

SUMMARY
The integrin receptor family plays important roles in cell-to-cell and cell-to-extracellular matrix (ECM) interactions through the recruitment of accessory molecules. One of them is the integrin cytoplasmic domain-associated protein-1 (ICAP-1), which specifically interacts with the cytoplasmic domain of β1 integrin subunit and negatively regulates its function in vitro. To address the role of ICAP-1 in vivo, we ablated the Icap-1 gene in mice. Here we report an unexpected role of ICAP-1 for osteoblast function during bone development. Icap-1-deficient mice suffer from a reduced osteoblast proliferation and delayed bone mineralization, giving rise to a retarded formation of bone sutures. In vitro studies revealed that primary and immortalized Icap-1-null osteoblasts display enhanced adhesion and spreading on extracellular matrix substrates likely due to an increase in β1 integrin activation. Finally, we provide evidence that ICAP-1 promotes differentiation of osteoprogenitors by supporting their condensation through modulating the integrin high affinity state.
Keywords: Animals; Antigens, CD29; metabolism; Bone Development; Calcification, Physiologic; Cell Adhesion; Cell Differentiation; Cell Proliferation; Cells, Cultured; Craniofacial Abnormalities; genetics; Dwarfism; genetics; Intracellular Signaling Peptides and Proteins; genetics; metabolism; Mice; Mice, Knockout; Osteoblasts; cytology; physiology; Osteogenesis; Protein Subunits; genetics; metabolism; Skull; embryology; growth & development; physiology; Stem Cells; cytology; physiology
Keywords: ICAP-1, integrin, cell differentiation, cell adhesion, osteoblast
INTRODUCTION
Cell anchorages to extracellular matrix (ECM) and surrounding cells control shape, migration, survival and proliferation. Integrins are a large family of adhesion receptors which mediate cell-matrix and cell-cell interactions (Brakebusch et al., 2002; Hynes, 1992; Hynes, 2002). Integrins are bi-directional signaling molecules, which switch between a low (inactive) and a high affinity (active) state. The switch to the high affinity state is controlled by intracellular signals, which act on the cytoplasmic domain of integrins and induce rapid and reversible changes in the conformation of the integrin extracellular domains (inside-out signal; (Calderwood, 2004)). Following activation, integrins bind their ligands, merge into large clusters, recruit a multitude of proteins to form so called focal adhesions (FAs), and transmit signals to various subcellular compartments (outside-in signal).
ICAP-1 (Integrin Cytoplasmic domain Associated Protein-1) is an ubiquitously expressed protein identified in a yeast-2 hybrid screen as a β1 integrin cytoplasmic interacting protein (Chang et al., 1997). Human cells express two Icap-1 isoforms which are generated by alternative splicing. The large isoform associates with the cytoplasmic tail of β1 integrin. The small isoform lacks a C-terminally located integrin binding site (Chang et al., 1997) and is therefore unable to interact with β1 integrin. Overexpression of ICAP-1 negatively regulates β1 integrin function by diminishing adhesion strength to, and enhancing cell migration on fibronectin (FN) (Bouvard and Block, 1998; Bouvard et al., 2003; Zhang and Hemler, 1999). How ICAP-1 exerts its functional properties is still unclear. One study proposed direct competition with talin for β1 integrin binding (Bouvard et al., 2003). Talin is a large, cytoplasmic protein that binds and activates several integrins, and links them to the actin cytoskeleton (Calderwood et al., 2002; Vinogradova et al., 2002). Recruitment of ICAP-1 on β1 integrin would dislodge talin and thereby reduce the affinity state of β1 integrins leading to FA disassembly (Bouvard et al., 2003). In line with this hypothesis is the finding that ICAP-1 is absent from FAs. A second study suggests that ICAP-1 may act as a guanine dissociation inhibitor (GDI) for the small GTPases Rac1 and Cdc42 (Degani et al., 2002). A reduced Rac1 and/or Cdc42 activity could also explain the spreading defects of cells overexpressing ICAP-1 (Bouvard et al., 2003; Degani et al., 2002).
Finally, the identification of additional binding partners such as Krit1 and the nucleotide diphosphate kinase NM23-H2 (Fournier et al., 2002; Zawistowski et al., 2002; Zhang et al., 2001) linked ICAP-1 to additional signaling pathways. Loss-of-function mutations in the Krit1 gene cause a human disease called Cerebral Cavernous Malformation type I (Laberge-le Couteulx et al., 1999) characterized by abnormalities of the brain vasculature. Krit1 has been shown to bind microtubules and the small GTPase Rap1A (Gunel et al., 2002; Serebriiskii et al., 1997). Rap1A can reverse the transformed phenotype of Ras-overexpressing cells and modulate integrin-mediated cell adhesion on FN (Bos et al., 2001). NM23-H2 is a protein with nucleoside disphosphate kinase activity that has been linked to a variety of cellular activities including suppression of metastasis and cell motility of tumour cells in vitro. NM23-H2 can bind to the promoter sequences of the PDGF-A and c-myc genes, modulate the activity of small GTPases such as Rad and Rac1, and localizes in cell ruffles upon integrin ligation (Fournier et al., 2002).
To directly test the function of ICAP-1 in vivo, we generated Icap-1-deficient mice. Most of the mutant mice are born and develop cranio-facial dysmorphism and dwarfism caused by abnormal proliferation and differentiation of osteoblasts leading to a delayed closure of calvarial sutures. Furthermore, we show that ICAP-1 regulates β1 integrin activity and the condensation of pre-osteoblastic cells, an absolute requirement for proper bone development.
RESULTS
Generation of Icap-1-deficient mice
Three PAC clones containing the mouse Icap-1 gene (also called Bodenin or Itgb1bp1) were isolated and characterized. The Icap-1 gene spans over 20 kb and comprises 7 exons (Fig. 1A). The transcription initiation and stop codon are located in exons 2 and 7, respectively. In human, two ICAP-1 isoforms have been identified: ICAP-1α corresponding to the full-length protein (200-amino acids) and ICAP-1β representing a shorter, 150-amino acid long protein. The short isoform results from alternative splicing of exon 6, which contains the β1 integrin binding site (Chang et al., 1997). In mice, we were unable to detect the short isoform using RT-PCR amplification of RNA isolated form different adult tissues (data not shown). Furthermore, searches of EST and UCSC genome annotated databases also did not provide evidence of a mouse Icap-1β. By computer blast search, the gene encoding ICAP-1 was localized on mouse chromosome 12 and on human chromosome 2, respectively.
Fig. 1. Disruption of mouse Icap-1 gene.
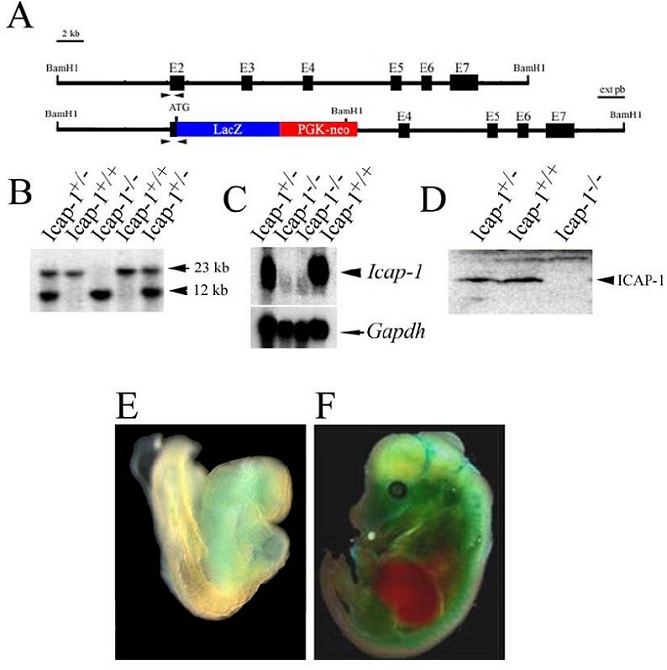
(A) Partial structure of the mouse Icap-1 gene and the targeted allele after homologous recombinaison. Black boxes represent exons (E2 to E7). The initiation codon (ATG) is located in exon 2. The expected fragment size for wild-type and recombinant alleles are 20 and 10 kb, respectively following digestion with BamH1 and hybridization with the indicated external probe (ext pb).
(B) Southern blot analysis of tail DNA isolated from Icap-1+/+, Icap-1+/− and Icap-1−/− mice.
(C) Northern blot analysis of total RNA derived from adult kidney of Icap-1+/+, Icap-1+/− and Icap-1−/− mice. The filter was hybridized with probes specific for Icap-1 and Gapdh, respectively.
(D) Western blot analysis of Icap-1+/+, Icap-1+/− and Icap-1−/− brain extracts.
(E-F) Whole mount lac Z staining of Icap-1+/− embryos at E8.5 (E) and E14.5 (F).
To study the in vivo function of ICAP-1, we generated an Icap-1 null allele by homologous recombination. The targeting strategy made use of a lacZ gene inserted in frame with the endogenous ATG and deleted exons 2 and 3 preventing the expression of a functional ICAP-1 protein (Fig. 1). Three correctly targeted embryonic stem cell (ES) clones were used to generate germline chimeric males. The null mutation was confirmed by Southern, Northern and Western blot analyses (Fig. 1B,C,D). Neither the Icap-1 mRNA nor the ICAP-1 protein were detected in tissues derived from homozygous mutant (Icap-1−/−) mice.
