

Abstract
Familial hemiplegic migraine type 1 (FHM-1) is caused by mutations in CACNA1A; the gene encoding for the Cav2.1 subunit of voltage-gated calcium channels. Although various studies attempted to determine biophysical consequences of these mutations on channel activity, it remains unclear exactly how mutations can produce a FHM-1 phenotype. A lower activation threshold of mutated channels resulting in increased channel activity has been proposed. However, hyper-activity may also be caused by a reduction of the inhibitory pathway carried by G-protein coupled receptor activation. The aim of this study is to determine functional consequences of the FHM-1 S218L mutation on direct G-protein regulation of Cav2.1 channels. In HEK 293 cells, DAMGO activation of human μ-opioid receptors induced a 55% Ba2+ current inhibition through both wild-type and S218L mutant Cav2.1 channels. In contrast, this mutation considerably accelerates the kinetic of current deinhibition following channel activation by 1.7- to 2.3-fold depending on membrane potential values. Taken together, these data suggest that the S218L mutation does not affect G-proteins association onto channel in the closed state, but promotes its dissociation from the activated channel thereby decreasing the inhibitory G-protein pathway. Similar results were obtained with the R192Q FHM-1 mutation, although of lesser amplitude, which seems in line with the less severe associated clinical phenotype in patients. Functional consequences of FHM-1 mutations appear thus as the consequence of the alteration of both intrinsic biophysical properties and of the main inhibitory G-protein pathway of Cav2.1 channels. The present study furthers molecular insight in the physiopathology of FHM-1.
Keywords: Animals; Calcium Channels; genetics; metabolism; Calcium Channels, N-Type; genetics; metabolism; Cell Line; Enkephalin, Ala(2)-MePhe(4)-Gly(5)-; pharmacology; GTP-Binding Protein alpha Subunits, Gi-Go; metabolism; Genotype; Humans; Ion Channel Gating; drug effects; Kinetics; Membrane Potentials; Migraine with Aura; genetics; metabolism; Mutation; Phenotype; Rats; Receptors, Opioid, mu; agonists; metabolism; Transfection
Keywords: Familial hemiplegic migraine, S218L mutation, R192Q mutation, Cav2.1 type calcium channel, Cav2.1 subunit, P/Q current, G-protein, G-protein coupled receptor, μ-opioid receptor, β-subunit.
Introduction
Familial hemiplegic migraine (FHM) is a rare, severe, monogenic subtype of migraine with aura, characterised by at least some degree of hemiparesis during the aura (1, 2). FHM-1 is caused by missense mutations in the gene CACNA1A encoding the Cav2.1 protein, the pore-forming subunit of Cav2.1 (formerly known as P/Q) calcium channels (14). To date, 19 different mutations have been described that are distributed over the four homologous domains of the Cav2.1 subunit. They generally affect structural determinants that are essential for channel activity including (i) the S4 transmembrane segments, thought to be voltage-sensor elements controlling channel activation, (ii) the S6 transmembrane segments, involved in the control of channel inactivation, and (iii) the P loops which form the ionic pore. Eleven of FHM-1 mutations have been characterized at the biophysical level (15), but only for one (the R192Q mutation that is associated with relatively mild, pure, FHM phenotype in patients (14)) the effects on G-protein regulation was reported (12). Ten out of 11 characterized mutations induce a hyperpolarizing shift of the voltage-dependence of channel activation, generally resulting in an increased channel opening probability already at mild potential values. However, additional effects have been observed on channel inactivation kinetics, current density, unitary conductance, open probability and selectivity (6, 8, 9, 13, 17, 18). A recently developed knock-in model expressing human pathogenic FHM-1 mutation R192Q revealed increased Ca2+ influx through mutant Cav2.1 channels and a decreased threshold and increased velocity of cortical spreading depression (CSD) (19, 20). CSD is the mechanism underlying migraine aura (10), and, in animal experiments, was shown to activate headache mechanisms (2). However, the exact molecular events leading to an increased susceptibility of CSD remain largely unresolved. A neuronal hyper-excitability due to a hyper-activity of Cav2.1 calcium channels seems most likely (15, 19, 20). The lower activation threshold of mutant Cav2.1 channels, associated with increased opening probability, may well trigger such hyper-activity. Alternatively, however, a reduction of inhibitory pathways may come in addition to produce a similar effect.
One important inhibitory regulation affecting Cav2.1 channels is produced by G-protein coupled receptor activation (for review see (3, 16)). This inhibition is based on the direct binding of the Gβγ signalling complex directly onto the Cav2.1 subunit (4). Recently, it was evidenced that this binding produces only the silencing of channel activity (“ON” effect), whereas Gβγ unbinding which follows channel activation induces an apparent set of biophysical modifications (“OFF” effects) that include: (i) a slowing of activation and inactivation kinetics, and (ii) a depolarizing shift of the voltage-dependence of channel activation (21). In that context, the kinetics of Gβγ dissociation from the channel is a critical factor in determining the probability that an inhibited channel recovers from G-protein inhibition during a burst of action potentials. Thereby, it should also influence the strength of persistence of a G-protein-related inhibitory pathway on synaptic signaling. Two factors were found to be essential in determining the speed of Gβγ dissociation: the voltage-dependence of channel activation and the kinetics of channel inactivation during membrane depolarization (21, 23). Indeed, activation at lower membrane potential values and faster inactivating channels were shown to be affected by the FHM-1 S218L mutation in the Cav2.1 subunit (18). This mutation is localized in the S4-S5 linker of the first domain and is associated with a particularly severe clinical phenotype of FHM and sometimes fatal delayed cerebral edema after minor head trauma in some mutation carriers (5, 7).
