

Abstract
The genetic and molecular approaches to heterosis usually do not rely on any model of the genotype–phenotype relationship. From the generalization of Kacser and Burns’ biochemical model for dominance and epistasis to networks with several variable enzymes, we hypothesized that metabolic heterosis could be observed because the response of the flux towards enzyme activities and/or concentrations follows a multi-dimensional hyperbolic-like relationship. To corroborate this, we used the values of systemic parameters accounting for the kinetic behaviour of four enzymes of the upstream part of glycolysis, and simulated genetic variability by varying in silico enzyme concentrations. Then we “crossed” virtual parents to get 1,000 hybrids, and showed that best-parent heterosis was frequently observed. The decomposition of the flux value into genetic effects, with the help of a novel multilocus epistasis index, revealed that antagonistic additive-by-additive epistasis effects play the major role in this framework of the genotype–phenotype relationship. This result is consistent with various observations in quantitative and evolutionary genetics, and provides a model unifying the genetic effects underlying heterosis.
Electronic supplementary material
The online version of this article (doi:10.1007/s00122-009-1203-2) contains supplementary material, which is available to authorized users.
Introduction
Most of the present studies on heterosis, or hybrid vigour, focus on QTL detection to untangle the genetic effects underlying the phenomenon (Hua et al. 2003; Meyer et al. 2009), or on the search for non-additive expression of transcripts or proteins in hybrids to identify possible molecular mechanisms accounting for heterosis for macroscopic traits (Paschold et al. 2009). Despite the interest of these descriptive, model-free approaches, they can hardly provide a general framework to comprehend a universal phenomenon which has many evolutionary and agronomical implications.
In this paper, we propose a fundamentally different way to tackle the problem of heterosis, by using a systemic approach based on metabolic network modelling, which provides a biologically realistic genotype–phenotype relationship.
As soon as 1934, Wright proposed a general explanation for the prevalence, in natural populations, of the dominance of wild alleles upon deleterious alleles. He considered the relationship between the activity of one enzyme in a linear metabolic pathway and the steady-state rate of production of the product of the chain, i.e. the flux. Because the product of one enzyme is the substrate for the next, the effect of changing the activity of a particular enzyme depends on the activities of all the others enzymes of the pathway. Even though at this time the biochemical theory of metabolic fluxes was not very developed, he predicted a hyperbolic relationship between enzyme activity and flux. Thus, if the wild-type enzyme activity is at the plateau of the flux curve and the enzyme activity of the heterozygote is intermediate, null or deleterious mutations will be recessive. This idea was brightly confirmed by the theoretical developments of Kacser and Burns (1981) for chains of Michaelian reversible enzymes. They also showed that epistasis is inherent to this non-linear model of genotype–phenotype relationship: an allelic substitution at one locus will change the effect of allelic substitution at all other loci. Actually, the hyperbolic-like relationship between enzyme activity and flux seems to be valid for most of the networks, regardless their complexity (see Fiévet et al. 2006, for a discussion). In addition Fiévet et al. (2006) reconstructed in vitro the first part of glycolysis, and varied in turn the concentration of the successive enzymes, the concentrations of the other enzymes being fixed. In spite of regulation and branching in the system they used, in all cases they observed a quasi-hyperbolic ascending curve.
More or less directly, fluxes affect all macroscopic traits, including agronomically or horticulturally important traits: seed/fruit weight depends on lipid, starch and/or sugar content, fruit ripening on ethanol synthesis, resistance against herbivores is related to glycosinolate profile, flowering date to hormonal balance, flower colour to anthocyanins, etc. Therefore fluxes through metabolic networks can be considered as model quantitative traits, depending on all the genes coding and/or regulating the enzymes of the network. In this framework, the genetically variable enzyme parameters represent the genotype, whereas the flux is the phenotype. As activity and/or concentration of several enzymes may vary together, the genotype–phenotype relationship can be modelled according to a multidimensional hyperbolic surface. Relying on this biologically realistic modelling, we simulated series of crosses between parents differing for the concentrations of enzymes of the upstream part of glycolysis. Best-parent heterosis was frequently observed, and the decomposition of the flux into genetic effects revealed a tight relationship between heterosis and antagonistic (“less-than-additive”) additive-by-additive epistasis.
Theoretical developments
The metabolic model
Linear pathways
Let us consider a linear pathway of unimolecular reversible reactions catalysed by n Michaelian enzymes far from saturation:
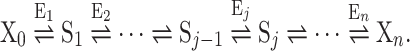 |
X0 and Xn are respectively the initial substrate and the final product of the pathway (external metabolites), S1,…, Sj are the successive substrates of the pathway (internal metabolites), and E1,…, En are the enzymes. At the steady state, the flux through the pathway is (Kacser and Burns 1973):
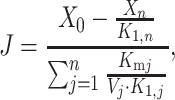 |
where X 0 and X n are respectively the concentrations of X0 and Xn, K 1,j (resp. K 1,n) is the product of the equilibrium constants of the reactions from X0 to Sj (resp. Xn), K mj is the Michaelis–Menten constant of enzyme j and V j is the maximum velocity of enzyme j.
With 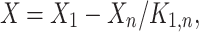 and to make apparent the enzyme concentration E
j, we can alternatively write:
and to make apparent the enzyme concentration E
j, we can alternatively write:
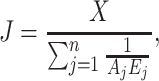 |
1 |
where 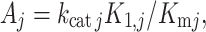 with k
catj the catalytic constant of enzyme j. In this paper, we will assume that the A
j’s are not genetically variable, so all the variability in enzyme activity is due to the variability on the concentrations E
j. However the formal developments would be identical if the A
j’s were genetically variable.
with k
catj the catalytic constant of enzyme j. In this paper, we will assume that the A
j’s are not genetically variable, so all the variability in enzyme activity is due to the variability on the concentrations E
j. However the formal developments would be identical if the A
j’s were genetically variable.
Networks: the general genotype–phenotype relationship
The previous developments do not apply to pathways that contain non-Michaelian and regulated enzymes, and/or to branched pathways, which require specific derivations of flux expressions based on the detailed kinetic equations of the individual reactions. However, we showed recently that a simple modification of the previous modelling allowed reliable predictions of the flux values for regulated and branched systems (Fiévet et al. 2006). The idea was based on data from the literature showing that increasing the concentration of a given enzyme in a system of any complexity, the concentration of other enzymes being fixed, usually results in quasi-hyperbolic response curve of the flux (saturation curve). In addition, an experimental system fully corroborated this view. We reconstructed in vitro the first part of glycolysis, from hexokinase to glycerol 3-phosphate dehydrogenase (therefore with the TPI branching), and included a cycle to regenerate ATP from ADP with creatine kinase. Increasing from 0 the concentrations of phosphoglucose isomerase (PGI), phosphofructokinase (PFK), fructose-1,6-bisphosphate aldolase (FBA) or triosephosphate isomerase (TPI), the concentrations of other enzymes being fixed, resulted in all cases in quasi-hyperbolic ascending saturation curves (Fiévet et al. 2006).