To determine the expression pattern of ICAP-1, heterozygous animals (Icap-1+/−) were collected at various embryonic and adult stages and subjected to LacZ histochemistry (Fig. 1E,F). At embryonic day 8.5 (E8.5), whole-mount staining demonstrated a faint LacZ activity in the developing heart and facial mesenchyme (Fig. 1E). At later stages, LacZ activity became gradually visible all over the embryo with exception of the liver (Fig. 1F). On tissue sections, only a moderate LacZ activity was observed in liver, spleen, thymus and intestinal epithelial cells, whereas other tissues expressed high LacZ levels (data not shown). These results were in agreement with previously published expression data (Faisst and Gruss, 1998). Mice heterozygous for the mutation appeared normal. Southern blot genotyping of newborn mice from heterozygous intercrosses resulted in normal number of homozygous mutants suggesting that ICAP-1 has no rate limiting function until birth. However, when the Mendelian ratio of 4-week old litters from Icap-1+/− × Icap-1+/− and Icap-1+/− × Icap-1−/− intercrosses was evaluated, 20% of Icap-1−/− mice were missing (Table I). The reason for the perinatal lethality is unknown.
Table I.
Mendelian distribution of Icap-1 progeny.
| Genotype | Wild-type | Heterozygous | Icap-1 null |
|---|---|---|---|
| Crossing | |||
| (+/−) × (+/−) | |||
| Expected (%) | 25 | 50 | 25 |
| P0 (%) (n=128) | 27 | 49 | 24 |
| P28 (%) (n=213) | 27 | 53 | 20 |
| (+/−) × (−/−) | |||
| Expected (%) | - | 50 | 50 |
| P28 (%) (n=34) | - | 60 | 40 |
P, postnatal day
Growth retardation and skeletal defects in Icap-1−/−mice
Homozygous mutant mice were slightly smaller at birth compared to wild-type littermates (Fig. 2A), and this disparity in size increased progressively postnatally. Fourteen days after birth (P14), Icap-1−/− mice were 5%-10% shorter and weighed 20%-50% less than control mice. These differences were maintained throughout life (Fig. 2B, and data not shown). At around three weeks of age, Icap-1−/− mice developed obvious cranio-facial abnormalities characterized by a domed skull, a shortened and broadened snout and bulged eyes (Fig. 2C). X-ray analysis of 5.5 month-old mutant mice revealed several additional abnormalities (Fig. 2D). The vault of the skull was shortened and rounded, the processus spinosus of the vertebrae was poorly ossified (Fig. 2D and insert). In the appendicular skeleton, the long bones had apparently normal bone density but were about 12%–23% shorter than in control mice. These morphological defects were observed in outbred (mixed C57Bl6/Sv129J) as well as inbred (CD1 and C57Bl6) lines indicating that they occurred independently of the genetic background.
Fig. 2. Growth delay, craniofacial malformation and delayed bone mineralization in Icap-1 - deficient mice.
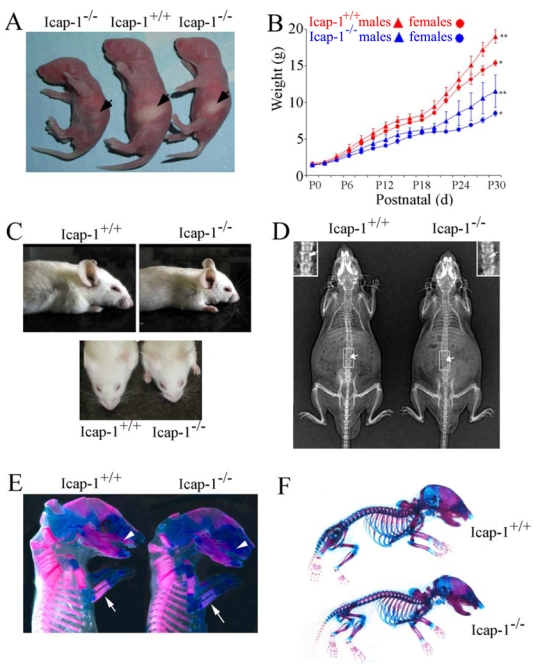
(A) Gross appearance of Icap-1+/+ and Icap-1−/−. P0 litter mates. Note that Icap-1−/− mice are smaller in size than their control littermate. A subset of the Icap-1−/− offsprings has an empty stomach (arrow). (B) Growth curves of control (Icap-1+/+or Icap-1+/−) versus Icap-1−/− (male and female) offspring of two pooled representative littermates. Mice were weighted every other day over a period of 34 days. Each point represents the mean ± S. D. (C) Lateral view (upper panel) or top view (lower panel) of 30-day-old wild-type and Icap-1-deficient mice. Note the abnormal shape (short nose and bulged-head) of the Icap-1-deficient skull compared to wild-type. (D) X-ray analysis of Icap-1+/+ and Icap-1−/− 5.5-month-old mice. Note that in the Icap-1-null mouse the skull shape is severely affected, long bones are shorter, and vertebrae are only poorly ossified (arrows and inserts for higher magnification). (E) Alizarin red/alcian blue staining of E16.5 skeletons. Reduced alizarin red staining intensity of the maxilla (arrowheads), the radius and ulna of the forelimb (arrows) in Icap-1-null tissues. (F) Alizarin red/alcian blue skeletal staining at newborn stage. No obvious difference in staining intensity or patterning could be noticed between the different genotypes.
Delayed ossification in Icap-1−/− embryos
To analyze cartilaginous and bony tissues, the skeletons of homozygous mutant and wild-type animals were stained at various developmental stages with alcian blue (stains cartilage) and alizarin red (stains calcified tissue). The alcian blue staining was indistinguishable between mutant and control embryos throughout development, suggesting that cartilage formation is not grossly affected in Icap-1-deficient mice (Fig. 2E,F). The alizarin red staining, however, was reduced in the skeleton of Icap-1−/− mice as early as E14.5 indicating a defect in ossification. The reduced alzarin red staining was most pronounced in the parietal and frontal bones of the calvaria and in the maxillary and mandibular components of the facial skeleton (not shown). At E16.5, the reduction in alizarin red staining became even more prominent in the skull and in the bony collar surrounding the long bones of the appendicular skeleton (Fig. 2E).
At the newborn stage, alizarin red staining of long bones was similar between control and Icap-1−/− animals (Fig. 2F) suggesting that the ossification of the collar started delayed but was catching up at later stages in Icap-1−/− mice. In the skull region, the postnatal development of the chondrocranium (the majority of the base of the skull) was normal in Icap-1−/− mice. This was shown by the normal ossification of the exoccipital, basioccipital, basisphenoid and presphenoid bones (Fig. 3A) and normal formation and development of the synchondrosis of the skull base in newborn, P15, P21 and P60 control Icap-1−/− animals (Fig. 3A and data not shown). The ossification defect of calvarial bones, however, was obvious in newborn Icap-1−/− mice (Fig. 3B). The frontal, parietal and interparietal (supraoccipital) bones were reduced in size giving rise to enlarged anterior and posterior fontanelles and to widened sagittal and metopic (interfrontal) sutures (Fig. 3B). At the age of 2 months, control mice had completed the ossification of the metopic sutures but still had patent lambdoid, sagittal and coronal sutures (Fig. 3C,F). In mutant mice of the same age the posterior part of the metopic suture was still un-ossified (open) (Fig. 3D,E,G,H). In some Icap-1−/−, non-ossified, alcian blue-positive areas were observed extending from the posterior metopic suture to the frontal bones (Fig. 3E,H). Furthermore, the parietal bones were hypoplastic in mutant calvarias leading to shortened sagittal and V-shaped coronal sutures. The bone defect was also evident in other parts of the skeleton such as in the vertebrae where 15-day old mutant mice showed non-fused vertebral arches (Fig. 3I) and in the pelvic bone where mutant mice showed delayed fusion of the pubis and the ischial bone (Fig. 3J).
Fig. 3. Defect of calvarial ossification in Icap-1 mutant mice.