Therefore, we argued that, direct G-protein inhibition of mutated S218L Cav2.1 channels may be altered. Here we tested this hypothesis by transiently expressing, in HEK-293 cells, human Cav2.1 wild-type (Cav2.1WT) or S218L mutant (Cav2.1S218L) along with β4 and α2δ-1b auxiliary subunits, and the human μ-opioid receptor (hMOR). Direct G-protein regulation was induced by (D-Ala2, N-Me-Phe4, glycinol5)-Enkephalin (DAMGO) application. Application of DAMGO induces a maximal current inhibition that is similar for both wild-type and mutated Cav2.1 channels, suggesting that the FHM-1 S218L mutation does not alter the association of Gβγ onto the closed state of the channel. However, the recovery from G-protein inhibition following channel activation is considerably accelerated for Cav2.1S218L channels, which is in accordance with the previously reported negative shift in the voltage-dependence of channel activation and the faster channel inactivation. In light of these data, G-protein regulation of the R192Q mutant (Cav2.1R192Q, that is associated with a less severe clinical phenotype) was also investigated with similar protocols, and comparable alteration of G-protein regulation were observed, albeit less pronounced. Taken together, these observations indicate that the FHM-1 S218L and R192Q mutations promote Cav2.1 current recovery from direct G-protein inhibition in line with their respective degree of clinical phenotype, and thus decrease the inhibitory influence of this signalling pathway. These effects on G-protein regulation should contribute, along with the main biophysical effects on channel activation, to render the neuronal network hyperexcitable, possibly as a consequence of reduced presynaptic inhibition, and help explain the pathophysiology of FHM.
Materials and Methods
Material
The cDNAs used in this study were human Cav2.1 (GenBank accession number AF004883), rat β4 (L02315), rat α2δ-1b (NM012919) and human μ-opioid receptor (hMOR, obtained from the UMR cDNA Resource Center www.cdna.org) (AY521028). The cDNA encoding for the Cav2.1S218L mutant protein is described in (18). The cDNA encoding for the Cav2.1R192Q mutant was kindly provided by Dr. J. Striessnig, University of Innsbruck, Austria. (D-Ala2, N-Me-Phe4, glycinol5)-Enkephalin (DAMGO) was purchased from Bachem (Budendorf, Germany).
Transient expression in HEK-293
Human embryonic kidney 293 (HEK-293) cells were grown in a Dulbecco’s modified Eagle’s culture medium containing 10% foetal bovine serum and 1% penicillin/streptomycin (all products were purchased from Invitrogen) and maintained under standard conditions at 37°C in a humidified atmosphere containing 5% CO2. Cells were transfected using the jetPEI™ transfection regent (Qbiogene, OH, USA) according to the protocol provided in the kit with cDNAs encoding Cav2.1WT or Cav2.1S218L mutant subunit along with β4, α2δ-1b, hMOR and the enhanced green fluorescent protein plasmid (pEGFP; Clontech, CA, USA). Two days following transfection, cells were briefly split at 10% confluence using PBS without calcium and magnesium and patch-clamp recording was performed 4 hours later from fluorescent cells.
Patch-clamp recordings
Ba2+ currents were recorded in the whole-cell configuration of the patch-clamp technique at room temperature (22–24°C) in a bathing medium containing (in millimolar): BaCl2 5, KCl 5, MgCl2 1, NaCl 128, TEA-Cl 10, D-glucose 10, HEPES 10 (pH 7.4 with NaOH). Patch pipettes were filled with a solution containing (in millimolar): CsCl 110, Mg-ATP 3, Na-GTP 0.5, MgCl2 2.5, D-glucose 5, EGTA 10, HEPES 10 (pH 7.4 with CsOH), and had a resistance of 2–4 MΩ. Whole-cell patch-clamp recording were performed using an Axopatch 200B amplifier (Axon Instruments, Union City, CA). Acquisition and analyses were performed using pClamp 6 and Clampfit 9 software, respectively (Axon Instruments). All traces were corrected on-line for leak and capacitance currents, digitized at 10 KHz and filtered at 2 KHz. DAMGO was applied at 10 μM by superfusion of the cells at 4 mL/min and all recordings were performed within 1 min after DAMGO produced maximal current inhibition in order to minimized voltage-independent G-protein regulation and hMOR desensitization.
Analyses of the parameters of G-protein regulation
The method used to extract all biophysical parameters of G-protein regulation (GIt0, the initial extent of G-protein inhibition before the start of depolarisation, τ, the time constant of G-protein unbinding from the channel, and RI, the extent of recovery from inhibition at the end of a 200 ms test pulse) was described in (22). Briefly, subtracting IDAMGO from IControl results in ILost, the evolution of the lost current under G-protein activation. IControl and ILost are then extrapolated to t = 0 ms (the start of the depolarisation) by fitting traces with an exponential function in order to determine GIt0, the maximal extend of G-protein inhibition. IDAMGO without unbinding (IDAMGO wo unbinding) represents an estimate of the amount of control current that is present in IDAMGO and is obtained by the following equation: IDAMGO without unbinding = IControl × (1 − (ILostt0/IControlt0)). Subtracting IDAMGO wo unbinding from IDAMGO results in IG-protein unbinding with inactivation, the evolution of inhibited current that recovers from G-protein inhibition following depolarisation. IG-protein unbinding with inactivation is divided by the fit trace (normalized to 1) describing inactivation kinetics of the control current in order to reveal the net kinetics of G-protein dissociation (IG-protein unbinding) from the channels. A fit of IG-protein unbinding by a mono-exponential decrease provides the time constant τ of G-protein dissociation from the channel. The percentage of recovery from G-protein inhibition (RI) after 200 ms test pulse is measured as RI = (IDAMGO – IDAMGO wo unbinding)/(IControl – IDAMGO wo unbinding) × 100.
Mathematical and statistical analyses
Current-voltage relationships (I/V) were fitted with the modified Boltzmann equation I(V) = (Gmax × (V − E))/(1 + exp (−(V − V1/2)/k)) where I(V) represents the maximal current amplitude in response to a depolarization at the potential V, Gmax is the maximal conductance, E is the inversion potential of Ba2+, and k is a slope factor. All data are given as mean ± S.E.M for n number observations and statistical significance (p) was calculated using Student’s t test.
