks based on the pattern of LD observed in this region using Phases I and II HapMap Caucasian samples (http://www.hapmap.org). Finally, we extended the region to fully include the three kallikrein genes: KLK15, KLK3 and KLK2.
Primers, sequence capture and sequencing
Thirteen sets of long-range PCR primers were designed to cover the 56 kb region targeted. Amplicon size ranged from 3,442 to 5,099 bps and primer sets overlapped, on average, 377 bps with the adjacent amplicon. Primers were designed using Primer3 (http://frodo.wi.mit.edu) (Rozen and Skaletsky 2000) and then quality checked in silico for uniqueness, potential sequence paralogy and DNA repeat sequences using the BLAT feature of the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgBlat). Next, NetPrimer (http://www.premierbiosoft.com/netprimer/index.html) was used to check for secondary structures and PCR efficiencies. Primers were ordered from Integrated DNA Technologies (Coralville, IA). All primers and coordinates are supplied in Supplemental Table 1.
Biotinylated solution capture probe pools targeting the regions of interest were generated for sequence capture (Roche NimbleGen, Madison, WI) (Albert et al. 2007). Capture was performed in solution in 0.2 ml PCR strip tubes on a thermal cycler. After ~72 h, the capture probe/sample duplexes were bound to streptavidin magnetic beads. Captured samples were then amplified directly off the beads and prepped for sequencing (454 linker ligation). After long-range PCR, all sequencing protocols were followed in accordance with standard kits for the 454 GS FLX system (http://www.454.com/products-solutions/productlist.asp).
Alignment and detection of polymorphisms
We developed an automated computational pipeline to process sequence reads generated by 454 FLX Genome Sequencers. Whenever applicable, sequence reads from the same sample were pooled based on barcodes provided by Roche/454. Quality check (QC) was performed using vendor-supplied software and sequence reads that passed QC were aligned to the target genomic region (chr19:56,019,829–56,076,043 bps) using the MOSAIK software (http://bioinformatics.bc.edu/marthlab/Mosaik). The resulting assembly was analyzed using a column by column approach and potential polymorphic sites and most likely genotypes were called based on a set of heuristic rules. The minimal sequence coverage depth was set to 20 reads for each nucleotide position. In addition, the ratio (r) of forward and reverse reads was determined. To avoid directional bias, an optimal range of r was set between 10 and 90%. Homozygous genotype calls were made when the most frequent allele was present in at least 85% of the reads. Heterozygous genotype calls were made when the two most frequent alleles were represented in 30–70% of reads. Genotype calls were not made if the above criteria were not met. Manual inspections aided by the NextGENe software (http://www.softgenetics.com) and Consed (http://bozeman.mbt.washington.edu/consed/consed.html) were performed to quality assurance (QA) and to resolve ambiguous cases.
Supplemental Table 2 includes flanking sequences for polymorphic loci that passed QC and had over 50% completion rates.
Concordance analysis
Concordance analysis between the re-sequencing data and CGEMS PLCO scan data was assessed for 74 individuals (cases and controls). Genotype data for the region (chr19:56,019,829–56,076,043 bps) was downloaded from HapMap (http://www.hapmap.org/) and the 1000 Genomes Project (http://www.1000genomes.org/; imputed genotypes were not included; Data release: May, 2009) to evaluate genotype concordance between re-sequencing data and HapMap or 1000 Genomes Project data, respectively. Twenty-one HapMap samples were used to calculate concordance between the present study and HapMap data. Ten HapMap samples were used to calculate concordance between the present study and the 1000 Genomes Project data. 102 HapMap samples, included in the 1000 Genomes Project data set were used to calculate concordance between HapMap and 1000 Genomes Project data. Genotype concordance was computed using the GLU software package (http://code.google.com/p/glu-genetics/).
A two group χ2 test of equal proportions (Newcombe 1998) was performed to evaluate the differences in minor allele frequencies (MAFs) for each of 243 SNPs detected both in this study and the CEU population available in the 1000 Genomes Project data. To correct for multiple testing, a q value was calculated using the QVALUE software package (Storey and Tibshirani 2003) for each test. This method measures significance in terms of a false discovery rate (Storey and Tibshirani 2003). Statistical analyses were performed using the R statistical software (http://www.r-project.org/).