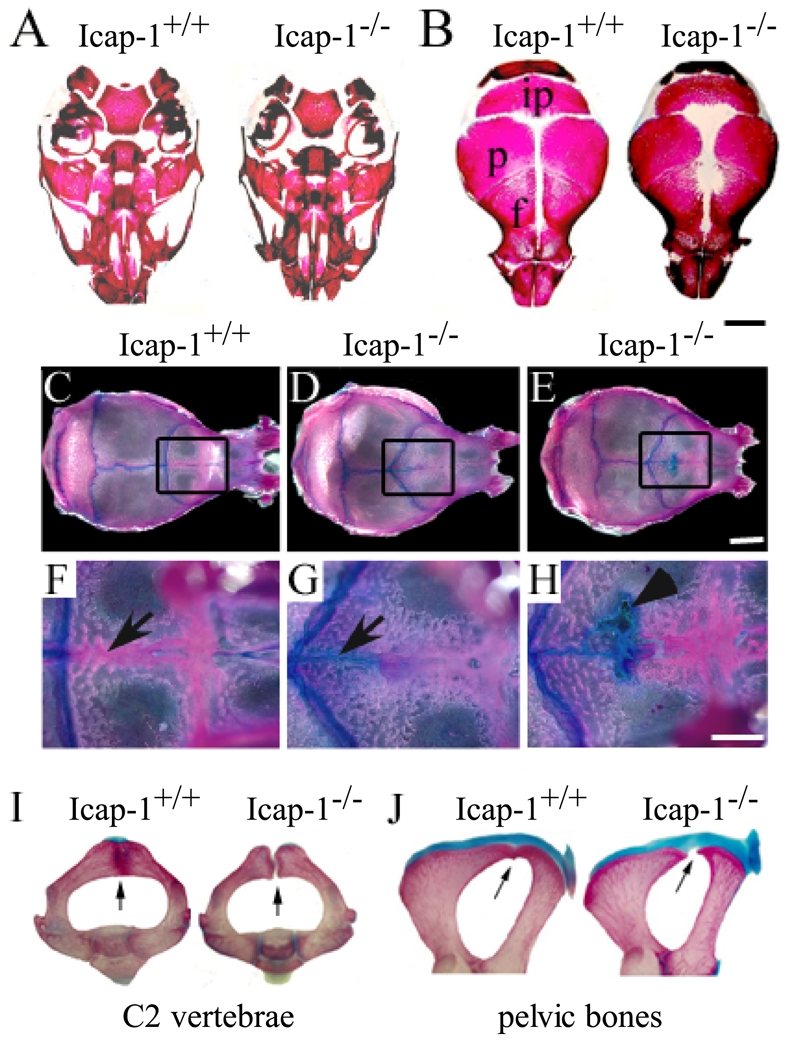
(A–B) Whole-mount alizarin red staining of the skulls of Icap-1+/+ and Icap-1−/− newborn mice. (A) Mineralization of the skull base is comparable in wild-type and mutant animals. (B) Mineralization of the skull vault. The mineralized areas of the interparietal (ip), parietal (p) and frontal bones (f) are smaller and fontanelles are open in Icap-1−/− mice. Bar, 2 mm. (C-H) Whole-mount alizarin red/alcian blue staining of wild-type and Icap1-null 2-month-old calvariae. The area of the metopic suture is boxed in C-E, and displayed at high magnification in F-H. In Icap1-null (D, E) the sagittal suture is shorter, the coronal sutures are V-shaped and irregular when compared with wild-type litter mates (C). At this age, the metopic suture (arrow) is closed in wild-type (F), whereas in Icap-1-deficient mice the posterior part of the metopic suture is not ossified and stained with alcian blue (G, H). Some Icap-1−/− mice display non-mineralized, alcian blue-positive areas in the frontal bones (arrowhead in H). Bars are 5 mm (C–E) and 2 mm (F–H), respectively. (I, J) Whole-mount skeletal staining of the axis (I) and the pelvic region (J) of 21-days-old wild-type (wt) and Icap-1−/− (mt) mice. Arrows point to the fusion defect observed in the Icap-1-deficient mice while fusion is completed in control animals.
Reduced proliferation of osteoprogenitors in calvarial bones
Since the skull vault is severely affected in Icap-1−/− mice, we decided to focus our analysis on the development of the calvaria. To test where ICAP-1 is expressed in the developing skull bones, we performed LacZ staining (Fig. 4A). Whole-mount staining of mutant calvariae revealed strong LacZ expression at the leading edges of the calvarial bones and in the suture regions (Fig. 4A). Frontal sections through the parietal region of the skull showed that the paired parietal bones from normal mice were separated at the midline by a narrow sagittal suture (Fig. 4B). However, in mutant mice the distance between the two parietal bones was wider without the typical suture organization (Fig. 4C). The sagittal suture is a fiber-rich stripe of mesenchyme flanked by condensed bulges of tissue, which are called osteogenic fronts and contain the osteoprogenitors that differentiate into osteoblasts and lay down bone (Fig. 4D). In mutant mice, the osteogenic fronts were thin and contained very few cells resulting in an impaired growth of the parietal bones and widening of the intervening mesenchyme (Fig. 4C,E). LacZ staining revealed strong Icap-1 promoter activity in mature osteoblasts of the parietal bone, in osteoprogenitors of the osteogenic fronts, and in mesenchymal cells between the two fronts (Fig. 4G). As expected, comparable tissue sections from wild-type mice showed no LacZ expression (Fig. 4F).
Fig. 4. Defective formation of the osteogenic front in Icap-1−/− calvaria.
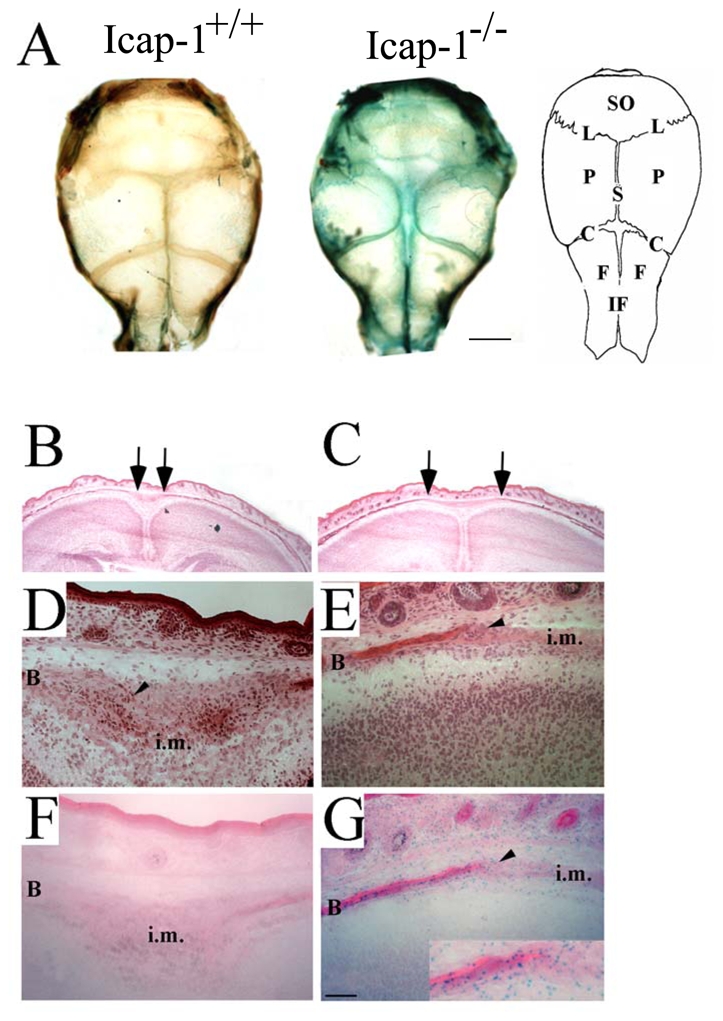
(A) Whole-mount LacZ staining of newborn calvariae of Icap-1+/+ and Icap-1−/− animals. Strong LacZ activity, visualizes Icap-1 expression in the sutural regions and the edges of the bony plates of the Icap-1-deficient calvaria. Bars are 2 mm. (B–C) Hematoxylin-eosin stained frontal section of the parietal region. The distance between the ossified ends of the pariatal bones (arrows) is wider in Icap-1−/− (C) compared to wild-type mice (B). (D–E) Frozen frontal sections through the sagittal suture of wild-type (D, F) and Icap-1−/− (E, G) animals stained with hematoxylin and eosin (D, E) or for LacZ activity (F, G). Note that the wild-type suture presents a typical condensed cell population corresponding to the osteogenic front (arrowheads) that is severely reduced in the Icap-1−/− mice. LacZ staining indicates extensive expression of ICAP-1 in this region. B, bone; i.m, intersutural mesenchyme; SO, supraoccipital bone; P, parietal bone; F, frontal bone; S, saggital suture; L, lambdoid suture; C, coronal suture; if, interfrontal suture. Bar is 50 μm.
The morphogenesis of the calvaria depends on the number of osteoprogenitors and their differentiation into osteoblasts at the margins of the suture. To test whether the reduced number of osteoprogenitors in mutant osteogenic fronts was caused by aberrant cell survival and/or proliferation we performed apoptosis assays, BrdU incorporation assays and Ki67 immunostaining in newborn mice. The TUNEL assay and staining of activated caspase-3 showed very few apoptotic cells similar in number for normal and mutant osteogenic fronts (data not shown). The labelling indexes for Ki67 and BrdU, however, were reduced in mutants by 40% and 64%, respectively, indicating a pronounced proliferation defect in the Icap-1-deficient osteoprogenitor cell population (Fig. 5A,B). Proliferation of osteoblast progenitor cells is tightly regulated by ECM interactions and growth factors. In order to determine whether the diminished proliferation at the osteogenic fronts are cell autonomous defects or caused by impaired secretion of growth factors and/or ECM components by the surrounding tissue, we isolated primary osteoblasts from the calvaria of control and mutant mice, immortalized them by retroviral transduction of the SV40 large T antigen, and tested their proliferation rates. Similarly to the in vivo observations, Icap-1−/− cells (SV2.1-Icap-1−/−) displayed a lower BrdU incorporation rate compared to control cells (SV6.5-Icap-1+/+) in vitro (Fig. 5C). Retroviral infection of the full length Icap-1 cDNA into SV2.1-Icap-1−/− osteoblasts (SV2.1-Icap-1resc) restored normal proliferation (Fig. 5C) corroborating the link of ICAP-1 loss with the proliferation defect. To further characterize the proliferation defect at the molecular level, we performed Western blot analysis of cyclin D1 expression. Adhesion of wild-type or rescued osteoblasts on FN leads to increased cyclin D1 expression 5 hours of after seeding. Conversely, Icap-1−/− preosteoblasts cultured under identical experimental conditions show a significant reduction of cyclin D1 expression (Fig. 5D). These results indicate that the decreased (pre)osteoblast proliferation observed in Icap-1−/− mice is cell autonomous.