Results
The FHM-1 S218L mutation alters the biophysical properties of Cav2.1/α2δ-1b/β4 channels expressed in HEK-293 cells
To characterize the biophysical impact of the FHM-1 S218L mutation, human Cav2.1WT or Cav2.1S218L mutant channels were transiently expressed in HEK-293 cells along with neuronal β4 (the predominant β-subunit associated with Cav2.1 channels in the brain) and α2δ-1b auxiliary subunits. Whole-cell barium (Ba2+) currents were recorded two days after transfection from cells expressing the two various channel subunit combinations. Average current traces are shown in Fig. 1a in response to a 500 ms membrane depolarisation ranging between −40 and 40 mV from a holding potential of −90 mV. The voltage-dependencies of activation of wild-type and mutant Cav2.1 channels were determined (Fig. 1b). The mean half-activation potential of the mutant Cav2.1 channel is significantly shifted (p < 0.001) towards more negative voltages by 4.8 mV on average from −8.2 ± 0.4 mV (n = 13, wild-type) to −13.0 ± 0.6 mV (n = 13, mutant). No significant differences in current densities were observed between both channel types (Fig. 1b). However, there is a slight tendency to observe larger current densities for the mutant channel at potential values inferior at 0 mV. Given the shift to the left of the voltage-dependence of activation of the mutant channel, these results suggest that the mutant channel expressed as well as the wild-type one (Fig. 1b). In contrast, significant differences were observed in activation and inactivation kinetics. The activation kinetics of the currents carried by S218L mutant channels were significantly accelerated (p < 0.001) at all voltages studied as evidenced by the 3.2- (10 mV) to 4.2-fold (30 mV) shorter time to peak compared to wild-type channels (Fig. 1c). Channel inactivation kinetics is best fitted by a mono-exponential decay in the case of the wild-type channel, whereas two exponential components are required to fit the mutant channel inactivation kinetics (Fig. 1d, dashed line). The slow component of the inactivation kinetics of the mutant channel is significantly faster by 2.6- (0 mV, p < 0.001) to 1.4-fold (20 mV, p = 0.003) than the inactivating component of the wild-type channel for membrane potential values equal or under 20 mV; no significant difference is observed above 20 mV. The fast inactivating component of the mutant channel is between 5.1- (−10 mV, p < 0.001) and 24.2-fold (40 mV, p < 0.001) significantly faster than its slow component. The contribution of each component to the extent of inactivation is shown in Fig. 1e. The fast component of the mutant channel represents a minor fraction of the inactivating current that decreases from 18.9 ± 2.0% (−10 mV) to 6.8 ± 0.1% (40 mV). The total amount of current inactivation rises from 50.2 ± 7.5% (−10 mV) to 74.3 ± 4.0% (40 mV) for the wild-type channel compared to 63.6 ± 5.9% (−10 mV) to 89.7 ± 2.6% (40 mV) for the mutant channel, a difference that is not significant except at −10 and 40 mV probably due to the difficulty to precisely measure this parameter at these two extreme membrane potentials. Analysis of integrated whole cell currents reveals that the S218L mutation differentially affects calcium entry depending of membrane potentials (Fig. 1f). While the mutation leads to an increase in calcium entry during the time course of the current for potential values below −10 mV, at −10 mV it promotes calcium influx only during the initial phase of current activation and has an opposite effect for potential values under −10 mV due to the faster inactivation of the mutant. Taken together, these results indicate that the FHM-1 S218L mutation produces three major biophysical modifications of Cav2.1/β4/α2δ-1b channels at the whole-cell level: a hyperpolarizing shift of the voltage-dependence of channel activation, and faster channel activation and inactivation. These findings confirm earlier observations in experiments that expressed Cav2.1WT or Cav2.1S218L channels together with other auxiliary β- (β2e or β1b) and α2δ-1b subunits (18). Since we have previously shown that these parameters critically affect the direct G-protein inhibition of voltage-gated calcium channels (21, 23), this study was pursued to investigate how the FHM-1 S218L mutation could affect the inhibition of Cav2.1 channels.
Fig. 1.

The familial hemiplegic migraine type 1 (FHM-1) mutation S218L induces kinetic and voltage alterations of Cav2.1 calcium channel activity. a Average set of whole cell current traces for Cav2.1WT/β4/α2δ-1b (n = 13) and Cav2.1S218L/β4/α2δ-1b (n = 13) channels obtained by 500 ms depolarization values ranging between −40 and 40 mV. Holding potential is −90 mV. b Average current-voltage relationships for currents measured at their peak values. Inset: average half-activation potentials for wild-type (filled circles) and mutant (open circles) channels. c Representative current traces at 0 mV showing differences in activation kinetics (top panel). Symbols illustrate the time to peak. Average current time to peak values as a function of test potentials for wild-type (filled circles, n = 13) and mutant (open circles, n = 13) channels (lower panel). d Representative current traces at 0 mV for wild-type and mutant channels showing differences in inactivation kinetics (top panel). Inactivating currents were fitted (dashed line) by a mono-exponential equation (wild-type channels, filled circle) or a bi-exponential equation (mutant channels, open square for fast component and open circle symbol for slow component). Average inactivation time constants for both conditions (n = 13 for control and n = 13 for mutation) as a function of test potential (lower panel). The y-axis presents a break point for display purposes. e Average proportion of each inactivating component for wild-type (filled bars, n = 13) and mutants channels (open bars, n = 13). f Average integrated whole cell currents for wild-type (filled circle, n = 13) and S218L mutant (open circle, n = 13) channels at −20 mV, −10 mV, 0 mV and 20 mV. Vertical scale: 500, 1000, 2000 and 1000 pA. ms. pF−1 respectively. Data are presented as mean ± S.E.M for n studied cells. Statistical t test: NS, non significant; *, p ≤ 0.05; ***, p ≤ 0.001.
The FHM-1 S218L mutation does not affect maximal current inhibition by G-proteins
It is essential to measure the extent of G-protein inhibition at the start of the depolarisation to avoid the important, but confounding, fraction of recovery from inhibition that has already occurred when current amplitudes are measured at their peak. Representative current traces for wild-type and mutant channels are shown at 0 and 20 mV in the top panels of Fig. 2a and 2b both before (IControl) and during DAMGO application (IDAMGO). According to our recently developed method of analysis of G-protein regulation (22, 23), the Lost current traces were extracted (ILost) by subtracting IDAMGO from IControl. ILost provides the time course of the lost current following G-protein activation, affected by both the recovery from G-protein inhibition following channel activation and by the inactivation process. Hence, current inhibition measured from the levels of ILost and IControl when extrapolated at t = 0 ms provides the net maximal G-protein inhibition (GIt0) before these processes (current recovery and channel inactivation) have taken place (Fig. 2a, b, lower panels). Average GIt0 values were reported as a function of membrane potential (Fig. 2c). As expected for an inhibition at t = 0 ms, the maximal current inhibition by G-proteins is not voltage-dependent. This inhibition varies between 55.8 ± 3.8% (−10 mV) and 51.2 ± 5.3% (30 mV) for the wild-type channel and is not significantly different for the mutant channel (between 53.1 ± 2.3% (0 mV) and 49.9 ± 3.3% (20 mV)). More importantly, also no significant differences were observed in the maximal current inhibition achieved by DAMGO application between wild-type and mutant channels at all membrane potentials studied. This suggests that the FHM-1 S218L mutation does not affect binding of Gβγ onto the closed state of Cav2.1 channels (“ON” effect).