So we used the following approximate general expression of the flux:
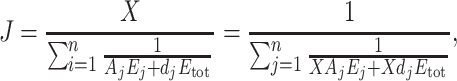 |
2 |
where X is a constant, A
j is a parameter accounting for the kinetic behaviour of enzyme j within the system (therefore its expression is more complex than previously defined), and d
j is a parameter accounting for the “dispensability” of enzyme j (if there is branching in the pathway, removing some enzymes does not drive the flux to 0), and E
tot is the sum of enzyme concentrations 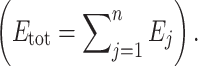 Note that in the Fiévet et al.’s paper, this expression was a little bit simpler, with the parameter p
j = d
j
E
tot, i.e. E
tot did not appear explicitly. Here we introduced E
tot for more generality, both to take into account the fact that E
tot may vary (in the cited paper E
tot was fixed), and to derive easily the summation property of the control coefficients (not shown).
Note that in the Fiévet et al.’s paper, this expression was a little bit simpler, with the parameter p
j = d
j
E
tot, i.e. E
tot did not appear explicitly. Here we introduced E
tot for more generality, both to take into account the fact that E
tot may vary (in the cited paper E
tot was fixed), and to derive easily the summation property of the control coefficients (not shown).
This is the relation we have used for modelling the genotype–phenotype relationship. The “phenotype” is flux J and the “genotype” is the vector of enzyme concentrations E j’s genetically variable. In silico or in vitro, it is thus possible to simulate genetic variability by varying enzyme concentrations E j, and for each genotype to calculate or measure the flux value from Eq. 2.
Genotype construction and flux computation
To run simulations with realistic values, we considered the network of the upstream part of glycolysis, with four variable enzymes, and we used the parameter values published by Fiévet et al. (2006), who estimated XA j and Xd j E tot by hyperbolic fitting of the titration curves obtained by varying in turn the concentration of each enzyme. The values were XA PGI = 499.4 s−1, XA PFK = 115.5 s−1, XA FBA = 22.5 s−1, XA TPI = 22,940 s−1, Xd PGI E tot = 0, Xd PFK E tot = 0, Xd FBA E tot = 0, Xd TPI E tot = 61.8 μM/s. As in these experiments E tot was equal to 2.82 μM, we get Xd TPI = 21.9 s−1. Thus for any set of E j values, that is to say for any virtual genotype, a flux (“phenotype”) value could be computed from the equation:
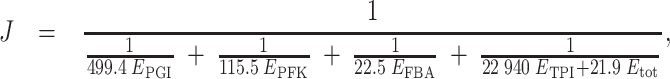 |
3 |
where 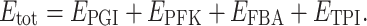
Thousand pairs of virtual parental genotypes were created. As the total enzyme amount allocated to the system was variable, but should be necessarily limited to remain biologically realistic (Lion et al. 2004), we proceeded in the following way to choose the enzyme concentrations. For each of the four enzymes, 10 concentration values evenly distributed from 0 to 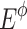 were defined (excluding of course these two extreme values), with
were defined (excluding of course these two extreme values), with 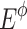 the sum of the physiological concentrations of the enzyme estimated in the yeast strain S288C (Fiévet et al. 2004) (PGI: 9.1 mg/l, PFK: 10.4 mg/l, FBA: 60.1 mg/l and TPI: 22.3 mg/l). The proportions of the three remaining enzymes were drawn at random using beta distributions (α = 1,
the sum of the physiological concentrations of the enzyme estimated in the yeast strain S288C (Fiévet et al. 2004) (PGI: 9.1 mg/l, PFK: 10.4 mg/l, FBA: 60.1 mg/l and TPI: 22.3 mg/l). The proportions of the three remaining enzymes were drawn at random using beta distributions (α = 1, 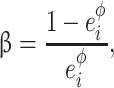 with
with 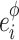 the physiological proportion of enzyme i), to cover a large range of variability of enzyme concentrations. Twenty-five independent drawings were performed for each concentration of the target enzyme, resulting in 1,000 parental distributions (4 enzymes × 10 concentrations × 25 drawings). Each of them was randomly associated to another one to get 1,000 pairs of parents. The total enzyme concentration varied from 0.07 to 2.73 μM.
the physiological proportion of enzyme i), to cover a large range of variability of enzyme concentrations. Twenty-five independent drawings were performed for each concentration of the target enzyme, resulting in 1,000 parental distributions (4 enzymes × 10 concentrations × 25 drawings). Each of them was randomly associated to another one to get 1,000 pairs of parents. The total enzyme concentration varied from 0.07 to 2.73 μM.
The predicted flux of each parental genotype was computed according to Eq. 3. The flux of the 1,000 hybrids was computed assuming that (i) there is additivity of all enzyme concentrations, or (ii) there is positive or negative non-additivity of concentrations of PFK and/or FBA (FBA is the most abundant enzyme), with the hybrid concentrations remaining within the range of parental concentrations.
Additivity writes:
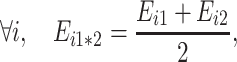 |
so the hybrid flux is:
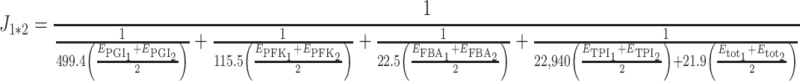 |
To analyse non-additivity, we considered five values of “coefficients of inheritance”: 1, 0.8, 0.5, 0.2 and 0, defined respectively as follows:
-
(i)
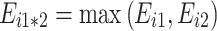 : “complete positive non-additivity” (the hybrid concentration is equal to the highest parental concentration).
: “complete positive non-additivity” (the hybrid concentration is equal to the highest parental concentration). -
(ii)
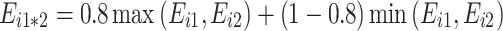 : “partial positive non-additivity”.
: “partial positive non-additivity”. -
(iii)
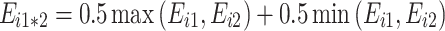 : additivity (reference case).