Descriptive statistics
Genotype completion, MAF estimations, deviations from fitness for Hardy–Weinberg proportion (HWP), pairwise linkage disequilibrium (LD) and tag SNP selection were computed using the GLU software package. Inheritance check analysis was performed on samples from HapMap families in Haploview (Barrett et al. 2005). Data from 78 unrelated individuals of European ancestry (66 cases/controls and 12 HapMap CEU) was used for SNP tagging using the GLU software package. LD was visualized in Haploview (Barrett et al. 2005).
In silico genomic analysis
The presence of highly conserved regions, copy number variation and predicted regulatory elements was assessed by using publicly available databases and bioinformatics tools: the UCSC genome browser (http://genome.ucsc.edu), the Copy Number Variation database at the Children’s Hospital of Philadelphia (http://cnv.chop.edu), the Database of Genomic Variants (http://projects.tcag.ca/variation/) and the VISTA Enhancer database (http://pipeline.lbl.gov/cgi-bin/gateway2).
Results
Sequence coverage and depth
The average depth and coverage in the genomic region sequenced (chr19:56,019,829–56,076,043 bps) is shown in Fig. 1. No gaps were observed in the coverage and the average depth was 70-fold (range 1- to 136-fold). Low coverage (<20-fold) was seen in six small regions (cumulative length 1,393 bp) listed in Supplemental Table 3.
Fig. 1.
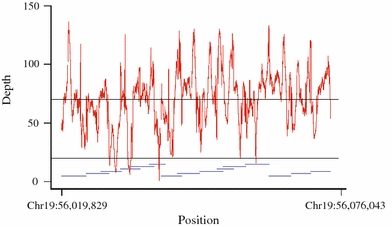
Coverage and sequence depth over the 56 kb region sequenced on chromosome 19q13.33. The horizontal line at 70-fold represents the average depth and the line at 20-fold represents the cutoff for low coverage. The blue horizontal lines represent primer amplicon
Polymorphism discovery and quality control assessment
Genotypes were called for 652 possible SNPs and indels in 74 samples from prostate cancer patients and controls from the PLCO Cancer Screening Trial (Gohagan et al. 2000), 18 HapMap CEU samples and 6 HapMap YRI samples. During data quality control assessment, samples were excluded when genotype completion was less than 50% or genotypes showed discordance with CGEMS PLCO or HapMap genotype data. After excluding four samples with 75% or less genotype concordance with the CGEMS PLCO data, concordance for the remaining samples was 100%. No samples were excluded based on low concordance with HapMap data (overall concordance 99.8%). Loci were excluded based on departures from Hardy–Weinberg equilibrium (P < 0.001, n = 1) or if they were monomorphic (n = 92) in our samples (Supplemental Table 4). No loci were dropped due to low completion rates. For inheritance check analysis, only members of the HapMap families were analyzed. No samples or SNPs were excluded on the basis of Mendelian errors (overall Mendelian error rate 0%).
A comparison of our dataset with an early build of the 1000 Genomes Project data showed 95.6% concordance rate, which is not surprising based on the preliminary nature of the data release. The concordance between HapMap data and 1000 Genomes data was 97.1%. We did not observe significant differences in MAFs for 243 SNPs detected both in this study and the CEU population available in the 1000 Genomes Project data set (average difference in MAFs = 0.008, range of q values 0.22–1; median 1). The total number of SNPs present in the 1000 Genomes Project in the targeted region was 365 as compared to 373 in the present study.
The final dataset for individuals of European ancestry contained genotypes from 78 individuals (66 PLCO and 12 unrelated HapMap samples) and 555 polymorphic loci (Table 1). These include 116 new SNPs, 182 new indels and 257 loci previously described in NCBI’s dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/). The latter number includes 81 HapMap SNPs (http://www.hapmap.org). The average locus call (completion) rate was 74.4% (range 2.6–100%, median 87.2%) as shown in Supplemental Fig. 1. MAF estimates were computed for each locus and were on average 13.2% (range 0.6–50%, median 6.8%). The number of new SNPs and indels with MAF >5% was 85. Supplemental Fig. 2 shows the distribution of MAFs for new and known polymorphisms detected in the study. Since our indel calling algorithm is still being refined, these low-frequency variants should be treated as preliminary data and require validation. Hence, for the subsequent analysis we only included loci with completion rates >50% which included 444 polymorphic loci (112 new SNPs, 96 new indels and 236 loci previously described in NCBI’s dbSNP database).