Fig. 2.
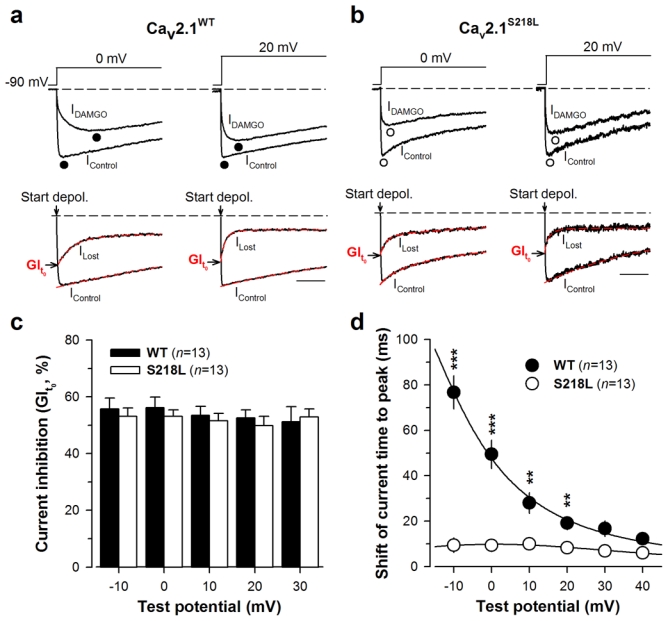
The S218L mutation does not affect the maximal G-protein inhibition of Cav2.1 currents. a Representative normalized current traces elicited at 0 and 20 mV before (IControl) and under 10 μM DAMGO application (IDAMGO) for Cav2.1WT channels with β4 and α2δ-1b auxiliary subunits (top panel). Corresponding traces allowing the measurement of the maximal DAMGO inhibition at the start of the depolarisation (GIt0) (bottom panel). IControl and ILost (obtained by subtracting IDAMGO from IControl) were fitted (red dashed line) by a mono- and a double-exponential, respectively, in order to better estimate GIt0. The arrow indicates the start of the depolarisation. Scale: 50 ms. b Legend as in (a) but for cells expressing Cav2.1S218L channels with β4 and α2δ-1b auxiliary subunits. c Bar chart representation of GIt0 for Cav2.1WT (filled bars, n = 13) and Cav2.1S218L (open bars, n = 13) channels as a function of membrane potential. d Shift of the current time to peak induced by DAMGO application for Cav2.1WT/β4/α2δ-1b channels (filled circle, n = 13) and Cav2.1S218L/β4/α2δ-1b channels (open circle, n = 13) as a function of membrane potential. Data are presented as mean ± S.E.M for n studied cells. Statistical t test: NS, non-significant; **, p ≤ 0.01; ***, p ≤ 0.001.
Next we searched for possible “OFF” effects of G-protein regulation for Cav2.1S218L channels. One characteristic “OFF” effect is the apparent slowing of current activation kinetics under DAMGO inhibition, which results from a channel opening- and time-dependent recovery from inhibition (21). The current traces shown in Fig. 2a, b (top panels) illustrate that DAMGO application produces an important slowing of activation kinetics for wild-type channels, whereas the extent of this effect appears greatly diminished for the FHM-1 S218L mutation. This was further quantified in Fig. 2d where the shift of the time to peak of the current follows an exponential decay from 76.8 ± 7.4 ms (−10 mV, n = 13) to 12.2 ± 1.5 ms (40 mV, n = 13) for the wild-type channel. Far less voltage-dependence is seen for the S218L mutant since the shift of current time to peak varies only between 10.0 ± 2.5 ms (10 mV, n = 13) and 6.9 ± 2.0 ms (30 mV, n = 13). The differences between the two channels remain statistical significant at all voltages (p < 0.01 or less) except at 30 and 40 mV where convergence between mean values is observed. These data strongly suggest a difference in the kinetics of recovery from G-protein inhibition.
The FHM-1 S218L mutation induces faster G-protein dissociation upon channel activation
The two parameters that characterize the “OFF” components of G-protein regulation of Cav2.1 voltage-gated calcium channels, i.e. the time constant of current recovery (τ) and the maximal extent of current recovery from inhibition (RI), were extracted from the data using our recently published method (22, 23). Representative current traces before and under DAMGO application are shown for wild-type and S218L mutant Cav2.1 channels at 0 and 20 mV step depolarisation from a holding potential of −90 mV (Fig. 3a, b, top panels). Corresponding current traces that describe the evolution of current recovery (IG-protein unbinding) were best fitted with a mono-exponential decrease providing τ values (Fig. 3a, b, middle panels, red dashed line). The RI values were extracted at 200 ms depolarization by comparing the level of current recovery from DAMGO traces (IDAMGO) to non-inhibited control currents (IControl) and the fraction of control currents under G-protein regulation (IDAMGO wo unbinding). According to the τ values reported below, measuring RI values at 200 ms ensures that the recovery process from G-protein inhibition is completed. Significantly faster time constants (i.e. average τ values, p = 0.028 or less) were observed for the Cav2.1S218L channel at all voltages compared to the Cav2.1WT channel (Fig. 3c): time constants decreased from 41.0 ± 5.4 ms (−10 mV, n = 13) to 6.6 ± 0.6 ms (30 mV, n = 13) for the wild-type channel, whereas a 1.7- to 2.3-fold faster τ values were observed for the mutant which ranged from 23.4 ± 4.0 ms (−10 mV, n=13) to 2.9 ± 0.5 ms (30 mV, n = 13). Also, significantly smaller RI values (p < 0.01 or less) were observed for the mutant channel at all membrane potentials except at −10 mV (Fig. 3d). Wild-type RI values ranged from 45.8 ± 5.5% (−10 mV, n = 13) to 64.2 ± 4.3% (20 mV, n = 13), whereas RI values for the S218L mutant show a diminished voltage-dependence and ranged from 30.6 ± 2.7% (20 mV, n = 13) to 35.6 ± 5.1% (30 mV, n = 13). The faster current recovery observed for the Cav2.1S218L channel, associated to a smaller extent of recovery, is consistent with previously published results, in which voltage-dependent channel inactivation not only promotes recovery from G-protein inhibition but also reduces the temporal window of the recovery process (23). Combined, these two parameters explain the reduced shift of the time to peak of the current of the S218L mutant channel under G-protein regulation.