: additivity (reference case). -
(iv)
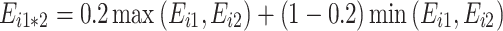 : “partial negative non-additivity”.
: “partial negative non-additivity”. -
(v)
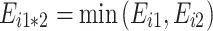 : “complete negative non-additivity” (the hybrid concentration is equal to the lowest parental concentration).
: “complete negative non-additivity” (the hybrid concentration is equal to the lowest parental concentration).
For instance, if for a pair of parents we have 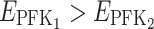 and
and 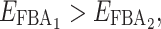 and if in their hybrid we have
and if in their hybrid we have 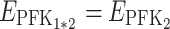 (case v, coefficient 0) and
(case v, coefficient 0) and 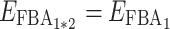 (case i, coefficient 1), the hybrid flux will be:
(case i, coefficient 1), the hybrid flux will be:
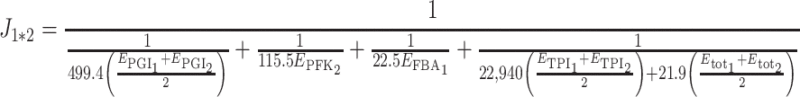 |
For each cross, the difference between the hybrid flux value and the higher parental flux value was computed as H = J 1*2 − max (J 1, J 2). For each pair of parents, the highest parental flux was noted J 2.
Decomposition of the genotypic values in the multilocus case
In order to decompose the flux values into a sum of genetic effects, we generalized the Hayman and Mather’s (1955) approach to any number of bi-allelic loci (Zeng et al. 2005). For a trait controlled by two biallelic loci A and B, the genotypic values G of the nine possible genotypes can be decomposed as a sum of nine genetic parameters, according to the so-called F ∞-metric model (Van Der Veen 1959):
| A 1 A 1 | A 1 A 2 | A 2 A 2 | |
|---|---|---|---|
| B 1 B 1 |
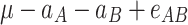
|
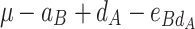
|
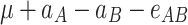
|
| B 1 B 2 |
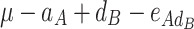
|
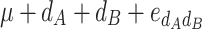
|
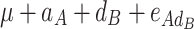
|
| B 2 B 2 |
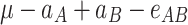
|
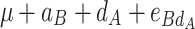
|
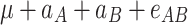
|
μ is the mean of the four homozygous genotypes, a
A and a
B are the additive effects of genes A and B, respectively, d
A and d
B are the dominance effects of genes A and B, respectively, e
AB is the additive-by-additive epistasis effect between A and B, 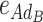 and
and 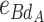 are the additive-by-dominance epistasis effects and
are the additive-by-dominance epistasis effects and 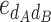 is the dominance-by-dominance epistasis effect. We chose the F
∞-metric model rather than the F
2-metric model (e.g. Melchinger et al. 2007) because it resulted in equations simpler and easier to interpret (see Yang 2004; Zeng et al. 2005, for discussions on these models).
is the dominance-by-dominance epistasis effect. We chose the F
∞-metric model rather than the F
2-metric model (e.g. Melchinger et al. 2007) because it resulted in equations simpler and easier to interpret (see Yang 2004; Zeng et al. 2005, for discussions on these models).
This decomposition can be generalized to L variable loci:
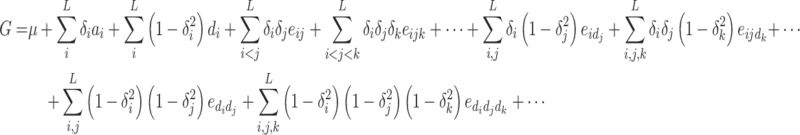 |
4 |
The indicator variable δ
i takes the value −1 for one of the homozygous genotypes, +1 for the other homozygous genotype, and 0 for the heterozygous genotype. μ is the mean of the homozygote genotypic values, a
i is the additive effect of gene i, d
i is the dominance of gene i, e
ij… is the additive-by-additive epistasis of any order, 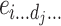 is the additive-by-dominance epistasis of any order, and
is the additive-by-dominance epistasis of any order, and 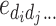 is the dominance-by-dominance epistasis of any order; the suspension points are for all the possible epistasis terms for the number L of loci considered. This model is completely determined: the number of parameters is equal to the number of genotypes (3L), so there is a complete specification of the genotypic values when the parameters are given, and vice versa. Let G the vector of the genotypic values, Δ the 3L × 3L matrix of the signs of the genetic parameters (determined from the indicator variables) and T the vector of the genetic parameters. We have
is the dominance-by-dominance epistasis of any order; the suspension points are for all the possible epistasis terms for the number L of loci considered. This model is completely determined: the number of parameters is equal to the number of genotypes (3L), so there is a complete specification of the genotypic values when the parameters are given, and vice versa. Let G the vector of the genotypic values, Δ the 3L × 3L matrix of the signs of the genetic parameters (determined from the indicator variables) and T the vector of the genetic parameters. We have
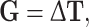 |
therefore
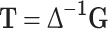 |
5 |
Thus all the genetic parameters can be determined provided that all the genotypic values are known.
Determining the genetic effects for the flux with four variable enzymes in the system
Each pair of parents is defined by two particular distributions of enzyme concentrations. The parent with the lowest (respectively highest) flux is given the virtual genotype A 1 A 1 B 1 B 1 C 1 C 1 D 1 D 1 (respectively A 2 A 2 B 2 B 2 C 2 C 2 D 2 D 2). With four biallelic loci, there are 81 possible genotypes (34). For each of them, the flux can be computed from Eq. 3. For instance, the flux of genotype A 1 A 2 B 1 B 1 C 1 C 1 D 1 D 2 if there is additivity of enzyme concentrations is
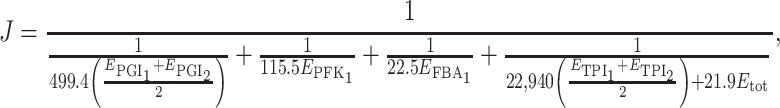 |
where 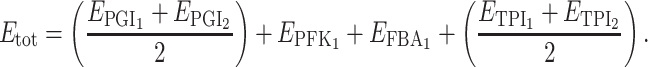
If there is not additivity, the flux is computed using the coefficients of inheritance as exemplified above.