Table 1.
Distribution of new and known SNPs and indels that were polymorphic in samples of European ancestry
| Category | SNPs | Indels | Total variants | Completion >50% | ||||
|---|---|---|---|---|---|---|---|---|
| MAF ≥1% | MAF ≥5% | All | MAF ≥1% | MAF ≥5% | All | |||
| dbSNP | 248 | 211 | 251 | 6 | 3 | 6 | 257 | 236 |
| HapMapa | 80 | 68 | 81 | 0 | 0 | 0 | 81 | 80 |
| Illuminab | 28 | 26 | 29 | 0 | 0 | 0 | 29 | 27 |
| Novel | 55 | 19 | 116 | 159 | 66 | 182 | 298 | 208 |
| All | 223 | 162 | 367 | 165 | 69 | 188 | 555 | 444 |
SNPs single nucleotide polymorphisms, indels insertion and deletion polymorphisms, MAF minor allele frequency
aHapMap phase I, II and III
bIllumina HumanHap610 assay. Note that HapMap and Illumina SNPs are also part of dbSNPs
Loci within the KLK15, KLK3 and KLK2 genes
Twenty polymorphic sites were identified in the KLK3 gene: five synonymous, five non-synonymous, one frameshift (resulting in a stop codon 23 amino acids downstream of the site) and nine variants that affect the 3′ untranslated (3′UTR) of different KLK3 isoforms. The proximal promoter contains three androgen-responsive elements (AREs), ARE I, ARE II and ARE III, centered at −170 bp, −394 bp and −4,200 bp, respectively from the transcription start site, that are known to influence KLK3 expression (Cleutjens et al. 1996; 1997; Schuur et al. 1996). The presence of two SNPs known to overlap with functionally validated regulatory elements for the KLK3 gene was confirmed (rs266882 in ARE I and rs925013 in an upstream enhancer that contains ARE III) (Cleutjens et al. 1996; 1997; Cramer et al. 2003; Schuur et al. 1996).
Four coding variants (three non-synonymous and one synonymous) were observed in the KLK15 gene. Three additional polymorphic sites were located in the 3′ UTR region of the gene. Five coding variants (1 non-synonymous, 2 synonymous and 2 frameshift) were observed in the KLK2 gene and eight sites were located within the 3′ UTR region. Supplemental Table 5 lists polymorphic loci observed in the three genes and the resulting changes in amino acid sequences of different KLK isoforms.
Linkage disequilibrium (LD) and tag SNP selection
Based on our sequence data, the map of LD of common variants (MAF >5%) demonstrates that there are two blocks of high LD in the telomeric part of the region from approximately 56,063–56,070 and 56,072–56,075 kb. On the other hand, the centromeric part of the region sequenced from ~56,020 to 56,063 kb has low LD (Fig. 2). The genetic map of this region has been refined compared to HapMap data (Supplemental Fig. 3); overall, the current sequence data corroborates the two HapMap recombination hotspots located at approximately 56,024–56,026 kb (recombination rate 54 cM/Mb) and 56,067–56,071 kb (recombination rate 31 cM/Mb), respectively.
Fig. 2.
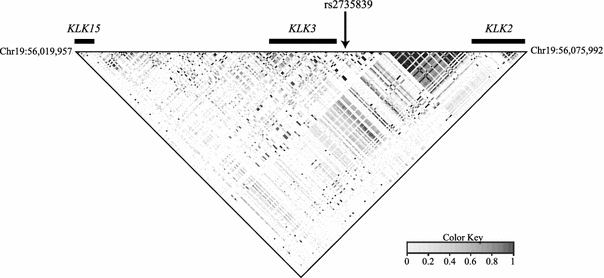
Linkage disequilibrium (LD) plot across the KLK locus on chromosome 19q13.33 as measured by r 2. Polymorphisms in this study with MAF >5% and completion rates >50% are included. Relative location of KLK15, KLK3, KLK2 and rs2735839 are shown. Coordinates are based on NCBI genome build 36.3
The main SNP associated with prostate cancer from Eeles et al. (2008b) and with PSA levels from Ahn et al. (2008), rs2735839, is located at 56,056 kb in an approximately 37 kb region of relatively low LD. Tagging the whole region with an r 2 threshold of 0.8 and using rs2735839 as an obligate included marker yielded a total of 144 tag SNPs that are necessary to cover the 357 loci with a MAF >1% (Table 2). The bin containing rs2735839 contains two additional SNPs located 0.4 kb (rs2569735, MAF 15.6%) and 1.2 kb centromeric (rs1058205, MAF 17.3%) on chromosome 19. At a MAF of 5% or higher, 86 tags are needed to cover 227 loci (Table 2). Supplemental Table 6 lists the bin and tag SNP information using thresholds of MAF ≥1% or 5% and r 2 ≥ 0.8. SNPs exhibiting high pairwise LD (r 2 ≥ 0.8) with rs2735839 are listed in Table 3.