Fig. 3.
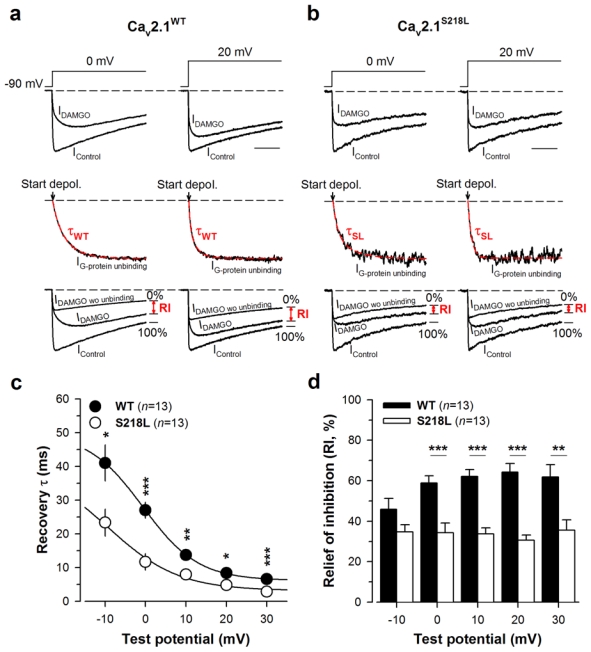
The FHM-1 S218L mutation promotes current recovery from G-protein inhibition. a Representative normalized current traces for Cav2.1WT/β4/α2δ-1b channels elicited at 0 and 20 mV before (IControl) and during DAMGO application (IDAMGO) (top panel). Corresponding normalized IG-protein unbinding traces fitted by a mono-exponential decrease (red dashed line) allowing the determination of the time constant τ of current recovery from G-protein inhibition (middle panel). The arrow indicates the start of the depolarisation. Traces that allowed the measurements of RI value (in red) are also shown (bottom panel). b Legend as in (a) but for cells expressing Cav2.1S218L/β4/α2δ-1b channels. c Time constant τ of recovery from G-protein inhibition as a function of membrane potential for Cav2.1WT/β4/α2δ-1b channels (filled circle, n = 13) and Cav2.1S218L/β4/α2δ-1b channels (open circle, n = 13). d Bar chart representation of RI values for Cav2.1WT/β4/α2δ-1b channels (filled bars, n = 13) and Cav2.1S218L/β4/α2δ-1b channels (open bars, n = 13) measured after 200 ms depolarization as a function of membrane potential. Data are presented as mean ± S.E.M for n studied cells. Statistical t test: NS, non significant; *, p ≤ 0.05, **, p ≤ 0.01; ***, p ≤ 0.001.
The FHM-1 R192Q mutation behaves similarly to the S218L mutation with regard to G-protein regulation
In order to extend the observations made with the S218L mutation, another FHM-1 mutation, R192Q, was also investigated in the same experimental conditions. Fig. 4 summarizes the main biophysical properties of Cav2.1R192Q/β4/α2δ-1b channels. Representative average current traces (n = 11) of Cav2.1R192Q mutant channels expressed along with β4 and α2δ-1b are shown in Fig. 4a. Average I-V relationships are shown in Fig. 4b and confirm earlier findings (6) that the mutation induces an increase in current density (average stimulation of 1.9-fold at 0 mV). As for the S218L mutation, an average hyperpolarizing shift of channel activation is observed although of lesser amplitude (half-activation potential of −10.7 ± 0.9 mV (n = 11) compared to −8.2 ± 0.4 mV (n = 13) for wild-type). A slight decrease in time to peak of the current is observed although this effect remains non significant (Fig. 4c). With regard to inactivation kinetics, currents follow a single exponential decrease similarly to wild-type currents, with faster inactivation kinetics at potential values below 10 mV (1.8-fold decrease at 0 mV) (Fig. 4d). Also, the inactivating component is accentuated at lower voltages for the R192Q mutation (for instance 15% more of inactivation at 0 mV) (Fig. 4e). Average integrals of current densities are shown at −20 mV, −10 mV, 0 mV and 20 mV for Cav2.1R192Q channels in Fig. 4f and indicate that contrary to the S218L mutation, the R192Q mutation promotes calcium influx at all membrane potential values tested. Taken together, these results indicate that this mutation shares similarities with the S218L mutation regarding the voltage-dependence of activation, and activation and inactivation kinetics, although all effects are less pronounced. One major difference concerns the current density which was previously interpreted as being due to an increase in the opening probability of the Cav2.1R192Q mutated channel (6). Nevertheless, all the biophysical changes are susceptible to alter G-protein regulation in a similar manner than that observed for S218L.
Fig. 4.