For each pair of parents, the 81 flux values were computed, from which we derived the 81 genetic effects using Eq. 5: the mean μ, 4 additive effects (a
A
,
a
B, a
C, a
D), 4 dominance effects (d
A
,
d
B, d
C, d
D), 11 additive-by-additive epistasis effect (6 e
AB-type, 4 e
ABC-type, 1 e
ABCD), 50 additive-by-dominance epistasis effects (12 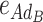 -type, 12
-type, 12 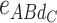 -type, 4
-type, 4 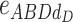 -type, 12
-type, 12 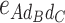 -type, 6
-type, 6 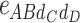 -type, 4
-type, 4 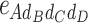 -type) and 11 dominance-by-dominance epistasis effect (6
-type) and 11 dominance-by-dominance epistasis effect (6 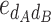 -type, 4
-type, 4 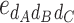 -type, 1
-type, 1 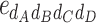 -type). Thus we got 1,000 vectors of genetic effects.
-type). Thus we got 1,000 vectors of genetic effects.
Expressing heterosis in terms of genetic effects
Let J 1 and J 2 the fluxes of two parents genetically different, P1 and P2. We assumed that J 2 > J 1. There is best-parent heterosis if the flux of the hybrid, J 1*2, is higher than J 2, or if
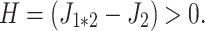 |
The difference H can be expressed as a function of the genetic parameters previously defined. Consider that the two lines P1 and P2 differ for L genes, and note their genotypes A 1 A 1 B 1 B 1 C 1 C 1…L 1 L 1 and A 2 A 2 B 2 B 2 C 2 C 2…L 2 L 2, respectively. The genotypic value G 2 is, from Eq. 4:
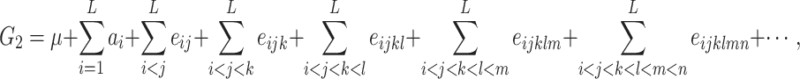 |
which depends on the additive effects of the genes and on the additive-by-additive epistasis effect of any order, hereafter noted e add.
The genotype of the hybrid between P1 and P2 is A 1 A 2 B 1 B 2…L 1 L 2, and its genotypic value, noted G1*2, is:
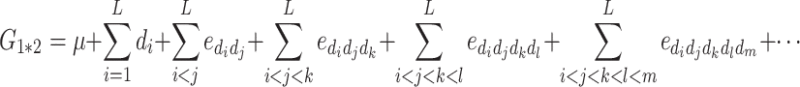 |
G 1*2 depends only on dominance and on dominance-by-dominance epistasis effects of any order, hereafter noted e dom.
Therefore H = G 1*2 − G 2 writes
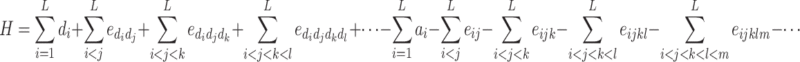 |
or, in a more condensed writing:
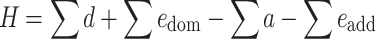 |
6 |
There is heterosis if H is positive, or if
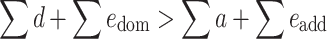 |
Thus we had two ways to compute H for the 1,000 crosses: from Eq. 6 or from the difference J 1*2 − J 2. We checked that both values were identical.
A generalized epistasis index
To assess the possible weight of epistasis in heterosis, we defined a generalized epistasis index derived from the “interaction index” proposed by Keightley (1996) in the haploid case. Consider two haploid genotypes P1 and P2 differing for only two loci, P1 with the “low” alleles at both loci, and P2 with the “high” alleles at both loci. The extent and the type of epistasis will affect the value of the genotypic difference between the two genotypes, noted G hh − G ll (subscripts h and l for high and low, respectively). Define
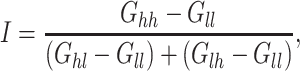 |
where G hl and G lh are the genotypic values for genotypes with one high and one low allele. If I = 1, there is additivity, i.e. the difference between G hh and G ll is just accounted for by the sum of the effects of every individual allelic substitution on the flux. The epistasis is synergistic if I > 1 and antagonistic I < 1. This index is identically valid for diploid homozygote genotypes (pure lines), as considered below.
It is possible to generalize this index to the multilocus case, with lines defined as particular combinations of “high” and “low” alleles. The genotypic difference between two lines P1 and P2 displaying specific combinations of alleles (G 2 − G 1 , with G 2 ≥ G 1) may be compared to the sum of the differences generated by individually substituting each allele of P1 for the allele from P2. Thus we defined the index:
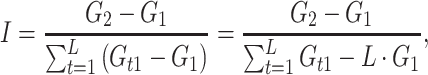 |
7 |
where 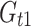 is the genotypic value of a line with the P2 allele for gene t and the P1 alleles for all other genes.
is the genotypic value of a line with the P2 allele for gene t and the P1 alleles for all other genes.
From the previous derivations, we get (see Electronic Supplementary Material)
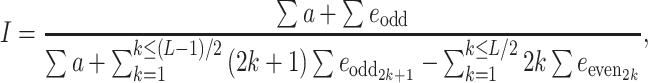 |
8 |
where e odd and e even stand for additive × additive epistasis of any order involving an odd and an even number of genes, respectively.
In the particular case where the genotypic value is a flux through a network, the hyperbolic relation between the flux and the enzyme parameters results in a necessarily positive value for I, even if some differences  are negative (see Electronic Supplementary Material). So there is synergistic epistasis if I > 1, antagonistic epistasis if 0 < I < 1, and additivity if I = 1.
are negative (see Electronic Supplementary Material). So there is synergistic epistasis if I > 1, antagonistic epistasis if 0 < I < 1, and additivity if I = 1.
The I values could be computed either from the flux values (Eq. 7) or from the genetic effects (Eq. 8). We checked that they were identical.
Results
A geometric view of heterosis
In the framework of the metabolic model of genotype–phenotype relationship, we assumed that the response of flux J with respect to the variations of enzyme concentrations E j follows a multidimensional hyperbolic surface (Kacser and Burns 1981; Fiévet et al. 2006):
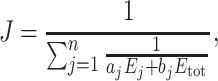 |
9 |
a j and b j are systemic parameters accounting for the kinetic behaviour of enzyme j in the network, and E tot is the total enzyme amount of the network (see Theoretical developments).
In case of additivity of the enzyme concentrations in the hybrids, the convexity of the surface generates inevitably mid-parent heterosis for the flux, i.e. the hybrid flux J 1*2 is higher than the mean parental flux (J 1 + J 2)/2. This can be seen geometrically on the two-dimensional hyperbolic flux response surface obtained for two variable enzymes (Fig. 1, parents P1 and P2). More interesting, best-parent heterosis can be observed, i.e. hybrid flux J 1*2 is higher than the best parental flux: J 1*2 > max (J 1 , J 2). Best-parent heterosis is expected for the flux whenever the parents are complementary for the “high” and “low” alleles of various enzymes (Fig. 1, parents P3 and P4).