Fig. 5. Reduced proliferation of calvarial osteogenic cells in Icap-1 null mice.
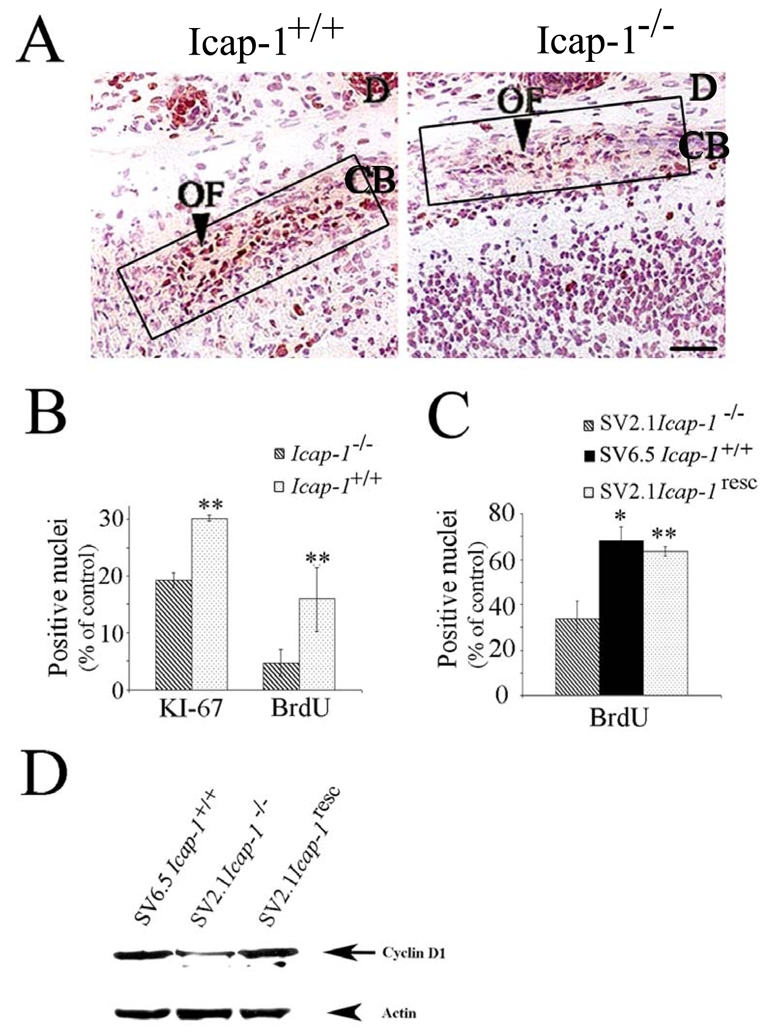
(A) Immunodetection of the proliferation marker Ki67 in the sagittal sutural region of newborn Icap-1+/+and Icap-1−/− calvaria. The osteogenic front of the Icap-1−/− mice displays a reduced number of Ki67-positive cells. Boxes indicate regions used for KI-67 and BrdU quantification in (B). Bar is 25 μm. (B) Quantification of Ki67- and BrdU-positive cells in the osteogenic front of control and mutant animals. Error bars represent S.D. Asterisks indicate a statistically significant difference between Icap-1+/+and Icap-1−/− (**, P<0.0001). (C) Immortalized calvarial osteoblasts. ICAP-1-deficient cell (SV2.1-Icap-1−/−) show significantly reduced BrdU-labeling index compared to wild-type cells (SV6.5-Icap-1+/+). Retroviral transfection of the Icap-1 cDNA into the Icap-1−/− cells rescues the proliferation defect (SV2.1-Icap-1resc) (**, P<0.0001). (D) SV2.1 and SV2.1-Icap-1resc cells were cultured for 24 h in 1% FCS before replating them onto 10 μg/ml FN. After 5 h of spreading, cells were washed with PBS and directly lysed onto Petri dishes with RIPA buffer. An amount of 30 μg of proteins per lane was gel separated and then transferred onto PVDF membrane before processing for Western blotting with the anti-cyclin D1 antibody. The same gel was blotted with anti-actin polyclonal antibodies for normalizing protein loading.
Calvarial osteogenesis is perturbed in Icap-1-deficient mice
The differentiation of osteoprogenitors into mature osteoblasts is characterized by the deposition of “bone-specific” extracellular matrix molecules, as well as the spatially and temporally coordinated expression of growth and transcription factors. To study the cranial skeletogenic differentiation in Icap-1−/− mice, we first evaluated the expression of osteogenic markers such as alkaline phosphatase (AP), osteonectin and collagen type I (Col1) on frontal sections of newborn calvariae (Fig. 6). In wild-type, AP activity was confined to the osteoid region of the developing parietal bones and to pre-osteoblasts at the osteogenic front of the sagittal suture (Fig. 6A). Osteonectin expression overlapped with sites of AP activity (Fig. 6B,C) and was strong along the bone surface and weak in cells at the osteogenic front. In mutant tissue, the cell number with AP activity was reduced at the bone-suture margins defining a smaller osteogenic front area committed to osteoblastic differentiation (Fig. 6A). Osteonectin deposition was also reduced at the osteoid surface and was almost absent at the osteogenic front (Fig. 5B,C). Similarly, collagen I immunostaining was markedly reduced on the bone surface and at the suture margin in mutant compared to wild-type (Fig. 6D,E).
Fig. 6. Osteogenic differentiation is abnormal in Icap-1−/− calvaria.
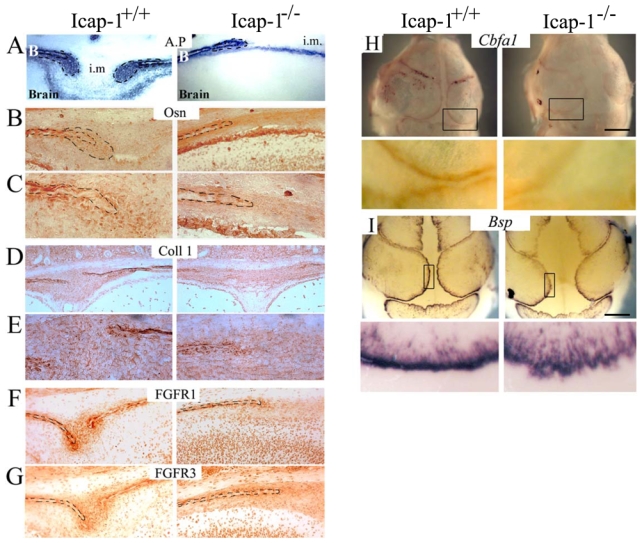
Frontal sections through the parietal bones and the sagittal suture of Icap-1+/+ and Icap-1−/− newborn mice were stained for (A) AP, (B, C) osteonectin, (D, E) Col1 (F) FGFR1 and (G) FGFR3. (C, E) are (400X) magnification views of (B) and (D) osteogenic front region, respectively. Dashed lines represent bone borders and osteogenic front area. B, bone; i.m., intersutural mesenchyme. Note the reduced expression of the osteogenic and differentiation markers in Icap-1−/− tissue.
Whole mount in situ hybridization on calvaria of E17.5 embryo was performed to detect either Cbfa1/Runx2 (H) or Bsp transcript (I) on Icap-1+/+and Icap-1−/− embryos. The top panel is an overview of the full calvaria, the bottom panel is a closer view of the osteogenic front (boxed in the overview). Bars are 2 mm.
Since the expression patterns of FGF-receptors correlate with the osteogenic differentiation process (Iseki et al., 1999; Rice et al., 2000) we compared the expression of FGFR1 and FGFR3 in wild-type and mutant newborn calvariae. In control tissue, both proteins were detected in the osteoblasts of the calvarial bones and in cells of the osteogenic front and weakly in the sutural mesenchyme (Fig. 6F,G). In Icap-1−/− calvariae, FGFR1 and FGFR3 immunolabelling was fainter than in wild-type, and this difference was particularly pronounced at the osteogenic fronts (Fig. 6F,G). In addition, whole mount in situ hybridization on E17.5 heads reveals that the expression of both the early bone marker Cbfa1/Runx2 and the later marker bone sialic protein (Bsp) are reduced in mutant mice (Fig. 6H,I). Again, a weaker signal is observed at the edge of the bony region reflecting the marked reduction of cells committed into the osteoblastic lineage within the osteogenic front. Altogether these results indicate that osteogenic front is not normally formed in mutant animals.