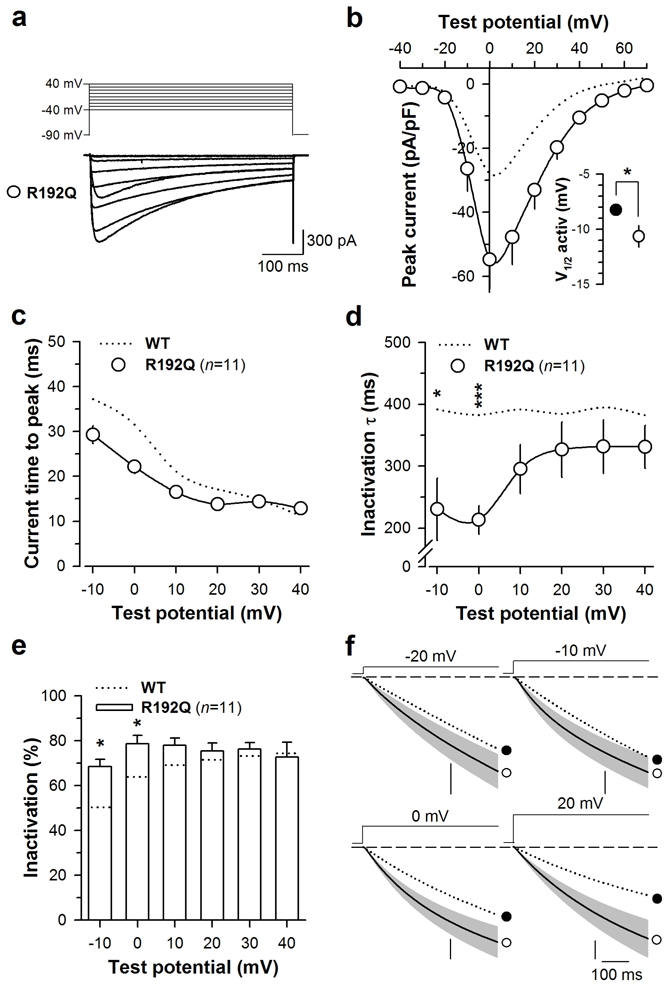
Biophysical properties of Cav2.1R192Q mutated channel. a Average set of whole-cell current traces for Cav2.1R192Q/β4/α2δ-1b (n = 11) channel obtained by 500 ms depolarization values ranging between −40 and 40 mV. Holding potential is −90 mV. b Average current voltage relationships for currents measured at their maximal amplitude. Inset: average half-activation potentials for wild-type (filled circles) and mutant (open circles) channels. c Average current time to peak values as a function of test potentials for Cav2.1R192Q mutated channel (n = 11). d Average inactivation time constants of Cav2.1R192Q channels (n = 11) as a function of test potential. The y-axis presents a break point for display purposes. e Average proportion of the inactivating component for Cav2.1R192Q channels (n = 11). f Average integrated whole cell currents for Cav2.1R192Q channels (n = 11) at −20 mV, −10 mV, 0 mV and 20 mV. Vertical scale: 400, 1500, 3000 and 2000 pA. ms. pF−1 respectively. Data are presented as mean ± S.E.M for n studied cells. The dotted line indicates values for the Cav2.1 wild-type channel. Statistical t test: NS, non significant; *, p ≤ 0.05; ***, p ≤ 0.001.
This regulation was investigated as shown in Fig. 5. Representative traces allowing the measurements of maximal G-protein inhibition (GIt0) are shown in Fig. 5a at 0 mV (left panel) and 20 mV (right panel). As for the S218L mutation, maximal G-protein inhibition (“ON” effect) of Cav2.1R192Q channels is similar to that of the wild-type channel (Fig. 5b). In contrast, a significant reduction was observed in the shift of the current time to peak induced by DAMGO application on Cav2.1R192Q channels (Fig. 5c) suggesting an alteration of G-protein dissociation from the channel. For instance, a 2.2-fold reduction was observed at 0 mV in the shift of the current time to peak. The parameters of G-protein dissociation (time constant of dissociation, τ, and the extent of recovery from inhibition, RI) were extracted as shown in Fig. 5d. Average τ values obtained for Cav2.1R192Q channels are shown in Fig. 5e as a function of membrane potentials. As for the S218L mutation, the dissociation of G-proteins from Cav2.1R192Q mutated channel is accelerated particularly for potentials below 10 mV (1.4-fold at 0 mV, compared to wild-type). In contrast to the S218L mutation however, the R192Q mutation does not affect the maximal extent of recovery from G-protein inhibition. This observation is consistent with the mild effect of this mutation on channel inactivation.
Fig. 5.
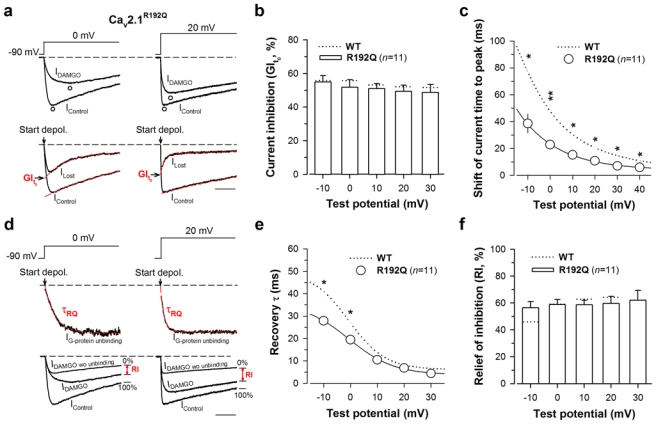
The FHM-1 R192Q mutation also promotes recovery from G-protein inhibition. a Representative normalized current traces elicited at 0 mV (left panel) and 20 mV (right panel) before (IControl) and under 10 μM DAMGO application (IDAMGO) for Cav2.1R192Q/β4/α2δ-1b channels (top panel). Corresponding traces allowing the measurement of the maximal DAMGO inhibition at the start of the depolarisation (GIt0) (bottom panel). The arrow indicates the start of the depolarisation. Scale: 50 ms. b Bar chart representation of GIt0 for Cav2.1R192Q/β4/α2δ-1b channels (n = 11) as a function of membrane potential. c Shift of the current time to peak induced by DAMGO application for Cav2.1R192Q/β4/α2δ-1b channels (n = 11) as a function of membrane potential. d Normalized IG-protein unbinding traces at 0 mV (left panel) and 20 mV (right panel) fitted by a mono-exponential decrease (red dashed line) allowing the determination of the time constant τ of current recovery from G-protein inhibition (top panel). The arrow indicates the start of the depolarisation. Traces that allowed the measurements of RI value (in red) are also shown (bottom panel). e Time constant τ of recovery from G-protein inhibition as a function of membrane potential for Cav2.1R192Q/β4/α2δ-1b channels (n = 11). f Bar chart representation of RI values for Cav2.1R192Q/β4/α2δ-1b channels (n = 11) measured after 200 ms depolarization as a function of membrane potential. Data are presented as mean ± S.E.M for n studied cells. The dotted line shows position of values for Cav2.1 wild-type channels. Statistical t test: *, p ≤ 0.05, **, p ≤ 0.01; ***, p ≤ 0.001.