Fig. 1.

Heterosis for the flux (J) through a linear metabolic pathway of Michaelian enzymes far from saturation. The flux is represented as a function of the activities of two enzymes, with the same arbitrary values of kinetic parameters. P1 and P2, and P3 and P4, are two pairs of parents. The hybrids have mid-parental concentration/activity for both enzymes (points in the middle of the curves relating the parental points). In the P1*P2 cross, there is only mid-parent heterosis for the flux because parent P2 has a flux close to the maximum due to high concentration/activity of both enzymes. In the P3*P4 cross, the hybrid displays best-parent heterosis because the parents have low flux values due to low concentration/activity of enzyme 2 (parent P3) or enzyme 1 (parent P4)
If there is not additivity, the hybrid point is no more mid-way on the line relating the parental points, but is on the part of the surface defined by the upper and lower limits of enzyme concentrations.
In silico heterosis: data from the upstream part of glycolysis
The fluxes of 1,000 virtual parents and 1,000 of their possible hybrids were computed from Eq. 9 for the upstream part of glycolysis, with four variable enzymes. When there was additivity of enzyme concentrations, all hybrids displayed either mid-parent heterosis (600 occurrences), or best-parent heterosis (400 occurrences). The relative heterosis 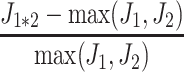 could reach very high values, since 55 hybrids had a flux 50% higher than the best-parental flux, and in two cases the hybrid flux was more than fourfold higher than the best-parental flux (Fig. 2).
could reach very high values, since 55 hybrids had a flux 50% higher than the best-parental flux, and in two cases the hybrid flux was more than fourfold higher than the best-parental flux (Fig. 2).
Fig. 2.

Histogram of the relative best-parent heterosis values when there is additivity of enzyme concentrations
When one enzyme, either PFK or FBA, displayed non-additive inheritance, the number of cases of heterosis depended on the direction of the non-additivity. As expected, more best-parent heterosis was observed with positive non-additivity and less with negative non-additivity (Table 1). From complete positive non-additivity to complete negative non-additivity, the numbers of occurrences of best-parent heterosis ranged from 512 to 126 for PFK and from 624 to 96 for FBA (Table 1). For mid-parent heterosis, the figures were respectively 488 to 372 and 376 to 305. When there was partial or complete negative non-additivity, some hybrids displayed neither best-parent nor mid-parent heterosis, but it is worth noting that, even with complete negative non-additivity of FBA concentration, more than 30% of the hybrids displayed mid-parent heterosis, and almost 10% best-parent heterosis.
Table 1.
Results of the simulations of 1,000 crosses between parents differing for the distribution of concentrations of four glycolytic enzymes
| Inheritance | Heterosis | R 2 | H > 0 and ∑e add < 0 | H > 0 and I < 1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PFK | FBA | No | Mid-parent | Best-parent | ∑a | ∑d | ∑e dom | ∑e add | Nb | % | Nb | % |
| 1 | 0.5 | 0 | 488 | 512 | 0.24 | 0.25 | 0.22 | 0.7 | 501 | 97 | 447 | 87 |
| 0.8 | 0.5 | 0 | 527 | 473 | 0.27 | 0.2 | 0.19 | 0.72 | 469 | 99 | 423 | 89 |
| 0.5 | 0.5 | 0 | 600 | 400 | 0.35 | 0.12 | 0.13 | 0.75 | 398 | 99 | 371 | 93 |
| 0.2 | 0.5 | 209 | 516 | 275 | 0.51 | 0.06 | 0.05 | 0.66 | 275 | 100 | 266 | 97 |
| 0 | 0.5 | 502 | 372 | 126 | 0.57 | 0.26 | 0.1 | 0.31 | 126 | 99 | 126 | 100 |
| 0.5 | 1 | 0 | 376 | 624 | 0.09 | 0.36 | 0.36 | 0.73 | 578 | 92 | 508 | 81 |
| 0.5 | 0.8 | 0 | 456 | 544 | 0.16 | 0.26 | 0.28 | 0.77 | 527 | 97 | 467 | 86 |
| 0.5 | 0.5 | 0 | 600 | 400 | 0.35 | 0.12 | 0.13 | 0.75 | 398 | 99 | 371 | 93 |
| 0.5 | 0.2 | 378 | 396 | 226 | 0.55 | 0.15 | 0.05 | 0.47 | 225 | 99 | 218 | 96 |
| 0.5 | 0 | 599 | 305 | 96 | 0.55 | 0.36 | 0.11 | 0.21 | 96 | 99 | 96 | 100 |
| 1 | 1 | 0 | 253 | 747 | 0.04 | 0.48 | 0.5 | 0.66 | 618 | 83 | 542 | 73 |
| 1 | 0 | 510 | 324 | 166 | 0.46 | 0.41 | 0.15 | 0.19 | 165 | 99 | 165 | 99 |
| 0 | 1 | 335 | 410 | 255 | 0.35 | 0.36 | 0.19 | 0.28 | 255 | 100 | 255 | 100 |
| 0 | 0 | 892 | 108 | 0 | 0.74 | 0.34 | 0.03 | 0.17 | 0 | – | 0 | – |
Columns “Inheritance”: coefficients of inheritance for enzymes PFK and FBA. Columns “Heterosis”: observed numbers of each type of inheritance. Columns R 2: fraction of explained variance in the regression of H to the sums of the different genetic effects. Last columns: numbers and percentages of cases where best-parent heterosis corresponded to a negative value of the sum of additive-by-additive epistasis effects (∑e add), or where best-parent heterosis corresponded to a value of the epistasis index I lower than unity
When both PFK and FBA had a non-additive inheritance, the number of cases of best-parent heterosis ranged from 748 (complete positive non-additivity for both enzymes) to 0 (complete negative non-additivity for both enzymes), but in the latter case, mid-parent heterosis was still observed 108 times (Table 1).
Translating flux heterosis into genetic effects: the major role of epistasis
The heterosis index H = J 1*2 − max(J 1, J 2) can be expressed as a sum of genetic effects:
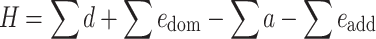 |
To evaluate the respective parts of the different genetic effects on heterosis, we calculated for each cross the sum of the additive effects (∑a), the sum of the dominance effects (∑d), the sum of the dominance-by-dominance epistasis effects (∑e dom) and the sum of the additive-by-additive epistasis effects (∑e add) (see Theoretical developments), and analysed their relationship with index H (Fig. 3).