To test whether committed cells normally differentiate, primary osteoblasts were isolated from newborn wild-type and Icap-1−/− calvariae and incubated in medium supplemented with ascorbic acid and β-glycerophosphate to induce osteoblast differentiation and the formation of mineralized bone nodules (Fig. 7A). After two weeks in the differentiation medium, AP activity was evident in almost all cells derived from control calvariae indicating their committment to the osteoblast lineage. Icap-1−/− calvarial cells also started to express AP albeit at lower levels (data not shown). After four weeks of culture, differentiating osteoblasts from Icap-1−/− calvariae contained fewer and smaller mineralized nodules as visualized by alizarin red (Fig. 7A) and von Kossa staining (data not shown). Similarly, immortalized Icap-1−/− cells (SV2.1-Icap-1−/−) showed a markedly reduced AP staining and mineralized nodule formation relative to wild-type (SV6.5-Icap-1+/+) and rescued cells (SV2.1-Icap-1resc) (Fig. 7B and data not shown). These data show that ICAP-1 loss impairs osteogenesis and identifies a role of ICAP-1 as a cell autonomous factor in osteoblast differentiation.
Fig. 7. Bone nodule formation by Icap-1−/− calvarial osteoblast is defective.

(A) Primary Icap-1+/+ and Icap-1-deficient osteoblasts were cultured for 4 weeks in inductive medium and stained with alizarin red for monitoring bone nodule formation. Icap-1−/− cultures show fewer and smaller mineralized nodules (arrows) compared with wild-type cultures (Icap-1+/+). The result is representative of at least 3 independent experiments from 3 different animals. Bar, 1 mm. (B) Immortalised osteoblasts SV2.1 Icap-1−/−, SV6.5 Icap-1+/+ or SV2.1-Icap-1resc were cultured in inductive medium for 3 weeks and mineralized nodules were identified by alizarin red staining. Icap-1−/− osteoblasts show a significantly reduced nodule formation compared to wild-type or Icap-1resc osteoblasts. The means and S.D. were calculated from 3 independent experiments (*, P<0.05, **, P<0.0001)
Since we routinely induced differentiation after cells derived from calvariae or immortalized osteoprogenitors have reached confluence in vitro, the numbers of neither the mutant (SV2.1-Icap-1−/−) nor the wild-type (SV6.5-Icap-1+/+) and rescued cells (SV2.1-Icap-1resc) significantly increased during the differentiation period (data not shown). Altogether these findings suggest that the differentiation block occurs in addition to the cell proliferation defect.
β1integrin is highly expressed and activated in osteogenic front
β1 integrins have been proposed to play a critical role during osteoblast proliferation and differentiation (Moursi et al., 1997; Zimmerman et al., 2000). Since ICAP-1 interacts with the cytoplasmic tail of the β1 integrin chain and modulates integrin function in vitro (Bouvard et al., 2003), we analyzed β1 integrin expression in wild-type and mutant calvariae in vivo. At newborn stage, frontal sections through the sagittal suture and the parietal bones were immunostained with an anti-β1 polyclonal antibody and with the monoclonal antibody 9EG7, which recognizes the ligand bound form of β1 integrins (Fig. 8A and data not shown). In sections from control mice, both antibodies strongly labeled the osteogenic fronts and the bone surfaces, while the intervening mesenchyme was faintly labeled. Conversely, sections from Icap-1-deficient mice showed clear β1 integrin staining of the bone surface but very faint staining of cells at the osteogenic front. This apparently diminished β1 staining was mainly due to the reduced osteogenic front population rather than a reduced expression on the cell surface of individual cells since FACS analysis showed only a slight reduction of β1 integrin expression on Icap-1−/− osteoblasts (Fig. 8B, see below).
Fig. 8. Increases in β1 integrin activity in Icap-1−/− cells.
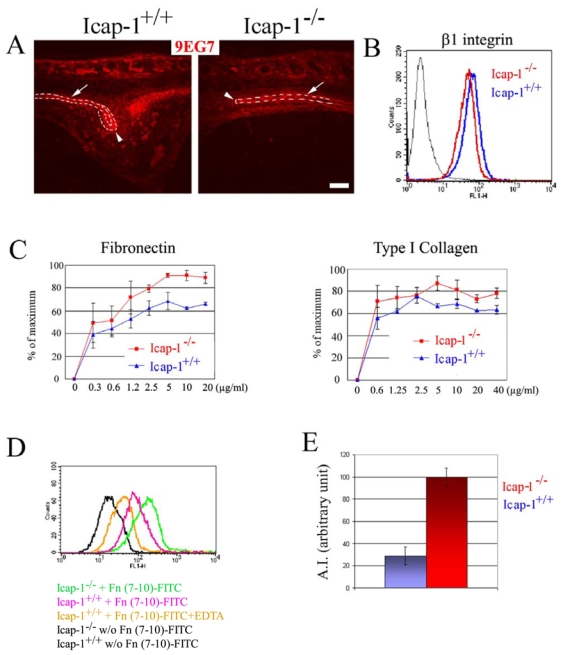
(A) Sagittal sections were stained with the 9EG7 monoclonal antibody (red) that recognizes ligand-bound β1 integrins. β1 integrins are highly expressed and strongly activated on wild-type cells (Icap-1+/+) at the osteogenic front (arrow heads) and at the surface of the bony plates (arrows). In Icap-1−/− tissues, the cells at the osteogenic front show a moderate staining for activated β1 integrin. Bar is 50 μm. (B) FACS analyses demonstrate a slight reduction in the surface expression of β1 integrins (assayed by the MB1.2 monoclonal antibody) on Icap-1−/− primary osteoblasts (red) compared to wild-type osteoblasts (blue). (C) Adhesion assays. The adhesion of Icap-1−/− primary osteoblasts to FN and Col1 is moderately increased compared to Icap-1+/+ cells. Adhesion is expressed as a percentage of the maximal adhesion and measured in duplicate in two independent experiments from two different animals (p<0.05). (D) FACS analysis demonstrates increased binding of FITC-Fn 7–10 fragment to Icap-1−/− osteoblasts (green). (E) The activation index (AI) of the β1 integrin is increased in Icap-1−/− osteoblats. The maximum AI obtained is used to normalize both genotype groups and referred as 100.
ICAP-1 controls β1 integrin activity and condensation of osteoprogenitors
To further analyze the consequence of ICAP-1 expression loss for the adhesion of osteoblasts assays were carried out using primary osteoblasts isolated from calvarial tissues. In spite of a small decrease in β1 expression (Fig. 8B), adhesion of Icap-1−/− osteoblasts to FN or Col1 was moderately but significantly increased compared to wild-type osteoblasts (Fig. 8C). This suggests that increased adhesion to ECM substrates resulted from the activation rather than an increased cell surface expression of β1 integrins on Icap-1−/− osteoblasts.
To investigate whether the loss of ICAP-1 expression interferes with integrin activation, we estimated the ligand-binding affinity of the FN receptor α5β1 integrin both in wild-type and Icap-1−/− primary osteoblasts. The cell binding domain of the FN corresponding to the type III repeats 7–10 (Fn 7–10) was expressed, purified and FITC-labeled. The capability of both mutant and wild-type primary osteoblast cells to interact with Fn 7–10 at a non saturable concentration was analyzed by FACS. As shown in Fig. 8D, we consistently observed an increase in Fn 7–10 binding to Icap-1−/− osteoblasts compared to wild-type cells. The activation index, which normalizes the specific binding of FITC-Fn 7–10 to the total β1 surface expression level, was elevated by approximately three-fold in Icap-1−/− cells compared to wild-type cells (Fig. 8E).
During in vitro differentiation of both primary and immortalized osteoblasts we constantly observed that the bone nodules formed by Icap-1−/− cells were fewer, smaller, and less compact than those formed by wild-type cells (Fig. 9A). Since differentiation of bone cells requires an initial step of cell condensation (Globus et al., 1998; Ornitz and Marie, 2002), a defect in this step might lead to the altered or delayed differentiation. To address this question we cultured immortalized osteoprogenitros in suspension using the hanging drop technique. Under these conditions, wild-type or rescued cells aggregate and form compact spheroids within 48 hours while spheroids formed by Icap-1−/− cells were less compacted (Fig. 9B and data not shown). Since ICAP-1 loss increases integrin affinity (Fig. 8), we investigated whether blocking of β1 integrins in their activated state would mimic ICAP-1 deficiency. To this end we complemented the culture medium with the integrin activating monoclonal antibody 9EG7 and then formed spheroids (Fig. 9B). While spheroid-triggered compaction of Icap-1−/− cells was not affected by the treatment with the 9EG7 antibody, compaction of control or rescued cells was consistently delayed. These findings indicate that integrin affinity modulation by ICAP-1 is required for proper compaction of osteoblastic cells.
Fig. 9. Defects in bone nodule formation and spheroid compaction with Icap-1−/− osteoblasts.
