

Abstract
The reaction of 5,10,15-triarylcorrole with 4-amino-4H-1,2,4-triazole provides another example of corrole ring expansion to give the corresponding 6-azahemiporphycene, a novel porphyrin analogue. The facile oxidation of the corrole ring is a required step for the ring expansion and for this reason the reaction fails in the case of corroles bearing meso-phenyl groups carrying electron-withdrawing substituents. Steric requirements also limited the scope of the reaction, which is not successful in the case of 2,6-disubstituted meso-aryl corroles. The occurrence of an initial oxidation is further supported by formation of the 6-azahemiporphycene derivative when the reaction is carried out under the same conditions, using a 5- or a 10-isocorrole as starting material. 1H NMR spectra and X-ray crystal characterization of 6-azahemiporphycene evidenced the presence of an intramolecular N–H··· N hydrogen bond in the inner core of the macrocycle, while photophysical characterization confirmed the aromatic character of the novel macrocycle, showing an intense Soret-like band around 410 nm in the absorption spectrum. The fluorescence emission is very modest, and 6-azahemiporphycene showed higher photostability than the corresponding corrole species. Different metal complexes of 6-azahemiporphycene were prepared following synthetic protocols usually exploited for the preparation of metalloporphyrins, demonstrating good coordination properties for the macrocycle. Both the free-base and metal derivatives were characterized by cyclic voltammetry and spectroelectrochemistry in dichloromethane and benzonitrile. To further detail the behavior of this novel macrocycle, density functional theory (DFT) calculations were carried out on the basic structure of 6-azahemiporphycene with the aim of assessing aromaticity and tautomerism, as well as calculating its stability with respect to the 5-aza isomer.
Introduction
Porphyrins are ubiquitous in Nature where they perform functions essential for life. This important role has been the springboard for a huge number of studies carried out on these macrocycles during the last two centuries.1 Researchers have focused on one hand on better understanding mechanisms of the biological systems and on the other in exploiting this knowledge to create synthetic platforms that are able to mimic the almost unique chemistry and physics of the natural systems. Particular attention in these efforts has been devoted to the close relationships that exist between the structures of the macrocycles and their inherent properties, with the consequent possibility of tuning both by subtle synthetic modifications. In this regard, Nature is a flawless teacher; iron porphyrins are used for oxygen binding and activation, while magnesium chlorins, which possess related macro-cycles, are catalysts in sunlight energy conversion. Starting from a similar molecular framework, a change of the central metal ion and/or saturation of a peripheral double bond can lead to completely different but equally vital functions.
Developing upon this approach, several pyrrolic macrocycles other than porphyrins have been prepared and fully characterized in recent years, with the aim both to study in detail structure–property relationships and to tune them for a particular targeted application.2 This effort has led to formation of the so-called “porphyrinoid” family, which comprises all polypyrrolic macrocycles having molecular frameworks similar to the parent porphyrin macrocycle.3 In this regard, the porphyrin family can be schematically divided into three groups: (1) contracted porphyrins, (2) isomeric porphyrins, and (3) expanded porphyrins, the exact assignment depending upon the structural variation present in the macrocyclic skeleton.3a
The above three types of macrocycles have been prepared by a variety of synthetic routes, which in several cases have been developed by variations of synthetic procedures already optimized for porphyrins. Progress in the synthetic organic chemistry of porphyrins has also stimulated extensions of the porphyrinoid family to give a variety of new macrocycles.4
In a new synthetic approach for the preparation of porphyrinoids, it was demonstrated that the macrocyclic skeleton is not a rigid block and can be modified even after its formation.5 It is important to note that this result was not unprecedented since examples were reported in the literature many years before, leading to porphyrins from nickel tetra-dehydrocorrin salts6 or homoporphyrins from a ring expansion of porphyrins.7 Only recently, however, it has become evident that these interconversions are not occasional,5 but rather are quite general and can be exploited for the preparation of macrocycles that may be difficult to prepare by direct or previously designed routes.