Fig. 3.

Relationship between the heterosis index H and the sum of the additive effects (a), the sum of the dominance effects (b), the sum of the dominance-by-dominance epistasis effects (c) and the sum of the additive-by-additive epistasis effects (d)
Additivity of enzyme concentrations
In case of additivity of enzyme concentrations, the sum of additive effects and the sum of additive-by-additive epistasis effects were negatively correlated to H, and the sum of dominance effects and the sum of dominance-by-dominance epistasis effects were positively correlated to H (Fig. 3). Due to the large number of data, these correlations were all significant (p < 0.001), but there were striking differences between the R 2 values (Table 1). While the R 2 was very weak for the sum of dominance effects and the sum of dominance-by-dominance effects (R 2 = 0.12 and 0.13 respectively), it was moderate for the sum of additive effects (R 2 = 0.35), and quite high for the sum of additive-by-additive epistasis effects (R 2 = 0.75) (Fig. 3d). With only two exceptions, the 400 positive H values (i.e. best-parent heterosis) corresponded to negative values of the sum of the additive-by-additive epistasis effects. Plotting the H value against the novel, general epistasis index we defined revealed that antagonistic epistasis is the main factor explaining best-parent heterosis, since 93% (371/400) of the positive H values corresponded to an epistasis index between 0 and 1 (Fig. 4).
Fig. 4.

Relationship between heterosis index H and epistasis index I (truncated at 5, the highest value being 16.82). The values on the left of the vertical dotted line of abscissa 1 correspond to antagonist epistasis. The positive H values correspond to best-parent heterosis
Non-additivity of enzyme concentrations
These results were quite robust with regard to non-additivity of concentration of one enzyme. In no case the sum of dominance effects and the sum of dominance-by-dominance epistasis effects had the highest R 2 values. The sum of additive-by-additive epistasis effects kept the highest values when there was positive non-additivity, and also when there was partial negative non-additivity of PFK (Table 1). More importantly, the association between best-parent heterosis and additive-by-additive epistasis was consistently very high: 92–100% of the cases of best-parent heterosis corresponded to negative ∑e add values, and 81% to 100% corresponded to antagonistic epistasis.
When there was non-additive inheritance for both enzymes, the highest R 2 was observed for ∑e add when there was complete positive non-additivity for both enzymes; otherwise the highest R 2 was obtained for the sum of additive effects. But again, the association between best-parent heterosis and additive-by-additive epistasis was very strong, with 83% to 100% of the cases corresponding to negative ∑e add values, and 73% to 100% to antagonistic epistasis (Table 1).
Discussion
The classical linear genotype–phenotype relationship of quantitative genetics has been very powerful for plant and animal breeding, but it is biologically questionable. Finding an explicit function to describe this relationship is of course out of reach, given the cellular complexity and the dramatic increase of the number of parameters with the size of the systems. For that reason, various modelling efforts based on conceptual shortcuts have been proposed to simulate complex cellular behaviours from a limited amount of biological data. In this connection, the metabolic control theory (MCT) proved to be quite powerful (Fell 1992, for a review). One of the outcomes of MCT has been to show that if the concentration/activity of an enzyme changes while the parameters of all other enzymes in the pathway are fixed, the flux displays a saturation curve (Kacser and Burns 1973; Heinrich and Rapoport 1974). Even though there are exceptions (see discussion in Fiévet et al. 2006), there are innumerable examples of such a behaviour (e.g. Kacser and Burns 1981; Fell 1997; Niederberger et al. 1992; Cronwright et al. 2002; Koebmann et al. 2005, etc.). Actually, as argued by Fiévet et al. (2006), the hyperbolic-like relationship between enzyme activity and flux could well be valid also for complex networks. These authors reconstructed in vitro a small network with regulation and branching, and observed such a relationship for the four enzymes the concentration of which was modified. More importantly, the hyperbolic-like relation can also be observed at other levels of cell/individual organization, from transcription to integrated phenotype. Rossignol et al. (2003) showed that the deleterious effects of a mitochondrial mutation were accounted for by a saturation curve at various levels of the expression of the mutation: translation, enzyme complex activity, respiratory flux, cell growth and clinical manifestations. Therefore, modelling the genotype–phenotype relying on a hyperbolic-like relationship could be biologically relevant for a large range of macroscopic traits.
To analyse heterosis in this framework, we approximated the flux through a network using a multidimensional hyperbolic modelling in which the kinetic behaviour of each enzyme was described by two systemic parameters. We decomposed the flux into genetic effects, and examined the relationships between these genetic effects and heterosis for glycolytic flux in a series of 1,000 virtual crosses between parents differing for their distribution of enzyme concentrations. We varied the concentrations because theoretical studies (Pettersson 1989) and experimental data suggested that enzyme concentrations are more likely to vary than their catalytic properties (Bulfield et al. 1978; Eanes et al. 1990; Tarun et al. 1998). In any case, introducing kinetic variable systemic parameters into the model is possible, and would not modify the theoretical framework. Concerning inheritance of enzyme concentrations, additivity is supported by classical observations (Kacser and Burns 1981). However, proteomic studies have shown that even though protein concentrations are indeed in majority additive, there are cases of non-additive inheritance (Leonardi et al. 1991; Kollipara et al. 2002; Hoecker et al. 2008). So we performed simulations assuming on the one hand additivity, on the other hand positive and negative non-additive inheritance for PFK and/or FBA, the latter being the most variable enzyme among the parents.
If there is additivity of enzyme concentrations, the convexity of the response of the flux towards enzyme concentrations makes mid-parent heterosis inevitable and, depending on the distribution of the parental enzyme concentrations, may result in best-parent heterosis. If there is partial dominance of the low allele, heterosis is no more inevitable. In all cases, the sum of the additive-by-additive epistasis effects, and in a lesser extent the sum of the additive effects, were negatively correlated to the difference between hybrid flux and the best-parent flux (H). Best-parent heterosis (H > 0) corresponded in almost all cases to negative values of the sum of the additive-by-additive epistasis effects. By contrast, and unexpectedly, the dominance and dominance-by-dominance epistasis effects did not seem to play a large role. Actually, this apparent paradox can be explained in the following way. As shown geometrically Fig. 1, dominance is sufficient to have mid-parent heterosis (parents P1 and P2), while both dominance and additive-by-additive epistasis are required to get best-parent heterosis (parents P3 and P4). It is mainly the level of additive-by-additive epistasis that drives the H value (Fig. 3d). The novel and general epistasis index we defined (I) does not include any dominance or dominance-by-dominance epistasis effect. When there was best-parent heterosis, index I took usually a value lower than unity, indicative of antagonistic (less-than-additive) epistasis. In other words, when the phenotypic difference between two parents is below the sum of the effects of every individual allelic substitution in the lowest genotype, their hybrid usually exhibits a high phenotypic value.