Discussion
This study shows that the FHM-1 S218L mutation of the human Cav2.1 subunit when expressed in HEK-293 cells along with β4 and α2δ-1b auxiliary subunits, affects both intrinsic biophysical properties of Cav2.1 channels and direct G-protein regulation of the channel. Similar observations are made for the FHM-1 R192Q mutation, although changes are less pronounced, which seems in line with the less severe clinical phenotype of that mutation (7, 14).
Biophysical consequences of the FHM-1 S218L mutation on Cav2.1 channel activity
It was previously shown that the S218L mutation, introduced in the human Cav2.1 subunit and expressed in HEK-293 cells or in cerebellar granule cells along with the β2e or β1b and α2δ-1b auxiliary subunits, (i) promotes channel activation at lower membrane potential and (ii) induces faster inactivation kinetics during relatively short depolarisation (< 200–300 ms) and introduces a component of current that inactivates very slowly and results in a lower extent of inactivation during long depolarisation (> 500 ms) (18). Here, the human Cav2.1WT or Cav2.1S218L channels were expressed in HEK-293 cells along with the preferentially neuronal associated β4 and α2δ-1b auxiliary subunits (11). Under these experimental conditions, the FHM-1 S218L mutation also induces a hyperpolarized shift of the voltage-dependence of Cav2.1 channel activation as well as faster inactivation kinetics at the whole-cell level. However, contrary to earlier studies, no statistical difference between wild-type and mutant channels was observed in the extent of inactivation, as extrapolated beyond the 500 ms depolarisation pulse. In addition, the FHM-1 S218L mutation is found here to induce faster activation kinetics. Whether this effect is specific of the presence of the β4 subunit or not remains to be seen. It was not reported in the presence of the β2e or β1b subunits (18), but a visual examination of the current traces from this earlier report seem to show that it is the case. These results indicate that although the FHM-1 S218L mutation produces some common biophysical alterations regardless of the type of β subunit used, some modifications might be β subunit-dependent.
The FHM-1 S218L mutation differentially affects “ON” and “OFF” G-protein regulation of Cav2.1 channels
One of the major inhibitory pathway controlling voltage-gated calcium channels at the synaptic level is mediated by G-protein coupled receptor activation. This inhibition is recognized by a strong ionic current inhibition (“ON” effect), whereas the process of channel activation produces deinhibition, which is characterized by slowed current kinetics and a more or less pronounced extent of current recovery (“OFF” effects). Hence, if current inhibition finally only represents an index of the total amount of channels potentially affected by direct G-protein inhibition, current deinhibition really reflects the importance of this regulation under channel activity. In this study “ON” and “OFF” G-protein regulation parameters were quantified using our recently developed method (22, 23). Hence, G-protein inhibition, measured at the start of the depolarisation, before the initiation of the process of recovery, is not affected by the FHM-1 S218L mutation. This suggests that the S218L mutation does not affect the binding of Gβγ onto the closed-state of the channel, which is consistent with the fact that this mutation is localized in the IS4-S5 linker of the Cav2.1 subunit. It is not expected to affect one of the structural channel determinants known to be involved in Gβγ binding (i.e. the I–II loop, the amino-terminus or the carboxy-terminal region of the Cav2.1 subunit). We did observe that the S218L mutation critically affects the “OFF” effects of Cav2.1 calcium channel regulation by G-proteins as the time constant of current recovery from inhibition (τ) was drastically accelerated by 1.7- to 2.3-fold for Cav2.1S218L channels depending on membrane potential values. In contrast, the maximal extent of current recovery, measured 200 ms after the start of the depolarisation, at a time point where the process of recovery is maximal according to time constant values, is clearly diminished by the mutation. These results are consistent with previous data showing that the molecular process of channel inactivation accelerates the recovery from inhibition, but reduces the temporal window in which the process can take place, thereby reducing the maximal extent of current recovery (23). We cannot exclude that the lower activation threshold of the mutant channel could also contribute to accelerate the recovery from G-protein inhibition, since we have previously shown that the time constant of G-protein dissociation follows the voltage-dependence of channel opening (21). Nevertheless, we can reasonably argue that, in a physiological context, i.e. during burst of action potentials, the main parameter that controls channel activity under G-protein inhibition is the kinetics of the recovery process. Hence, by promoting Cav2.1 channels deinhibition from G-proteins, the FHM-1 S218L mutation diminishes the strength of this negative feedback, and is therefore likely to contribute to the hyperactivity of Cav2.1 channels and to the neuronal hyperexcitability, that is an emerging theme in the pathophysiology of FHM.
The FHM-1 R192 mutation also affects direct G-protein regulation of Cav2.1 channels
Similarly to that we observed with the S218L mutation, the R192Q mutation does not affect the maximal G-protein inhibition of Cav2.1 channels (i.e., also suggesting that it does not affect G-proteins association to the channel), but promotes current recovery from G-protein inhibition. This conclusion contrasts with that of a similar study on the R192Q mutation, for which a reduced G-protein inhibition was reported (12). However, in that study, G-protein inhibition levels were measured at the peak of the currents, at time points where significant recovery from G-protein inhibition may already have occurred for the mutant channel if this mutation also accelerates G-protein dissociation, thereby artificially providing the impression that the “ON” effect is diminished.
Potential implication in the pathogenesis of the FHM-1
Cortical spreading depression (CSD) is commonly believed to underlie the migraine aura, and probably headache mechanisms (2, 10, 19). However, what occurs at the very first stages of CSD is largely unclear. Neuronal silencing in CSD follows a short period of intense neuronal firing. It is conceivable that Cav2.1 calcium channels could be the actors initiating the process. The diminution of the channel activation threshold induced by the FHM-1 S218L mutation (and R192Q mutation) represents a potential intrinsic factor, which may easily contribute to neuronal hyperexcitability in patients. Moreover, the hyperactivity of Cav2.1 channels could also be triggered by a diminution of the inhibitory pathway carried by G-proteins. Thus, the FHM-1 S218L mutation contributes to maintain calcium influx through Cav2.1 channels especially during high synaptic activity by promoting channel deinhibition. Hence, the combined diminution of channel activation threshold and decrease of the inhibitory pathway may represent the initiating molecular events of CSD, ultimately leading to migraine.