One group of well-characterized porphyrinoids includes the corroles or contracted porphyrins, which have assumed in the past few years a protagonist role.8 The behavior of these macrocycles can be quite different from the related porphyrins with the same metal ions and this has made the corroles especially promising for specific applications.9 For example, one characteristic of the corroles is that the central metal ion is often stabilized in a formal oxidation state which is higher than that of the corresponding porphyrins, and this property may be related to the fact that some corroles are superior to porphyrins as catalysts in specific reactions.10
The corrole is also a protagonist in switching between different macrocycle structures, through ring-opening and/or ring-closure reaction sequences. For example, it has been reported that corroles can undergo a ring-expansion to porphyrins11 and that they can also be formed by ring-contraction of a porphyrin.12 However, the versatility of the corrole is not confined to the direct pyrrole–pyrrole link formation or expansion demonstrated by the fact that corroles can be transformed into hemiporphycenes by reaction with CI4 where the ring-expansion reaction occurs at one meso carbon bridge.13
Most recently we discovered an additional example of corrole ring-expansion that leads to the formation of 6-azahemiporphycene upon reaction of the corrole with 4-amino-4H-1,2,4-triazole.14 As a follow up to this initial communication, we now present a detailed characterization of 6-azahemiporphycenes with different central metal ions, together with the electrochemistry of these compounds and a computational study with the aim of assessing aromaticity and tautomerism of these new macrocycles. Although the synthetic chemistry of corroles was greatly improved after initial reports for the preparation of meso triarylcorroles, the reactions described in the present paper will open up a facile route to the preparation of these novel porphyrinoids.
Experimental Section
Reagents and solvents (Sigma-Aldrich, Fluka and Carlo Erba Reagenti) were of synthetic grade and used without further purification. Silica gel 60 (70–230 mesh) and neutral alumina (Brockmann grade III) were used for chromatography. Chloroform Uvasol was obtained from Merck; triflic acid and tributylamine were obtained from Aldrich. Benzonitrile (PhCN) was purchased from Aldrich Chemical Co. and distilled over P2O5 under vacuum prior to use. Tetra-n-butylammonium perchlorate (TBAP, 99%) was purchased from Fluka Chemical Co. and used as received.
1H NMR spectra were recorded on a Bruker AV300 (300 MHz) spectrometer. Chemical shifts are given in parts per million relative to tetramethylsilane (TMS). UV–vis spectra were measured on a Cary 50 spectrophotometer. Mass spectra were recorded on a VGQuattro spectrometer in the positive-ion mode, using m-nitrobenzyl alcohol (NBA, Aldrich) as a matrix (FAB), or on a Voyager DE STR Biospectrometry workstation in the positive mode, using α-cyano-4-hydroxycinnamic acid as a matrix (MALDI).
Electronic spectra were measured in chloroform using a Perkin-Elmer Lambda 40 spectrophotometer. Corrected emission and excitation spectra (450 W Xe lamp) and excited state lifetimes were obtained with a modular UV–vis–NIR Spectrofluorimeter Edinbourg, equipped with a single photon counting apparatus. Corrections for instrumental response, inner filter effects, and phototube sensitivity were performed.15 Ru(bpy)32+ in aerated H2O (Φ = 0.028) was used as a standard for florescence quantum yields. Emission spectra in a rigid, transparent 2-methylcyclohexane matrix at 77 K were recorded using quartz tubes immersed in a quartz Dewar filled with liquid N2. A correction for a difference in the refraction index was introduced when necessary.
Diffraction data were collected on a Nonius KappaCCD diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) and an Oxford Cryostream low-temperature device.
Cyclic voltammetry was carried out with an EG&G model 173 potentiostat-galvanostat. A homemade three-electrode electrochemistry cell was used and consisted of a glassy carbon working electrode, a platinum wire counter electrode, and a saturated calomel reference electrode (SCE). The SCE was separated from the bulk of the solution by a fritted-glass bridge of low porosity that contained the solvent/supporting electrolyte mixture. All potentials are referenced to the SCE.
UV–visible spectroelectrochemical experiments were performed with an optically transparent platinum thin-layer electrode of the type described in the literature.16 Potentials were applied with an EG&G Model 173 potentiostat-galvanostat. Time-resolved UV–visible spectra were recorded with a Hewlett-Packard Model 8453 diode array rapid-scanning spectrophotometer.
Corroles 1–7 (see Chart 1) were prepared following literature methods.17
Figure 12.