It is possible to set up a bridge between our general approach of heterosis and a classical result of Mendelian genetics. As underlined by Phillips (2008) in his recent review on epistasis, it was very early shown (Bateson et al. 1905), and repeatedly illustrated in many plants (Sinnott and Dunn 1939; Dooner et al. 1991, etc.), that crossing two individuals with colourless flowers may result in hybrids with purple flowers. The explanation is well known: the parents have each a mutation inactivating a particular enzyme of the anthocyanin biosynthesis pathway, which disrupts the flux, and in the hybrid the flux is restored because both enzymes are active. In more formal terms, consider two biallelic loci A/a and B/b, a and b being recessive, and leading to a flux equal to zero when homozygous. We get the following table of possible phenotypic values (0 and 1 for absence and presence of flux, respectively):
| AA | Aa | aa | |
|---|---|---|---|
| BB | 1 | 1 | 0 |
| Bb | 1 | 1 | 0 |
| bb | 0 | 0 | 0 |
The cross between AAbb and aaBB, which have no flux, will produce the hybrid AaBb, with a restored flux. This situation represents the simplest possible case for determining the genetic effects and indices we defined. We have H = 1 (best-parent heterosis), ∑e add = e AB = −0.25 and I = 0 (maximum antagonistic epistasis). In an F2 progeny, this case corresponds to the classical 9:7 segregation, where dominance and epistasis occurs (with dominance alone, we would have a 9:6:1 segregation).
Heterosis appears as an emergent property of the system, because the properties of each enzyme separately are not sufficient to account for the phenomenon. With total enzyme amount half-way between its parents, hybrids can display best-parent heterosis, which corresponds to a better exploitation of cell resources than in the parents. This conclusion is consistent with the negative correlation between energy cost of growth and mean individual heterozygosity classically described in marine animals (e.g. Koehn 1991; Bayne and Hawkins 1997; Danzmann et al. 1987).
Antagonistic epistasis, which is supposed to be favoured by natural selection (Desai et al. 2007), is commonly observed in populations, as attested by experiments of accumulation of mutations which show that the decrease of fitness with the number of mutations is “less-than-additive” (e.g. Maisnier-Patin et al. 2005; Silander et al. 2007). Antagonistic epistasis has also been evidenced from the comparison of chromosome substitution strains, in plants and animals, which have revealed that the sum of the individual chromosomal effects often dramatically exceeded the difference between the parental strains (Redden 1991; Shao et al. 2008). Finally, marker-based studies have revealed less-than-additive interactions between QTL (Eshed and Zamir 1996; Ming et al. 2001). All these data are consistent with the frequent occurrence of heterosis observed in all species.
We may speculate that the common occurrence of both antagonistic epistasis and heterosis in natural populations reflects the non-linearity of the genotype–phenotype relationship for the vast majority of the phenotypic traits. Any evolutionary process that may stabilize the favourable epistatic interactions, su£ch as gene duplications or constraint on the recombination rate, should be selected as it reduces the genetic load inherent to heterosis in populations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Professor Olivier C. Martin for his help to demonstrate that the generalized epistasis index is positive.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Contribution to the special issue “Heterosis in Plants”.
References
- Bateson W, Saunders ER, Punnett RC, Hurst CC (1905) Reports to the Evolution Committee of the Royal Society, Report II (Harrison and Sons, London)
- Bayne BL, Hawkins AJ. Protein metabolism, the costs of growth, and genomic heterozygosity: experiments with the Mussel Mytilus galloprovincialis Lmk. Physiol Biochem Zool. 1997;70:391–402. doi: 10.1086/515848. [DOI] [PubMed] [Google Scholar]
- Bulfield G, Moore EA, Kacser H. Genetic variation in activity of the enzymes of glycolysis and gluconeogenesis between inbred strains of mice. Genetics. 1978;89:551–561. doi: 10.1093/genetics/89.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronwright GR, Rohwer JM, Prior BA. Metabolic control analysis of glycerol synthesis in Saccharomyces cerevisiae. Appl Environ Microbiol. 2002;68:4448–4456. doi: 10.1128/AEM.68.9.4448-4456.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzmann RG, Ferguson MM, Allendorf FW. Heterozygosity and oxygen-consumption rate as predictors of growth and developmental rate in rainbow trout. Physiol Zool. 1987;60:211–220. [Google Scholar]
- Desai MM, Weissman D, Feldman MW. Evolution can favor antagonistic epistasis. Genetics. 2007;177:1001–1010. doi: 10.1534/genetics.107.075812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooner HK, Robbins TP, Jorgensen RA. Genetic and developmental control of anthocyanin biosynthesis. Annu Rev Gene. 1991;25:173–199. doi: 10.1146/annurev.ge.25.120191.001133. [DOI] [PubMed] [Google Scholar]
- Eanes W, Katona L, Longtime M. Comparison of in vitro and in vivo activities associated with the g6pd allozyme polymorphism in Drosophila melanogaster. Genetics. 1990;125:845–853. doi: 10.1093/genetics/125.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed Y, Zamir D. Less-than-additive epistatic interactions of quantitative trait loci in tomato. Genetics. 1996;141:1147–1162. doi: 10.1093/genetics/143.4.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell DA. Metabolic control analysis: a survey of its theoretical and experimental background. Biochem J. 1992;286:313–330. doi: 10.1042/bj2860313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell DA. Understanding the control of metabolism. London: Portland Press; 1997. [Google Scholar]
- Fiévet J, Dillmann C, Lagniel G, Davanture M, Négroni L, Labarre J, de Vienne D. Assessing factors for reliable quantitative proteomics based on two-dimensional electrophoresis. Proteomics. 2004;4:1939–1949. doi: 10.1002/pmic.200300731. [DOI] [PubMed] [Google Scholar]