Table 2.
Tag SNP information, bins and coverage in samples of European ancestry
| Category | n a | MAF ≥1% and r 2 ≥ 0.8 | MAF ≥5% and r 2 ≥ 0.8 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bins monitored | Variants monitored | Coverage (%) | Bins monitored | Variants monitored | Coverage (%) | ||||||
| Yes | No | Yes | No | Yes | No | Yes | No | ||||
| dbSNP | 236 | 96 | 48 | 233 | 124 | 65 | 76 | 10 | 194 | 33 | 85 |
| HapMapb | 80 | 42 | 102 | 79 | 278 | 22 | 33 | 53 | 67 | 160 | 30 |
| Illuminac | 27 | 21 | 123 | 27 | 330 | 8 | 19 | 67 | 25 | 202 | 11 |
| Novel | 208 | 57 | 87 | 124 | 233 | 35 | 15 | 71 | 33 | 194 | 15 |
| All | 444 | 144 | 0 | 357 | 0 | 100 | 86 | 0 | 227 | 0 | 100 |
SNPs single nucleotide polymorphisms, MAF minor allele frequency. Only markers with completion rates >50% were used for tagging. Note that HapMap and Illumina SNPs are part of dbSNPs
aNumber of SNPs and indels used for tagging
bHapMap phase I, II and III
cIllumina HumanHap610 assay
Table 3.
SNPs with r 2 > 0.8 with rs2735839
| Locus | r 2 | MAF | Allele count |
|---|---|---|---|
| rs2569735 | 1.00 | 0.156 | 130|24 |
| rs1058205 | 0.91 | 0.173 | 129|27 |
SNPs single nucleotide polymorphisms, MAF minor allele frequency, Allele count major allele|minor allele
In silico genomic and copy number analysis
The Copy Number Variation project at the Children’s Hospital of Philadelphia (http://cnv.chop.edu) reports heterozygous deletions within the region sequenced. Deletions were reported in 20/1,320 (1.52%) individuals of European American ancestry and 4/694 (0.58%) individuals of African American ancestry. The CNVs map to 56,022,744–56,024,482 (1,739 bps) and 56,022,744-56,028,151 (5,408 bps) and are seen in 22 and 2 individuals, respectively (Shaikh et al. 2009). The Database of Genomic Variants (http://projects.tcag.ca/variation/) reports a loss of small genomic region about 10 kb telomeric (chr19:56,034,084–56,034,206 bps) but it is seen in one individual (Levy et al. 2007).
Substantial mammalian conservation was noted for the exons of all three genes using the UCSC browser (http://genome.ucsc.edu). Sequences with predicted regulatory potential (King et al. 2005) were seen upstream of all genes, and in some intronic regions, especially in the KLK15 gene. No experimentally validated or predicted enhancers are listed in the VISTA browser (http://pipeline.lbl.gov/cgi-bin/gateway2) for the region (data not shown) (Couronne et al. 2003).
Discussion
In this study, we have characterized common genetic polymorphisms (SNPs and indels) spanning a 56 kb region on chromosome 19q13.33 in 78 individuals (156 chromosomes) of European ancestry by using 454 next-generation sequencing (Rothberg and Leamon 2008) coupled with a novel solution-based sequence capture method. This capture method provides a reliable and less labor intensive alternative to long-range PCR when sequencing large genomic regions. We discovered 298 new polymorphisms (116 SNPs and 182 indels) and confirmed 257 previously known loci in the process and constructed a detailed LD map of the region. A large fraction (~65%) of the SNPs described here has also been observed in an early release of the 1000 Genomes Project. Many of the indel polymorphisms detected are rare and validation is required to conclusively establish allele frequencies. Our analysis provides a comprehensive inventory of common genetic variation in the region surrounding the KLK3 gene and allows for the selection of tag SNPs to be used in follow-up studies to thoroughly examine the association of genetic polymorphisms on chromosome 19q13.33 to prostate cancer risk and PSA levels. At an r 2 threshold of 0.8 and MAF of 1% or higher, 144 variants are necessary to tag the region, and at an r 2 threshold of 0.8 and MAF >5%, 86 loci are required. The resulting improvement in coverage is an additional 78% as compared to HapMap SNPs and 35% over variants known prior to this study (dbSNP).