Chart 1
5,10,15-Tris-(2,6-dimethoxyphenyl)corrole 8
A solution of pyrrole (5.6 mL, 80 mmol) and 2,6-dimethoxybenzaldehyde (1.32 g, 8.3 mmol) was degassed by bubbling nitrogen for 10 min, and trifluoroacetic acid (16 μL, 0.2 mmol) was then added. The mixture was stirred for 15 min at room temperature (RT) and then diluted with CH2Cl2 (25 mL). The reaction mixture was left at RT for 1 h; chloranil (1.74 g, 7.01 mmol) was added, and the mixture was stirred for another 10 min. The solvent and the residual pyrrole were removed under reduced pressure, the crude mixture dissolved in CH2Cl2 and then chromatographed on silica gel using a mixture of CH2Cl2/CHCl3 (1:1) as eluent. The red-purple fraction was recrystallized from CH2Cl2/hexane (yield 13%; 254 mg).
Spectroscopic data for 8. Found: C, 72.9; H, 5.5; N, 7.7 C43H38O6N4 requires C, 73.1; H, 5.4; N, 7.9%. UV-vis: λmax-(CHCl3), nm 408 (log ε, 4.94), 566 (4.13), 604 (3.97), and 632 (3.63); 1H NMR δ (CDCl3, J [Hz]) 8.80 ppm (2 H, d, J 4.0, β-pyrrole), 8.51 (2 H, d, J 4.6, β-pyrrole), 8.33 (2 H, d, J 4.7, β-pyrrole), 8.30 (2 H, d, J 4.1, β-pyrrole), 7.66 (3 H, t, J 8.4, phenyl), 6.98 (6 H, m, phenyl), 3.57 (12 H, s, −OCH3), 3.54 (6 H, s, −OCH3); MS (MALDI): m/z 707 (M+).
General Procedure for Preparation of 6-Azahemiporphycene Derivatives
Route A
In a 100 mL round-bottomed flask, with a stirring bar, the corrole (0.1 mmol) was dissolved in 22 mL of toluene/abs. ethanol mixture (10:1); NaOH (0.5 mmol) and 4-amino-4H-1,2,4-triazole (1.2 mmol) were added, and the mixture was refluxed for about 2 h, monitoring the progress of the reaction by UV–vis spectroscopy and TLC. After cooling to RT, the solvent was evaporated under vacuum, and the crude product dissolved in 50 mL of chloroform and washed three times with water. The organic phase was dried over sodium sulfate and the product purified by chromatography on silica gel, first using CH2Cl2 to elute traces of unreacted corrole. The other two fractions were then eluted using a CH2Cl2/CHCl3 (2:1) mixture. The second red eluted fraction corresponded to 6-azahemiporphycene, while the third blue-green fraction was characterized as an open-chain tetrapyrrole.
Route B
In a 50 mL round-bottomed flask, with a stirring bar, 5,10,15-tris-(4-methylphenyl)-5-hydroxy-isocorrole (a) [or 5,10,15-tris-(4-methylphenyl)-10-hydroxy-isocorrole (b)] (30 mg, 0.05 mmol) was dissolved in 11 mL of a toluene/ethanol absolute mixture (10:1); NaOH (10 mg, 0.25 mmol) and 4-ammo-4H-1,2,4-triazole (50.4 mg, 0.6 mmol) were then added and the mixture refluxed for about 30 min as the progress of the reaction was monitored by UV–vis spectroscopy and TLC. The reaction workup was carried out as described above. Yield: 54%, 16 mg (a), 60%, 17mg(b).
6-Aza-5,11,16-triphenylhemiporphycene 9
The red solid was recrystallized from CH2Cl2/methanol (yield 48%, 26 mg). Found: C, 82.2; H, 4.8; N, 12.9 C37H25N5 requires C, 82.3; H, 4.7; N, 13.0%. UV–vis: λmax(CHCl3), nm 412 (log ε, 5.12) and 571 (4.20); 1H NMR δ (CDCl3, J [Hz]) 8.99 ppm (1 H, d, J 4.6, β-pyrrole), 8.86 (1 H, d, J 4.4, β-pyrrole), 8.72 (1 H, d, J 4.8, β-pyrrole), 8.62 (1 H, d, J 4.3, β-pyrrole), 8.45 (4 H, m, β-pyrrole + phenyl), 8.31 (1 H, d, J 4.6, β-pyrrole), 8.16 (2 H, m, phenyl), 8.08 (1 H, m, β-pyrrole), 8.04 (2 H, m, phenyl), 7.73 (9 H, m, phenyl), 4.06 (1 H, s, NH), 2.94 (1 H, s, NH); MS (MALDI): m/z 540 (M+).