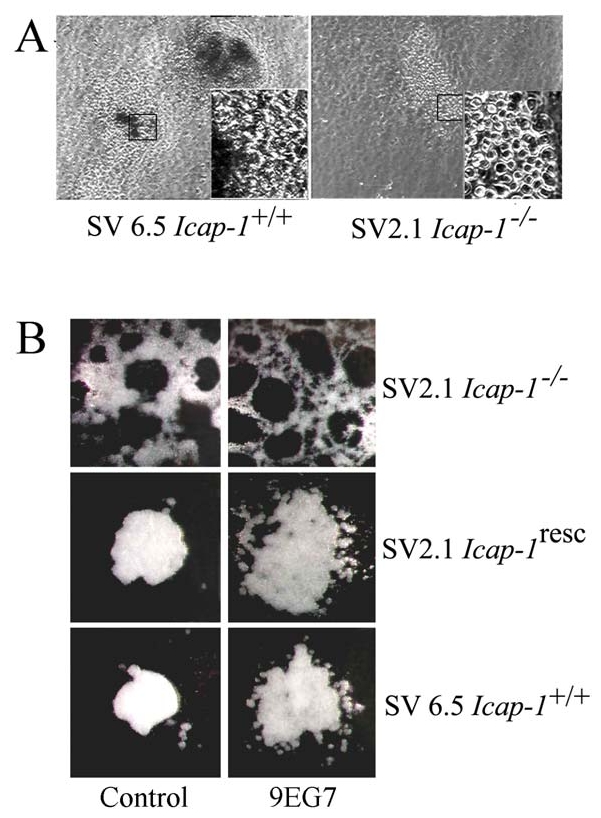
(A) Immortalised SV6.5 (wild-type) or SV2.1 (Icap-1−/−) pre-osteoblast were induced to differentiate in vitro for 15 days, then bone nodule formation and organization was visualized by phase contrast microscopy. Insert is a higher magnification view of the boxed region; note that SV2.1 cells are less cohesive than control cells. (B) Spheroids were formed from SV2.1, SV2.1 rescued with Icap-1 cDNA or SV6.5 pre-osteoblast and analyzed after 16 hours incubation at 37°C using a standard protocol for hanging drops assay. We used 25.000 cells per drop in each experimental condition. For antibody treatment 9EG7 or control antibodies (ctl) were added at a final concentration of 10 μg/ml during the condensation process in a medium supplemented with a FN-depleted serum.
DISCUSSION
In the present paper we report the molecular analysis of mice carrying a disrupted Icap-1 gene. Icap-1-deficient mice suffer from defective osteogenesis characterized by mild growth retardation and severe craniofacial dysmorphism. We focused our analyses on the calvarial abnormalities to obtain a clear view on osteoblast differentiation.
ICAP-1 plays an important role for osteogenesis
Bone formation (osteogenesis) involves the conversion of mesenchymal tissue into bone either directly (intramembranous ossification) or via cartilaginous intermediates (endochondral ossification) (Zelzer and Olsen, 2003). The first mechanism is responsible for the generation of the flat bones of the skull vault as well as for the formation of the bony collar around the diaphysis of the long bones. In Icap-1−/− mice, intramembranous ossification is affected, since both periosteal and calvarial osteogenesis are delayed. Alcian blue staining revealed that the formation of cartilaginous templates of endochondral bones is grossly undisturbed in mutant mice. Since ICAP-1 is also expressed in chondrocytes (data not shown), we cannot fully exclude mild differentiation and/or proliferation defects of mutant chondrocytes, which may also contribute to the small stature of Icap-1−/− mice. We focused our investigations on intramembranous ossification and show for the first time the important role of ICAP-1 during osteoblast differentiation. The ossification of calvarial bones (frontal, parietal and interfrontal) was dramatically delayed during embryonic development leading to open fontanels and wide sutures at birth. These symptoms mirror the calvarial abnormalities of mice carrying mutations for the transcription factors Cbfa1/Runx2 (Otto et al., 1997) and Atf4 (Yang et al., 2004), which have been identified as key regulators of osteoblast differentiation. The remarkable phenotypic similarities between the Icap-1−/− mice and these mouse models suggest that ICAP-1 is a new and important regulator of osteoblast development. Cranial sutures regulate bone expansion during postnatal life and remain non-ossified as long as the brain is growing (Opperman, 2000). Osteogenesis at the suture involves the differentiation of mesenchymal cells into preosteoblasts and into osteoblasts at the sutural margins and the subsequent deposition of a collagenous matrix along the bony plates. Increased growth of the calvarial bones leads to premature closure of the sutures (craniosynostoses), while delayed bone growth results in suture latency. ICAP-1 is expressed at the osteogenic fronts and its deficiency leads to a dramatic reduction in the osteoblast population at the osteogenic fronts, which in turn prevents correct temporal fusion of cranial sutures. The reduced osteoblast number at the osteogenic fronts is due to reduced proliferation and impaired osteoblast differentiation.
ICAP-1 regulates osteoblast proliferation
We have previously reported that ICAP-1 has a dual localization and is found in the cytoplasm/membrane and in the nucleus (Fournier et al., 2005). Loss of ICAP-1 expression in immortalized osteoblast considerably reduced cell proliferation and cyclin D1 expression. Therefore the reduced proliferation in the osteogenic front is likely due to a lack of ICAP-1 in the nucleus that would lead to a reduced cyclin D1 expression. This might explain the severe decrease in the cell population committed to the osteoblast lineage in the sutural region observed in the Icap-1 deficient animals.
ICAP-1 regulates integrin activity
Isolated Icap-1-null primary osteoblasts displayed an increased cell adhesion to FN and Col1. We have previously shown that overexpression of ICAP-1 in HeLa cells disrupts focal adhesions likely by inhibiting the association of the cytoplasmic tail of β1 integrin with talin (Bouvard et al., 2003). It was shown that talin binding to the β integrin domain is a key step in the regulation of integrin activation (Calderwood et al., 2004; Calderwood et al., 2002). In good agreement with this, we found an increase in Fn 7–10 binding to integrin in mutant cells which was not accompanied with any upregulation of β1 integrin expression. This confirms that ICAP-1 regulates β1 integrin affinity.
ICAP-1 regulates osteoblast differentiation
Osteoblast differentiation is a multistep process that first required an initial condensation of the mesenchymal cells to form the osteogenic front (Hall and Miyake, 2000). The cell population at the osteogenic front is visible during calvaria bone development at the edge of the expanding bone within the intervening mesenchyme. This condensed cell population further differentiates and expresses different osteoblastic markers such as Runx2/Cbfa1, AP, bone sialoprotein (BSP) and others. Our knowledge of how integrins or cell adhesion is implicated into this process is poorly documented, but recent reports suggested an unexpected role for the matrix stiffness for controlling early osteoblast differentiation (Engler et al., 2006; McBeath et al., 2004).
An important role of β1 integrins has also been reported for osteoblast differentiation in vitro. It was shown that Col1 interaction with α2β1 integrin regulates osteoblast-specific gene expression and osteoblast differentiation (Takeuchi et al., 1996; Xiao et al., 1998). Similarly, α5β1 integrin interaction with FN promotes differentiation (Moursi et al., 1997) and survival (Globus et al., 1998) of cultured calvarial osteoblasts. Finally, transgenic overexpression of a dominant-negative form of β1 integrin in osteocytes blocks β1 integrin function, reduces adhesion to collagen and FN, and diminishes bone mass in cortical and flat bones (Zimmerman et al., 2000). While these studies identify an important role of β1 integrins for osteogenesis they do not address the role of integrin affinity modulation for bone development. Our data demonstrate for the first time that not only β1 integrin affinity increase, but also affinity decrease are of paramount importance for proper bone development both in vivo and in vitro.
We consistently observed a marked reduction of osteoblast cells in Icap-1-deficient animals. Even the expression of the very early marker Runx2 at the osteogenic front region was significantly reduced. Furthermore, the in vitro differentiation assays revealed that the bone nodules formed by Icap-1 null immortalized osteoprogenitors were smaller and less compacted compared to nodules formed from wild-type or rescued cells. This reduced compaction of Icap-1-deficient cells was further confirmed with the hanging drop technique suggesting that ICAP-1 is required for osteoblast condensation which is a crucial and early step during osteoblast differentiation.
Our data suggest that the proliferation and the differentiation defects occur independently and that they both contribute to the abnormal osteogenesis. In support for this notion we observed that limited ICAP-1 expression in Icap-1-deficient preosteoblast cell lines fully restored their proliferation rate but only partially their potential to differentiate and form nodules in vitro. Furthermore, in our differentiation assay we routinely use confluent cells to rule out that the proliferation defect is influencing the formation of mineralized nodules. Indeed, we observed only a slight increase of cells during the differentiation period and this increase was similar in control and Icap-1-deficient cultures. Our data indicate that the condensation defect of the Icap-1-deficient preosteoblasts further limits the number of progenitors that will finally differentiate into mature osteoblasts (Fig. 9). The functional importance of the condensation of progenitors for osteoblast development has also been observed in connexin-43-deficient mice (Lecanda et al., 2000).
Previous work implicated β1 integrins in cell compaction (Robinson et al., 2004; Robinson et al., 2003). Our data suggest that ICAP-1 may control β1 integrin function during this process by regulating activation/deactivation cycles. To our knowledge this is the first direct evidence reporting that integrin affinity is important for cell cohesion and differentiation.