Chromosome 19q13.33 harbors a cluster of 15 kallikrein genes tandemly arranged over ~300 kb. Three genes that belong to this family of serine proteases are located within the region sequenced in this study: KLK15, KLK3 and KLK2. The KLK3 gene encodes PSA, a protein that is produced almost solely by the prostate gland. Small amounts of PSA are detectable in the bloodstream of healthy men (Lilja 1985). An increase in serum PSA levels in men with prostate cancer forms the basis of the PSA test, a widely used screening tool for prostate cancer. The lack of specificity and sensitivity of the test has led to questions about its usefulness as a screening tool for prostate cancer and two large prospective randomized trials are currently underway to directly assess its benefits: The Prostate, Lung, Colorectal and Ovarian Cancer Screening trial (PLCO) (Andriole et al. 2009) and the European Randomized Study of Screening for Prostate Cancer (ERSPC) (Schroder et al. 2009).
The KLK2 and KLK15 genes have also been implicated in prostate cancer etiology. The KLK2 gene is expressed in the prostate gland and has been proposed as a potential marker for prostate cancer. Like PSA, human kallikrein 2 (hK2) levels in the bloodstream are strongly associated with prostate cancer but do not increase the value of total PSA measurements for predicting risk of disease (Lilja et al. 2007). Interestingly, KLK3 and KLK2 share ~80% nucleotide sequence identity across exons, introns and non-coding regions of the two genes, suggesting a recent duplication event (Gan et al. 2000). PSA and hK2 also share ~80% amino acid identity (Gan et al. 2000). KLK15 is the next gene centromeric to KLK3 and shares considerable similarities to other kallikrein genes. It encodes yet another member of the kallikrein family, hK15. Expression of the KLK15 gene appears to be upregulated in a large percentage of prostate cancers and is possibly associated with a higher stage disease (Stephan et al. 2003; Yousef et al. 2001).
Previous association studies with candidate or tag SNPs have reported a number of SNPs in or near the KLK3 gene that appear to be associated with prostate cancer, PSA levels or both (Cramer et al. 2003; 2008; Pal et al. 2007). Results from GWAS and their follow-up studies are conflicting, and it appears that the association to prostate cancer may depend on how control individuals were selected. The SNP most significantly associated with prostate cancer risk (Eeles et al. 2008b) and PSA levels (Ahn et al. 2008), rs2735839, lies in a region of relatively low LD. We discovered two markers in high LD (r 2 ≥ 0.8) with rs2735839; thus, these variants are the most likely to be advanced in laboratory analyses designed to investigate the biological basis of the association signal(s).