- Fiévet JB, Dillmann C, Curien G, de Vienne D. Simplified modelling of metabolic pathways for flux prediction and optimization: lessons from an in vitro reconstruction of the upper part of glycolysis. Biochem J. 2006;396:317–326. doi: 10.1042/BJ20051520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman BI, Mather K. The description of genetic interactions in continuous variation. Biometrics. 1955;11:69–82. doi: 10.2307/3001481. [DOI] [Google Scholar]
- Heinrich R, Rapoport TA. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur J Biochem. 1974;42:89–95. doi: 10.1111/j.1432-1033.1974.tb03318.x. [DOI] [PubMed] [Google Scholar]
- Hoecker N, Lamkemeyer T, Sarholz B, et al. Analysis of nonadditive protein accumulation in young primary roots of a maize (Zea mays L) F(1)-hybrid compared to its parental inbred lines. Proteomics. 2008;8:3882–3894. doi: 10.1002/pmic.200800023. [DOI] [PubMed] [Google Scholar]
- Hua J, Xing Y, Wu W, Xu C, Sun X, Yu S, Zhang Q. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA. 2003;100:2574–2579. doi: 10.1073/pnas.0437907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H, Burns JA. The control of flux. Symp Soc Exp Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- Kacser H, Burns JA. The molecular basis of dominance. Genetics. 1981;97:639–666. doi: 10.1093/genetics/97.3-4.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P. Metabolic models of selection response. J Theor Biol. 1996;182:311–316. doi: 10.1006/jtbi.1996.0169. [DOI] [PubMed] [Google Scholar]
- Koebmann B, Solem C, Jensen PR. Control analysis as a tool to understand the formation of the las operon in Lactococcus lactis. FEBS J. 2005;272:2292–2303. doi: 10.1111/j.1742-4658.2005.04656.x. [DOI] [PubMed] [Google Scholar]
- Koehn RK. The cost of enzyme synthesis in the genetics of energy balance and physiological performance. Biol J Linn Soc. 1991;44:231–247. doi: 10.1111/j.1095-8312.1991.tb00618.x. [DOI] [Google Scholar]
- Kollipara KP, Saab IN, Wych RD, Lauer MJ, Singletary GW. Expression profiling of reciprocal maize hybrids divergent for cold germination and desiccation tolerance. Plant Physiol. 2002;129:974–992. doi: 10.1104/pp.000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi A, Damerval C, Gallais A, de Vienne D. Association of protein amount polymorphism (PAP) among Maize lines with performance of their hybrids. Theor Appl Genet. 1991;82:552–560. doi: 10.1007/BF00226790. [DOI] [PubMed] [Google Scholar]
- Lion S, Gabriel F, Bost B, Fiévet J, Dillmann C, de Vienne D. An extension to the metabolic control theory taking into account correlations between enzyme concentrations. Eur J Biochem. 2004;271:4375–4391. doi: 10.1111/j.1432-1033.2004.04375.x. [DOI] [PubMed] [Google Scholar]
- Maisnier-Patin S, Roth JR, Fredriksson A, Nyström T, Berg OG, Andersson DI. Genomic buffering mitigates the effects of deleterious mutations in bacteria. Nat Genet. 2005;37:1376–1379. doi: 10.1038/ng1676. [DOI] [PubMed] [Google Scholar]
- Melchinger AE, Utz HF, Piepho HP, Zeng ZB, Schön CC. The role of epistasis in the manifestation of heterosis: a systems-oriented approach. Genetics. 2007;177:1815–1825. doi: 10.1534/genetics.107.077537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RC, Kusterer B, Lisec J, Steinfath M, Becher M, Scharr H, Melchinger AE, Selbig J, Schurr U, Willmitzer L, Altmann T (2009) QTL analysis of early stage heterosis for biomass in Arabidopsis. Theor Appl Genet (this issue) [DOI] [PMC free article] [PubMed]
- Ming R, Liu SC, Moore PH, Irvine JE, Paterson AH. QTL analysis in a complex autopolyploid: genetic control of sugar content in sugarcane. Genome Res. 2001;11:2075–2084. doi: 10.1101/gr.198801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederberger P, Prasad R, Miozzari G, Kacser H. A strategy for increasing an in vivo flux by genetic manipulations. The tryptophan system of yeast. Biochem J. 1992;287:473–479. doi: 10.1042/bj2870473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschold A, Marcon C, Hoecker N, Hochholdinger F (2009) Molecular dissection of heterosis manifestation during early maize root development. Theor Appl Genet (this issue) [DOI] [PubMed]
- Pettersson G. Effect of evolution on the kinetic properties of enzymes. Eur J Biochem. 1989;184:561–566. doi: 10.1111/j.1432-1033.1989.tb15050.x. [DOI] [PubMed] [Google Scholar]
- Phillips PC. Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redden R. The effect of epistasis on chromosome mapping of quantitative characters in wheat. I. Time to spike emergence. Aust J Agric Res. 1991;42:1–11. doi: 10.1071/AR9910001. [DOI] [Google Scholar]
- Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Burrage LC, Sinasac DS, Hill AE, Ernest SR, O’Brien W, Courtland HW, Jepsen KJ, Kirby A, Kulbokas EJ, Daly MJ, Broman KW, Lander ES, Nadeau JH. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc Natl Acad Sci USA. 2008;105:19910–19914. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silander OK, Tenaillon O, Chao L. Understanding the evolutionary fate of finite populations: the dynamics of mutational effects. PLoS Biol. 2007;5:e94. doi: 10.1371/journal.pbio.0050094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott EW, Dunn LC (1939) Principles of genetics. McGraw-Hill Book Company, Inc., New-York and London
- Tarun A, Lee J, Theologis A. Random mutagenesis of 1-aminocyclopropane-1-carboxylate synthase: a key enzyme in ethylene biosynthesis. Proc Natl Acad Sci USA. 1998;95:9796–9801. doi: 10.1073/pnas.95.17.9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Veen JH. Tests of non-allelic interaction and linkage for quantitative characters in generations derived from two diploid pure lines. Genetica. 1959;30:201–232. doi: 10.1007/BF01535675. [DOI] [PubMed] [Google Scholar]
- Wright S. Molecular and evolutionary theories of dominance. Am Nat. 1934;68:24–53. doi: 10.1086/280521. [DOI] [Google Scholar]
- Yang RC. Epistasis of quantitative trait loci under different gene action models. Genetics. 2004;167:1493–1505. doi: 10.1534/genetics.103.020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng ZB, Wang T, Zou W. Modeling quantitative trait loci and interpretation of models. Genetics. 2005;169:1711–1725. doi: 10.1534/genetics.104.035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.