Acknowledgments
We warmly thank Dr. Daniela Pietrobon for helpful comments and discussion on the manuscript. We are very grateful to Dr. J. Striessnig (University of Innsbruck, Austria) for the gift of the Cav2.1R192Q cDNA. We acknowledge financial support of Inserm. Additional support was obtained from EU project EUROHEAD (LSHM-CT-2004–504837).
Abbreviations: The following abbreviations have been used
- DAMGO
(D-Ala2, N-Me-Phe4, glycinol5)-Enkephalin
- FHM-1
familial hemiplegic migraine type 1
- hMOR
human μ-opioid receptor
- GI
G-protein inhibition
- RI
recovery from inhibition
- CSD
cortical spreading depression
- NS
non statistically significant
References
- 1.The International Classification of Headache Disorders: 2nd edition. Cephalalgia. 2004;24(Suppl 1):9–160. doi: 10.1111/j.1468-2982.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 2.Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- 3.De Waard M, Hering J, Weiss N, Feltz A. How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol Sci. 2005;26:427–436. doi: 10.1016/j.tips.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 4.De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 5.Fitzsimons RB, Wolfenden WH. Migraine coma. Meningitic migraine with cerebral oedema associated with a new form of autosomal dominant cerebellar ataxia. Brain. 1985;108 (Pt 3):555–577. doi: 10.1093/brain/108.3.555-a. [DOI] [PubMed] [Google Scholar]
- 6.Hans M, Luvisetto S, Williams ME, Spagnolo M, Urrutia A, Tottene A, Brust PF, Johnson EC, Harpold MM, Stauderman KA, Pietrobon D. Functional consequences of mutations in the human α1A calcium channel subunit linked to familial hemiplegic migraine. J Neurosci. 1999;19:1610–1619. doi: 10.1523/JNEUROSCI.19-05-01610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, Love S, van den Maagdenberg AM, Haan J, Frants RR, Ferrari MD. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. 2001;49:753–760. doi: 10.1002/ana.1031. [DOI] [PubMed] [Google Scholar]
- 8.Kraus RL, Sinnegger MJ, Glossmann H, Hering S, Striessnig J. Familial hemiplegic migraine mutations change α1A Ca2+ channel kinetics. J Biol Chem. 1998;273:5586–5590. doi: 10.1074/jbc.273.10.5586. [DOI] [PubMed] [Google Scholar]
- 9.Kraus RL, Sinnegger MJ, Koschak A, Glossmann H, Stenirri S, Carrera P, Striessnig J. Three new familial hemiplegic migraine mutants affect P/Q-type Ca2+ channel kinetics. J Biol Chem. 2000;275:9239–9243. doi: 10.1074/jbc.275.13.9239. [DOI] [PubMed] [Google Scholar]
- 10.Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117 (Pt 1):199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- 11.Ludwig A, Flockerzi V, Hofmann F. Regional expression and cellular localization of the α1 and β subunit of high voltage-activated calcium channels in rat brain. J Neurosci. 1997;17:1339–1349. doi: 10.1523/JNEUROSCI.17-04-01339.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melliti K, Grabner M, Seabrook GR. The familial hemiplegic migraine mutation R192Q reduces G-protein-mediated inhibition of P/Q-type (Cav2.1) calcium channels expressed in human embryonic kidney cells. J Physiol. 2003;546:337–347. doi: 10.1113/jphysiol.2002.026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullner C, Broos LA, van den Maagdenberg AM, Striessnig J. Familial hemiplegic migraine type 1 mutations K1336E, W1684R, and V1696I alter Cav2.1 Ca2+ channel gating: evidence for β-subunit isoform-specific effects. J Biol Chem. 2004;279:51844–51850. doi: 10.1074/jbc.M408756200. [DOI] [PubMed] [Google Scholar]
- 14.Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 15.Pietrobon D. Migraine: new molecular mechanisms. Neuroscientist. 2005;11:373–386. doi: 10.1177/1073858405275554. [DOI] [PubMed] [Google Scholar]
- 16.Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- 17.Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D. Familial hemiplegic migraine mutations increase Ca2+ influx through single human Cav2.1 channels and decrease maximal Cav2.1 current density in neurons. Proc Natl Acad Sci U S A. 2002;99:13284–13289. doi: 10.1073/pnas.192242399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tottene A, Pivotto F, Fellin T, Cesetti T, van den Maagdenberg AM, Pietrobon D. Specific kinetic alterations of human Cav2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. 2005;280:17678–17686. doi: 10.1074/jbc.M501110200. [DOI] [PubMed] [Google Scholar]
- 19.van de Ven RC, Kaja S, Plomp JJ, Frants RR, van den Maagdenberg AM, Ferrari MD. Genetic models of migraine. Archives of neurology. 2007;64:643–646. doi: 10.1001/archneur.64.5.643. [DOI] [PubMed] [Google Scholar]
- 20.van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, van de Ven RC, Tottene A, van der Kaa J, Plomp JJ, Frants RR, Ferrari MD. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–710. doi: 10.1016/s0896-6273(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 21.Weiss N, Arnoult C, Feltz A, De Waard M. Contribution of the kinetics of G protein dissociation to the characteristic modifications of N-type calcium channel activity. Neurosci Res. 2006;56:332–343. doi: 10.1016/j.neures.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Weiss N, De Waard M. Introducing an alternative biophysical method to analyze direct G protein regulation of voltage-dependent calcium channels. J Neurosci Methods. 2007;160:26–36. doi: 10.1016/j.jneumeth.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Weiss N, Tadmouri A, Mikati M, Ronjat M, De Waard M. Importance of voltage-dependent inactivation in N-type calcium channel regulation by G-proteins. Pflugers Arch. 2007;454:115–129. doi: 10.1007/s00424-006-0184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]