Prostate cancer is the second leading cause of cancer deaths in the United States (Jemal et al. 2008). It shows both indolent and aggressive forms and it is difficult to distinguish patients that require aggressive therapy and management from those that should be left to watchful waiting. Although the benefits of PSA screening in detecting earlier stage cancers may be important, this leads to a significant intervention and unnecessary treatment. Evidence for or against the efficacy of PSA screening in reducing morbidity and mortality due to prostate cancer is eagerly awaited. Our effort to comprehensively describe common genetic variation in the KLK3 locus on chromosome 19q13.33 should enable a rational approach towards the follow-up analyses of the role genetic variation plays on PSA levels and prostate cancer risk.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Conflict of interest statement
All authors report no financial interests or potential conflicts of interests.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Ahn J, Berndt SI, Wacholder S, Kraft P, Kibel AS, Yeager M, Albanes D, Giovannucci E, Stampfer MJ, Virtamo J, Thun MJ, Feigelson HS, Cancel-Tassin G, Cussenot O, Thomas G, Hunter DJ, Fraumeni JF, Jr, Hoover RN, Chanock SJ, Hayes RB. Variation in KLK genes, prostate-specific antigen and risk of prostate cancer. Nat Genet. 2008;40:1032–1034. doi: 10.1038/ng0908-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, Weinstock GM, Gibbs RA. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia D, Church TR, Fouad MN, Gelmann EP, Kvale PA, Reding DJ, Weissfeld JL, Yokochi LA, O’Brien B, Clapp JD, Rathmell JM, Riley TL, Hayes RB, Kramer BS, Izmirlian G, Miller AB, Pinsky PF, Prorok PC, Gohagan JK, Berg CD. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009;360:1310–1319. doi: 10.1056/NEJMoa0810696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem. 1996;271:6379–6388. doi: 10.1074/jbc.271.11.6379. [DOI] [PubMed] [Google Scholar]
- Cleutjens KB, van der Korput HA, van Eekelen CC, van Rooij HC, Faber PW, Trapman J. An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol Endocrinol. 1997;11:148–161. doi: 10.1210/me.11.2.148. [DOI] [PubMed] [Google Scholar]
- Couronne O, Poliakov A, Bray N, Ishkhanov T, Ryaboy D, Rubin E, Pachter L, Dubchak I. Strategies and tools for whole-genome alignments. Genome Res. 2003;13:73–80. doi: 10.1101/gr.762503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SD, Chang BL, Rao A, Hawkins GA, Zheng SL, Wade WN, Cooke RT, Thomas LN, Bleecker ER, Catalona WJ, Sterling DA, Meyers DA, Ohar J, Xu J. Association between genetic polymorphisms in the prostate-specific antigen gene promoter and serum prostate-specific antigen levels. J Natl Cancer Inst. 2003;95:1044–1053. doi: 10.1093/jnci/95.14.1044. [DOI] [PubMed] [Google Scholar]
- Cramer SD, Sun J, Zheng SL, Xu J, Peehl DM. Association of prostate-specific antigen promoter genotype with clinical and histopathologic features of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:2451–2457. doi: 10.1158/1055-9965.EPI-08-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeles R, Giles G, Neal D, Muir K, Easton DF. Reply to “Variation in KLK genes, prostate-specific antigen and risk of prostate cancer”. Nat Genet. 2008;40:1035–1036. doi: 10.1038/ng0908-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, Donovan JL, Hamdy FC, Dearnaley DP, Muir KR, Smith C, Bagnato M, Ardern-Jones AT, Hall AL, O’Brien LT, Gehr-Swain BN, Wilkinson RA, Cox A, Lewis S, Brown PM, Jhavar SG, Tymrakiewicz M, Lophatananon A, Bryant SL, Horwich A, Huddart RA, Khoo VS, Parker CC, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Fisher C, Jamieson C, Cooper CS, English DR, Hopper JL, Neal DE, Easton DF. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- Gan L, Lee I, Smith R, Argonza-Barrett R, Lei H, McCuaig J, Moss P, Paeper B, Wang K. Sequencing and expression analysis of the serine protease gene cluster located in chromosome 19q13 region. Gene. 2000;257:119–130. doi: 10.1016/S0378-1119(00)00382-6. [DOI] [PubMed] [Google Scholar]