6-Aza-5,11,16-tris-(4-methylphenyl)hemiporphycene 10
The red solid was recrystallized from CH2Cl2/hexane (yield 51%; 30 mg). Found: C, 82.4; H, 5.3; N, 11.9 C40H31N5 requires C, 82.6; H, 5.4; N, 12.0%. UV–vis: λmax(CHCl3), nm 416 (log ε, 5.27) and 571 (4.33); 1H NMR δ (CDCl3, J[Hz]) 8.93 ppm (1 H, d, J 4.3, β-pyrrole), 8.80 (1 H, d, J 4.5, β-pyrrole), 8.68 (1 H, d, J 4.1, β-pyrrole), 8.59 (1 H, d, J 4.5, β-pyrrole), 8.37 (4 H, m, β-pyrrole + phenyl), 8.28 (1 H, d, J 4.5, β-pyrrole), 8.06 (3 H, m, β-pyrrole + phenyl), 7.91 (2 H, d, J 7.8, phenyl), 7.59 (4 H, m, phenyl), 7.49 (2 H, d, J 7.8, phenyl), 4.24 (1 H, s, NH), 3.13 (1 H, s, NH), 2.67 (6 H, s, −CH3), 2.62 (3 H, s, −CH3); MS (MALDI): m/z 582 (M+).
6-Aza-5,11,16-tris-(4-methoxyphenyl)hemiporphycene 11
The red solid was recrystallized from CH2Cl2/methanol (yield 56%; 35 mg). Found: C, 76.1; H, 5.1; N, 10.9. C40H31N5O3 requires C, 76.3; H, 5.0; N, 11.1%. UV–vis: λmax(CHCl3), nm 424(log ε, 5.00) and 574 (4.05); 1H NMR δ (CDCl3, J [Hz]) 8.88 ppm (1 H, d, J 4.3, β-pyrrole), 8.76 (1 H, d, J 4.4, β-pyrrole), 8.62 (1 H, d, J 4.3, β-pyrrole), 8.55 (1 H, d, J 4.4, β-pyrrole), 8.44 (2 H, d, J 8.6, phenyl), 8.35 (1 H, d, J 3.3, β-pyrrole), 8.29 (1 H, d, J 4.5, β-pyrrole), 8.22 (1 H, d, J 4.5, β-pyrrole), 8.09 (2 H, d, J 8.5, phenyl), 8.04 (1 H, m, β-pyrrole), 7.93 (2 H, d, J 8.4, phenyl), 7.32 (4H, d, J 8.3, phenyl), 7.23 (2 H, d, J 8.4, phenyl), 4.53 (1 H, s, NH), 4.07 (6 H, s, -OCH3), 4.04 (3 H, s, -OCH3), 3.81 (1 H, s, NH); MS (MALDI): m/z 630 (M+).
Crystal data for 11: dark red plate, monoclinic, space group C2/c, a= 37.860(12), b = 10.166(2), c = 17.081(5) Å, β = 94.528(6)°, V = 109.082(12) Å3, Z = 8, Dcalc = 1.346 g cm−3, μ =0.087 mm−1, T = 90.0(5) K, 22317 reflections collected with θmax < 25.5°, 5698 independent reflections (Rint = 0.072) which were used in all the calculations, 3334 data with I > 2σ(I). Final residuals (for 442 parameters) were R1 [I > 2σ(I)] = 0.059, wR2 (all data) = 0.140, CCDC 736599.