Materials and Methods
Generation of Icap-1-deficient mice
Five positive PAC clones were isolated from RPCI21 library and used to generate the targeting construct (for details email daniel.bouvard@ujf-grenoble.fr). Electroporation into passage 13 R1 ES cells was done as previously described (Talts et al., 1999). Animals were either genotyped by southernblot or by PCR.
Antibodies
Polyclonal anti ICAP-1 antibodies were previously described (Bouvard and Block, 1998). Monoclonal antibodies against actin, vinculin (h-Vin1) and talin (clone 8d4) were from Sigma-Aldrich (Germany). Polyclonal anti-β1 integrin serum was a gift from Dr. Johansson (Uppsala, Sweden). Monoclonal β1 antibodies 9EG7 and MB1.2 were from Pharmingen (France) and a gift from Dr. Bosco (Ontario, Canada), respectively. Polyclonal anti-collagen I, III and osteonectin/BM-40 were from Dr. R. Timpl (Martinsried, Germany). Polyclonal antibodies against cyclin D1, FGFR1 and 3 were from SantaCruz (USA), against Ki67 from Novocastra (UK), and against 5-bromo-2′-deoxyuridine (BrdU) from Roche (Germany).
Isolation and assays with primary osteoblats
A primary mouse osteoblast enriched cell population was isolated from newborn calvaria using a mixture of 0.3 mg/ml collagenase type I (Sigma) and 0.25% trypsin (Gibco BRL) as described previously (Bellows et al., 1986; Otto et al., 1996). Cells were grown in α-MEM medium containing 10% FCS.
In vitro differentiation of isolated osteoblasts was performed essentially as described (Globus et al., 1998). Briefly, 60.000 cells per well were plated in a 24 well tray. After 3 days of culture when cells were confluent, the medium was switched to differentiation medium (α-MEM, 10% FBS, 50 μg/ml ascorbic acid, 10 mM β-glycerophosphate) and changed every second day. The differentiation process was visualized by AP staining for osteoblast activity and by Alizarin Red S staining for calcium deposition.
For the adhesion assay, primary osteoblasts (passage 2) were seeded at 0.5×105 cells in a 96 well tray coated with various concentrations of FN or Col1. The cells were incubated for 1 hour at 37°C and then washed three times with PBS before staining with a crystal violet solution (0.1% crystal violet, 20% methanol) for 1 hour at room temperature. After three washes in water, cells were lysed in 0.1% SDS for 1 hour. The absorbance was read at 550 nm with a Beckman Coulter’s AD 340 Absorbance Detector.
Cell proliferation was estimated using BrdU assay as previously described (Fournier et al. 2003)
Immortalization of osteoblasts
Primary osteoblasts (passage 2) were infected with a retrovirus expressing the large SV40 T antigen (Fässler et al., 1995), cloned and tested for their ability to induce AP upon differentiation (Mansukhani et al., 2000). Clone SV2.1 from an Icap-1-deficient mouse and clone SV6.5 from a wild-type animal were used in this study. Rescue of ICAP-1 expression in SV2.1 cells was done via retroviral infection using the pCLMFG-Icap-IRES-EGFP vector. A homogeneous cell population was sorted based on EGFP fluorescence with a MoFlo cell sorter (Dako Cytomation). ICAP-1 expression was checked by Western blot, immunofluorescence and FACS using EGFP as a marker. This non-clonal cell population is referred as SV2.1-Icap-1resc hereafter.
Compaction assay in hanging drops
Immortalized cells were harvested by trypsin digestion and washed twice in DMEM media. Drops of 10 μl of DMEM-SVF medium containing 25,000 cells were spotted onto the cover lid of 10 cm Petri dishes inverted and placed on a Petri dish containing 8 ml of PBS. Spheroids compaction was then followed over a 72 hours incubation time and images were taken using a binocular microscope equipped with a digital camera.
Skeletal preparation, X-gal staining and X-ray analysis
Staining of whole-mount embryos with alcian blue/alizarin red (Aszodi et al., 1998) and X-Gal (Sakai et al., 2001) was carried out as described earlier. X-ray images have been obtained on a dual energy setup developed at CEA/LETI (Grenoble, France).
Whole mount in situ hybridization, histology, immunohistochemistry and in vivo cell proliferation
Whole mount in situ hybridization was performed as described (Rice et al., 2000). Histochemistry and immunostaining on tissue sections were carried out as previously described (Aszodi et al., 1998). In vivo cell proliferation was analyzed using either BrdU incorporation assay (Aszodi et al., 1998) or Ki67 immunohistochemistry. To detect cells with AP activity, calvarial cryosections were fixed for 10 minutes in 3% paraformaldehyde. After washing in PBS, the color reaction was developed in the BCIP/NBT substrate solution (Roche, France). Immunofluorescence staining and FACS analysis of primary osteoblasts were performed as previously described (Bouvard et al., 2003).
Activation index of β1 integrin on primary osteoblast
Activation index of β1 integrin was estimated essentially as previously described (Calderwood et al., 2004). Briefly, primary osteoblasts were isolated and passage 2 cells were aliquoted into two pools containing either Tyrode’s buffer alone or Tyrode’s buffer supplemented with 5 mM EDTA. After a 15 min incubation at 4°C, cells were incubated with or without the FITC labelled Fn 7–10 fragment for 45 min at 4°C in the presence or absence of 5mM EDTA, washed in ice cold Tyrode’s and analyzed on a FACScan (Becton Dickinson) flow cytometer. The collected data were analyzed using CellQuest software (Becton Dickinson). In parallel cells were analyzed for β1 expression using the MB1.2 monoclonal antibody to detect the level of β1 integrin on the cell surface. The activation index (AI) was calculated as follows: each specific mean intensity fluorescence (MFI) was calculated by subtracting the background obtained with Fn 7–10 fragment incubation in the presence of EDTA or without the primary antibody in the case of the MB1.2 labelling. AI=((MFI Fn 7–10)-(MFI Fn7–10+EDTA))/(MFI MB1.2)-(MFI MB1.2 control).
RNA and protein analyses
Total RNA was isolated from adult kidney using TRIzol reagent (GIBCO BRL) according to the manufacturer’s recommendations. For Northern analysis, 10 μg of total RNA was separated on a 1.2 % agarose-2.2 M formaldehyde gel, transferred to Hybond+ membrane (Amersham) and probed with a 32P-labelled Icap-1 cDNA.
For biochemistry, brains of adult mice were homogenized in RIPA buffer (10% w/v) and used for Western blotting as described (Bouvard et al., 1998).
Fig. 10. Schematic representation of bone formation in calvaria.
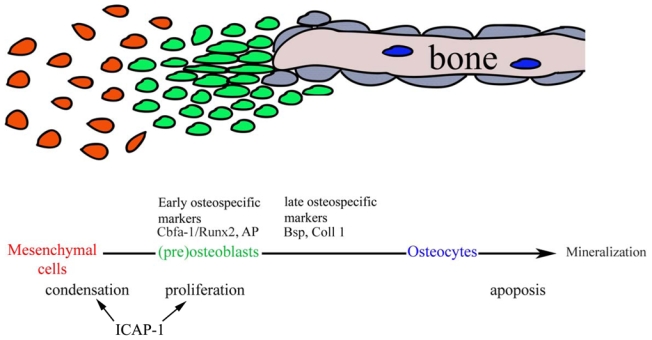
Bone formation is a multistep process that requires first an initial condensation stage. This step enables the cells to start the differentiation process where some early markers such as Cbfa1/Runx2, Osterix are expressed. At later stages, other osteogenic markers such as AP, BSP, osteocalcin and osteonectin are produced. Lack of ICAP-1 expression slows down proliferation and in addition the ability of mesenchymal cells to compact. Since the compaction is a very early event in the osteoblast differentiation pathway, expression of more distal maker are consequently reduced in Icap-1−/− animals.
Acknowledgments
We want to thank Drs R. Timpl, S. Johannsonn, B. Nieswandt, C. Bosco, G. Karsenty for generously providing antibodies and in situ probes, C. Robert-Coutant and J.M. Dinten for X-ray imaging of mice. V. Collin for cell sorting, and P. Marie, M. Pfaff and D. Pearton for their valuable discussion and critical reading of the manuscript. DB was supported by an E.C. Marie Curie long term fellowship (QLGA-CT-2000-52076), the Association pour la Recherche contre le Cancer (ARC), the CNRS and the Max Planck Society. RF is supported by the DFG, BMBF and the Max Planck Society.
This work is dedicated to the memory of Rupert Timpl, Günter Kostka, Martin Pfaff, and Christine Robert-Coutant.