- Gohagan JK, Prorok PC, Hayes RB, Kramer BS. The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial of the National Cancer Institute: history, organization, and status. Control Clin Trials. 2000;21:251S–272S. doi: 10.1016/S0197-2456(00)00097-0. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Kader AK, Sun J, Isaacs SD, Wiley KE, Yan G, Kim ST, Fedor H, DeMarzo AM, Epstein JI, Walsh PC, Partin AW, Trock B, Zheng SL, Xu J, Isaacs W. Individual and cumulative effect of prostate cancer risk-associated variants on clinicopathologic variables in 5, 895 prostate cancer patients. Prostate. 2009;69:1195–1205. doi: 10.1002/pros.20970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 2005;15:1051–1060. doi: 10.1101/gr.3642605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AW, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL, Venter JC. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja H. A kallikrein-like serine protease in prostatic fluid cleaves the predominant seminal vesicle protein. J Clin Invest. 1985;76:1899–1903. doi: 10.1172/JCI112185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja H, Ulmert D, Bjork T, Becker C, Serio AM, Nilsson JA, Abrahamsson PA, Vickers AJ, Berglund G. Long-term prediction of prostate cancer up to 25 years before diagnosis of prostate cancer using prostate kallikreins measured at age 44 to 50 years. J Clin Oncol. 2007;25:431–436. doi: 10.1200/JCO.2006.06.9351. [DOI] [PubMed] [Google Scholar]
- Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med. 1998;17:873–890. doi: 10.1002/(SICI)1097-0258(19980430)17:8<873::AID-SIM779>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Pal P, Xi H, Sun G, Kaushal R, Meeks JJ, Thaxton CS, Guha S, Jin CH, Suarez BK, Catalona WJ, Deka R. Tagging SNPs in the kallikrein genes 3 and 2 on 19q13 and their associations with prostate cancer in men of European origin. Hum Genet. 2007;122:251–259. doi: 10.1007/s00439-007-0394-3. [DOI] [PubMed] [Google Scholar]
- Punglia RS, D’Amico AV, Catalona WJ, Roehl KA, Kuntz KM. Effect of verification bias on screening for prostate cancer by measurement of prostate-specific antigen. N Engl J Med. 2003;349:335–342. doi: 10.1056/NEJMoa021659. [DOI] [PubMed] [Google Scholar]
- Punglia RS, D’Amico AV, Catalona WJ, Roehl KA, Kuntz KM. Impact of age, benign prostatic hyperplasia, and cancer on prostate-specific antigen level. Cancer. 2006;106:1507–1513. doi: 10.1002/cncr.21766. [DOI] [PubMed] [Google Scholar]
- Rothberg JM, Leamon JH. The development and impact of 454 sequencing. Nat Biotechnol. 2008;26:1117–1124. doi: 10.1038/nbt1485. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V, Kwiatkowski M, Lujan M, Lilja H, Zappa M, Denis LJ, Recker F, Berenguer A, Maattanen L, Bangma CH, Aus G, Villers A, Rebillard X, van der Kwast T, Blijenberg BG, Moss SM, de Koning HJ, Auvinen A. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320–1328. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- Schuur ER, Henderson GA, Kmetec LA, Miller JD, Lamparski HG, Henderson DR. Prostate-specific antigen expression is regulated by an upstream enhancer. J Biol Chem. 1996;271:7043–7051. doi: 10.1074/jbc.271.12.7043. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O’Hara R, Casalunovo T, Conlin LK, D’Arcy M, Franckelton EC, Geiger EA, Haldeman-Englert C, Imielinski M, Kim CE, Medne L, Annaiah K, Bradfield J, Dabaghyan E, Eckert A, Onyiah CC, Ostapenko S, Otieno FG, Santa E, Shaner JL, Skraban R, Smith RM, Elia J, Goldmuntz E, Spinner NB, Zackai EH, Chiavacci RM, Grundmeier R, Rappaport EF, Grant SF, White PS, Hakonarson H (2009) High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res 19:1682–1690 [DOI] [PMC free article] [PubMed]
- Stephan C, Yousef GM, Scorilas A, Jung K, Jung M, Kristiansen G, Hauptmann S, Bharaj BS, Nakamura T, Loening SA, Diamandis EP. Quantitative analysis of kallikrein 15 gene expression in prostate tissue. J Urol. 2003;169:361–364. doi: 10.1016/S0022-5347(05)64127-4. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL, Minasian LM, Ford LG, Lippman SM, Crawford ED, Crowley JJ, Coltman CA., Jr Prevalence of prostate cancer among men with a prostate-specific antigen level < or = 4.0 ng per milliliter. N Engl J Med. 2004;350:2239–2246. doi: 10.1056/NEJMoa031918. [DOI] [PubMed] [Google Scholar]
- Xu J, Isaacs SD, Sun J, Li G, Wiley KE, Zhu Y, Hsu FC, Wiklund F, Turner AR, Adams TS, Liu W, Trock BJ, Partin AW, Chang B, Walsh PC, Gronberg H, Isaacs W, Zheng S. Association of prostate cancer risk variants with clinicopathologic characteristics of the disease. Clin Cancer Res. 2008;14:5819–5824. doi: 10.1158/1078-0432.CCR-08-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- Yousef GM, Scorilas A, Jung K, Ashworth LK, Diamandis EP. Molecular cloning of the human kallikrein 15 gene (KLK15). Up-regulation in prostate cancer. J Biol Chem. 2001;276:53–61. doi: 10.1074/jbc.M005432200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.