6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycene 12
The spectroscopic data are reported in a previous paper.14
General Procedure for Preparation of 6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycene Metal Complexes
Mn(III) and Fe(III) Complexes
In a 50 mL round-bottomed flask, with a stirring bar, 6-aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycene (20 mg, 0.03 mmol) was dissolved in 10 mL of DMF and a 3-fold excess of MnCl2 or FeCl2 salt was added. The mixture was heated to reflux and the progress of the reaction followed by UV–vis spectroscopy and TLC. After disappearance of absorption peaks of the starting material, the solvent was evaporated under vacuum and the crude mixture purified by chromatography on neutral alumina using CHCl3/hexane (1:1) as eluent.
M(II) Complexes where M = Co, Ni, Cu, and Zn
In a 50 mL round-bottomed flask, with a stirring bar, 6-aza-5,11,16-tris (4-tert-butylphenyl)hemiporphycene (20 mg, 0.03 mmol) was dissolved in 10 mL of CHCl3, and a saturated solution of M(OAc)2 salt in methanol was added. The mixture was heated to reflux, and the progress of the reaction followed by UV–vis spectroscopy and TLC. After disappearance of absorption peaks for the starting material, the solvent was evaporated under vacuum, and the crude mixture purified by column chromatography.
Chloro[6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycenato]Manganese 13
Purification of the crude reaction mixture was performed on a neutral alumina column, eluting first with CH2Cl2 and then with CHCl3. The green fraction was recrystallized from CH2Cl2/hexane. Yield 73%, 18 mg. Found: C, 73.4; H, 6.1; N, 8.7 C49H49ClMnN5 requires C, 73.7; H, 6.2; N, 8.8%. UV–vis: λmax(CH2Cl2), nm 440 sh, 473 (log ε, 4.53), 542 (3.77) and 636 (3.84). MS (MALDI): m/z 798 (M+).
Chloro[6- Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycenato]Iron 14
Purification of the crude reaction mixture was performed on a neutral alumina column eluting with CH2Cl2/hexane (1:1). The green fraction was washed with 1 M HCl and recrystallized from methanol. Yield 61%, 15 mg. Found: C, 73.9; H, 6.1; N, 8.7 C49H49ClFeN5 requires C, 73.6; H, 6.2; N, 8.8%. UV–vis: λmax(CH2Cl2), nm 412 (log ε, 4.68) and 577 (3.63). MS (MALDI): m/z 799 (M+).
[6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycenato]Cobalt 15
Purification of the crude reaction mixture was performed on a neutral alumina column eluting with CH2Cl2/hexane (1:1). The green fraction was recrystallized from CH2Cl2/hexane. Yield 68%, 16 mg. Found: C, 76.9; H, 6.2; N, 9.2 C49H49CoN5 requires C, 76.7; H, 6.4; N, 9.1%. UV–vis: λmax(CH2Cl2), nm 413 (log ε, 4.97) and 584 (3.83); MS (MALDI): m/z 767 (M+).
[6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycenato]Nickel 16
Purification of the crude reaction mixture was performed on a silica gel column eluting with CH2Cl2. The green fraction was recrystallized from CH2Cl2/methanol. Yield 74%, 17 mg. Found: C, 77.1; H, 6.3; N, 8.9 C49H49N5Ni requires C, 76.8; H, 6.4; N, 9.1%. UV–vis: λmax(CH2Cl2), nm 412 (log ε, 4.84), 607 (3.85) and 648 (3.93); 1H NMR δ (CDCl3, J [Hz]) 8.85 ppm (1 H, d, J 4.6, β-pyrrole), 8.70 (2 H, dd, β-pyrrole), 8.60 (1 H, d, J 4.7, β-pyrrole), 8.57 (1 H, d, J 4.9, β-pyrrole), 8.53 (1 H, d, J 4.7, β-pyrrole), 8.49 (1 H, d, J 4.9, β-pyrrole), 8.24 (3 H, m, β-pyrrole + phenyl), 7.96 (2 H, d, J 8.1, phenyl), 7.87 (2 H, d, J 8.1, phenyl), 7.71 (6 H, m, phenyl), 1.58 (9H, s, tert-butyl), 1.57 (9 H, s, tert-butyl), 1.51 ppm (9 H, s, tert-butyl); MS (MALDI): m/z 767 (M+).