References
- Aszodi A, Chan D, Hunziker E, Bateman JF, Fassler R. Collagen II is essential for the removal of the notochord and the formation of intervertebral discs. J Cell Biol. 1998;143:1399–412. doi: 10.1083/jcb.143.5.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows CG, Aubin JE, Heersche JN, Antosz ME. Mineralized bone nodules formed in vitro from enzymatically released rat calvaria cell populations. Calcif Tissue Int. 1986;38:143–54. doi: 10.1007/BF02556874. [DOI] [PubMed] [Google Scholar]
- Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–77. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- Bouvard D, Block MR. Calcium/calmodulin-dependent protein kinase II controls integrin alpha5beta1-mediated cell adhesion through the integrin cytoplasmic domain associated protein-1alpha. Biochem Biophys Res Commun. 1998;252:46–50. doi: 10.1006/bbrc.1998.9592. [DOI] [PubMed] [Google Scholar]
- Bouvard D, Molla A, Block MR. Calcium/calmodulin-dependent protein kinase II controls alpha5beta1 integrin-mediated inside-out signaling. J Cell Sci. 1998;111 (Pt 5):657–65. doi: 10.1242/jcs.111.5.657. [DOI] [PubMed] [Google Scholar]
- Bouvard D, Vignoud L, Dupe-Manet S, Abed N, Fournier HN, Vincent-Monegat C, Retta SF, Fassler R, Block MR. Disruption of focal adhesions by integrin cytoplasmic domain-associated protein-1 alpha. J Biol Chem. 2003;278:6567–74. doi: 10.1074/jbc.M211258200. [DOI] [PubMed] [Google Scholar]
- Brakebusch C, Bouvard D, Stanchi F, Sakai T, Fassler R. Integrins in invasive growth. J Clin Invest. 2002;109:999–1006. doi: 10.1172/JCI15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood DA. Integrin activation. J Cell Sci. 2004;117:657–66. doi: 10.1242/jcs.01014. [DOI] [PubMed] [Google Scholar]
- Calderwood DA, Tai V, Di Paolo G, De Camilli P, Ginsberg MH. Competition for talin results in trans-dominant inhibition of integrin activation. J Biol Chem. 2004;279:28889–95. doi: 10.1074/jbc.M402161200. [DOI] [PubMed] [Google Scholar]
- Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, Ginsberg MH. The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem. 2002;277:21749–58. doi: 10.1074/jbc.M111996200. [DOI] [PubMed] [Google Scholar]
- Chang DD, Wong C, Smith H, Liu J. ICAP-1, a novel beta1 integrin cytoplasmic domain-associated protein, binds to a conserved and functionally important NPXY sequence motif of beta1 integrin. J Cell Biol. 1997;138:1149–57. doi: 10.1083/jcb.138.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degani S, Balzac F, Brancaccio M, Guazzone S, Retta SF, Silengo L, Eva A, Tarone G. The integrin cytoplasmic domain-associated protein ICAP-1 binds and regulates Rho family GTPases during cell spreading. J Cell Biol. 2002;156:377–87. doi: 10.1083/jcb.200108030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Fassler R, Pfaff M, Murphy J, Noegel AA, Johansson S, Timpl R, Albrecht R. Lack of beta 1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J Cell Biol. 1995;128:979–88. doi: 10.1083/jcb.128.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier HN, Dupe-Manet S, Bouvard D, Lacombe ML, Marie C, Block MR, Albiges-Rizo C. Integrin cytoplasmic domain-associated protein 1alpha (ICAP-1alpha) interacts directly with the metastasis suppressor nm23-H2, and both proteins are targeted to newly formed cell adhesion sites upon integrin engagement. J Biol Chem. 2002;277:20895–902. doi: 10.1074/jbc.M200200200. [DOI] [PubMed] [Google Scholar]
- Fournier HN, Dupe-Manet S, Bouvard D, Luton F, Degani S, Block MR, Retta SF, Albiges-Rizo C. Nuclear translocation of integrin cytoplasmic domain-associated protein 1 stimulates cellular proliferation. Mol Biol Cell. 2005;16:1859–71. doi: 10.1091/mbc.E04-08-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globus RK, Doty SB, Lull JC, Holmuhamedov E, Humphries MJ, Damsky CH. Fibronectin is a survival factor for differentiated osteoblasts. J Cell Sci. 1998;111 (Pt 10):1385–93. doi: 10.1242/jcs.111.10.1385. [DOI] [PubMed] [Google Scholar]
- Gunel M, Laurans MS, Shin D, DiLuna ML, Voorhees J, Choate K, Nelson-Williams C, Lifton RP. KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc Natl Acad Sci U S A. 2002;99:10677–82. doi: 10.1073/pnas.122354499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22:138–47. doi: 10.1002/(SICI)1521-1878(200002)22:2<138::AID-BIES5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Iseki S, Wilkie AO, Morriss-Kay GM. Fgfr1 and Fgfr2 have distinct differentiation- and proliferation-related roles in the developing mouse skull vault. Development. 1999;126:5611–20. doi: 10.1242/dev.126.24.5611. [DOI] [PubMed] [Google Scholar]
- Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–93. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- Lecanda F, Warlow PM, Sheikh S, Furlan F, Steinberg TH, Civitelli R. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J Cell Biol. 2000;151:931–44. doi: 10.1083/jcb.151.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biol. 2000;149:1297–308. doi: 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–95. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- Moursi AM, Globus RK, Damsky CH. Interactions between integrin receptors and fibronectin are required for calvarial osteoblast differentiation in vitro. J Cell Sci. 1997;110 (Pt 18):2187–96. doi: 10.1242/jcs.110.18.2187. [DOI] [PubMed] [Google Scholar]
- Nakayamada S, Okada Y, Saito K, Tamura M, Tanaka Y. Beta1 integrin/focal adhesion kinase-mediated signaling induces intercellular adhesion molecule 1 and receptor activator of nuclear factor kappaB ligand on osteoblasts and osteoclast maturation. J Biol Chem. 2003;278:45368–74. doi: 10.1074/jbc.M308786200. [DOI] [PubMed] [Google Scholar]
- Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal. 1999;11:545–54. doi: 10.1016/s0898-6568(98)00063-1. [DOI] [PubMed] [Google Scholar]
- Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219:472–85. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–65. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Otto TE, Nulend JK, Patka P, Burger EH, Haarman HJ. Effect of (poly)-L-lactic acid on the proliferation and differentiation of primary bone cells in vitro. J Biomed Mater Res. 1996;32:513–8. doi: 10.1002/(SICI)1097-4636(199612)32:4<513::AID-JBM3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, Thesleff I. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–55. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- Robinson EE, Foty RA, Corbett SA. Fibronectin matrix assembly regulates alpha5beta1-mediated cell cohesion. Mol Biol Cell. 2004;15:973–81. doi: 10.1091/mbc.E03-07-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson EE, Zazzali KM, Corbett SA, Foty RA. Alpha5beta1 integrin mediates strong tissue cohesion. J Cell Sci. 2003;116:377–86. doi: 10.1242/jcs.00231. [DOI] [PubMed] [Google Scholar]
- Sakai K, Hiripi L, Glumoff V, Brandau O, Eerola R, Vuorio E, Bosze Z, Fassler R, Aszodi A. Stage-and tissue-specific expression of a Col2a1-Cre fusion gene in transgenic mice. Matrix Biol. 2001;19:761–7. doi: 10.1016/s0945-053x(00)00122-0. [DOI] [PubMed] [Google Scholar]
- Serebriiskii I, Estojak J, Sonoda G, Testa JR, Golemis EA. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21–22. Oncogene. 1997;15:1043–9. doi: 10.1038/sj.onc.1201268. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Nakayama K, Matsumoto T. Differentiation and cell surface expression of transforming growth factor-beta receptors are regulated by interaction with matrix collagen in murine osteoblastic cells. J Biol Chem. 1996;271:3938–44. doi: 10.1074/jbc.271.7.3938. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Suzawa M, Kikuchi T, Nishida E, Fujita T, Matsumoto T. Differentiation and transforming growth factor-beta receptor down-regulation by collagen-alpha2beta1 integrin interaction is mediated by focal adhesion kinase and its downstream signals in murine osteoblastic cells. J Biol Chem. 1997;272:29309–16. doi: 10.1074/jbc.272.46.29309. [DOI] [PubMed] [Google Scholar]
- Talts JF, Brakebusch C, Fassler R. Integrin gene targeting. Methods Mol Biol. 1999;129:153–87. doi: 10.1385/1-59259-249-X:153. [DOI] [PubMed] [Google Scholar]
- Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas T, Plow E, Qin J. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110:587–97. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Xiao G, Wang D, Benson MD, Karsenty G, Franceschi RT. Role of the alpha2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. J Biol Chem. 1998;273:32988–94. doi: 10.1074/jbc.273.49.32988. [DOI] [PubMed] [Google Scholar]
- Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117:387–98. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA. KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum Mol Genet. 2002;11:389–96. doi: 10.1093/hmg/11.4.389. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Olsen BR. The genetic basis for skeletal diseases. Nature. 2003;423:343–8. doi: 10.1038/nature01659. [DOI] [PubMed] [Google Scholar]
- Zhang J, Clatterbuck RE, Rigamonti D, Chang DD, Dietz HC. Interaction between krit1 and icap1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum Mol Genet. 2001;10:2953–60. doi: 10.1093/hmg/10.25.2953. [DOI] [PubMed] [Google Scholar]
- Zhang XA, Hemler ME. Interaction of the integrin beta1 cytoplasmic domain with ICAP-1 protein. J Biol Chem. 1999;274:11–9. doi: 10.1074/jbc.274.1.11. [DOI] [PubMed] [Google Scholar]
- Zimmerman D, Jin F, Leboy P, Hardy S, Damsky C. Impaired bone formation in transgenic mice resulting from altered integrin function in osteoblasts. Dev Biol. 2000;220:2–15. doi: 10.1006/dbio.2000.9633. [DOI] [PubMed] [Google Scholar]