[6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycenato]Copper 17
Purification of the crude reaction mixture was performed on a neutral alumina column eluting with CH2Cl2/hexane (2:1). The green fraction was recrystallized from CH2Cl2/methanol. Yield 65%, 15 mg. Found: C, 76.5; H, 6.6; N, 8.8 C49H49CuN5 requires C, 76.3; H, 6.4; N, 9.1%. UV–vis: λmax(CH2Cl2), nm 431 (log ε, 5.01) and 615 (4.16); MS (MALDI): m/z 771 (M+).
[6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycenato]Zinc 18
Purification of the crude reaction mixture was performed on a silica gel column eluting first with CH2Cl2 and finally with CHCl3. The green fraction was recrystallized from CH2Cl2/methanol. Yield 86%, 20 mg. Found: C, 75.9; H, 6.5; N, 9.3 C49H49N5Zn requires C, 76.1; H, 6.4; N, 9.1%. UV–vis: λmax-(CH2Cl2), nm 436 (log ε, 4.99), 535 (3.69), 570 (3.78), and 618 (4.24); 1H NMR δ (CDCl3, J [Hz]) 8.99 ppm (1 H, d, J 3.8, β-pyrrole), 8.91 (1 H, d, J 4.0, β-pyrrole), 8.68 (2 H, m, β-pyrrole), 8.54 (1 H, d, J 4.2, β-pyrrole), 8.47 (1 H, m, β-pyrrole), 8.36 (1 H, d, J 4.3, β-pyrrole), 8.11 (4 H, d, J 7.6, phenyl), 7.80 (4 H, d, J 7.5, phenyl), 7.08 (5 H, br, β-pyrrole + phenyl), 1.71 (9 H, s, tert-butyl), 1.62 (9 H, s, tert-butyl), 1.30 ppm (9 H, s, tert-butyl); MS (MALDI): m/z 773 (M+).
Spectroscopic data of compounds 19–22 are reported in Table 1. Crystal data for 20: dark red parallelepiped, monoclinic, space group C2/c,a = 28.711(2), b= 13.3259(10), c = 18.9309(15) Å, β = 90.199(5)°, V= 7242.9(9) Å3, Z =8, Dcalc =1.321 gcm−3, μ = 2.621 mm−1, T = 90.0(5) K, 49650 reflections collected with θmax < 69.0°, 6591 independent reflections (Rint = 0.041) which were used in all the calculations, 5561 data with I > 2 σ(I). Final residuals (for 604 parameters) were R1[I > 2σ(I)] = 0.035, wR2 (all data) = 0.093. Two of the three MePh groups are disordered, each into two conformations. The chloroform solvent molecule is also disordered into two orientations, sharing a single Cl atom.
Table 1.
Spectroscopic Data of Open Chain Tetrapyrroles
| cpd | 1H NMRa | UV-visb | MSc | elem. analysis (%) |
|---|---|---|---|---|
| 19 | 10.87 (2H,br s, NH), 10.01 (1H, br s, NH), 7.80 (2H, d, J 7.2, ph), 7.46 (13H, m, ph), 7.09 (2H, m, β-py), 6.80 (1H, m, β-py), 6.69 (1H, m, β-py), 6.45 (2H, d, J 4.3, β-py), 6.37 (1H, d, J 4.2, β-py), 6.33 (1H, d, J 5.6, β -py) | 386 (4.55), 588 (4.28) | 559 [M]+ | found: C, 79.9; H, 4.5; N, 9.9; C37H26O2N4 requires C, 79.6; H, 4.7; N, 10.0 |
| 20 | 11.21 (2H,br s, NH), 10.36 (1H, br s, NH), 7.65 (2H, d, J 7.9, ph), 7.37 (2H, d, J 7.9, ph), 7.28 (2H, d, J 7.5, ph), 7.16 (8H, m, β-py + ph), 6.73 (1H, m, β-py), 6.64 (3H, m, β-py), 6.34 (2H, m, β-py), 2.47 (3H, s, –CH3), 2.42 (3H, s, –CH3), 2.40 (3H, s, –CH3) | 392 (4.67), 592 (4.39) | 601 [M]+ | found: C, 79.9; H, 5.5; N, 9.1; C40H32O2N4 requires C, 80.0; H, 5.4; N, 9.3 |
| 21 | 10.76 (2H, br s, NH), 9.90 (1H, br s, NH), 7.81 (2H, d, J 8.5, ph), 7.41 (2H, d, J 8.5, ph), 7.32 (2H, m, ph), 7.10 (2H, m, ph), 6.98 (4H, m, β-py + ph), 6.88 (2H, d, J 8.7, ph), 6.78 (1H, m, β-py), 6.69 (3H, m, β-py), 6.41 (1H, d, J 4.2, β-py), 6.31 (1H, d, J 5.4, β-py), 3.92 (3H, s, -OCH3), 3.87 (3H, s, -OCH3), 3.83 (3H, s, -OCH3) | 409 (4.43), 595 (4.12) | 649 [M]+ | found: C, 74.3; H, 5.2; N, 8.4; C40H32O5N4 requires C, 74.1; H, 5.0; N, 8.6 |
| 22 | 10.93 (2H, br s, NH), 10.15 (1H, br s, NH), 7.72 (2H, d, J 8.0, ph), 7.41 (10H, m, ph), 7.15 (2H, d, J 5.5, β-py), 6.79 (1H, m, β-py), 6.74 (2H, d, J 4.0, β-py), 6.69 (1H, m, β-py), 6.41 (1H, d, J 4.3, β-py), 6.30 (1H, d, J 5.4, β-py), 1.40 (9H, s, - tert-butyl), 1.35 (18H, s, tert-butyl) | 394 (4.62), 598 (4.33) | 727 [M]+ | found: C, 80.9; H, 6.7; N, 7.8; C49H50O2N4 requires C, 81.0; H, 6.9; N, 7.7 |
CDCl3, 300 MHz, δ, ppm.
CH2Cl2 λmax, nm (log ε).
FAB (m/z)
Results and Discussion
Synthesis
We previously described how 3-NO2-6-aza-hemiporphycene was serendipitously obtained during our studies involving the peripheral functionalization of 5,10,15-triarylcorroles.14 Our intention was to prepare the 2-amino-3-nitro substituted corrole by reacting the Ag(III) 3-nitrocorrole complex with 4-amino-4H-1,2,4-triazole through vicarious substitution directed by the nitro group.18 However under the basic reaction conditions utilized, the corrole was readily demetalated,19 and the resulting free-base corrole macrocycle reacted with the triazole leading to 6-azahemiporphycene.14 The nitro substituents had no effect in either promoting or orienting the reaction, and we obtained the same product by reacting the unsubstituted free-base corrole under the same conditions. This result led us to investigate the scope of the reaction, utilizing in this case, under the same reaction conditions, the free base 5,10,15-triarylcorroles shown in Chart 1.
The reaction was successful in the case of corroles 1–4, to give 6-azahemiporphycenes 9–12 with yields varying in a range of 42–58%. The reaction is regioselective and only the 6-azahemiporphycene isomer was obtained as the ring-expansion product. Spectral characterization of the 6-azahemiporphycenes confirmed the aromatic character of these macrocycles.
The lower symmetry of 6-azahemiporphycene compared with porphyrin and corrole is reflected in the 1H NMR spectrum by the β-pyrrole hydrogen resonances that are located in the range of 9.0–8.3 ppm. These resonances indicate the existence of a diatropic ring current effect, typical of an aromatic macrocycle, although the inner-core hydrogen atoms are present as broad singlets in the range of 4.5–3.0 ppm, respectively; these are significantly shifted downfield from those of porphyrins, corroles, and hemiporphycenes.3g A similar feature has been observed in the case of porphycenes3c and can be confidently attributed to N–H · · ·-N intramolecular hydrogen bonding and not to a reduced ring current effect due to a lower aromatic character of the macrocycle.
This hypothesis is corroborated by the X-ray crystal structure of 11, shown in Figure 1. The N–H hydrogen atoms are localized on the N atoms shown, rather than being disordered over the four pyrroles. Insertion of the additional N atom into the ring system leads to a seven-membered ring intramolecular hydrogen bond which has both a longer N · · · N distance, 2.843(3) Å, and a larger N–H · · · N angle, 146(2)°, as compared with the “normal” six-membered ring on the other side of the molecule which has corresponding values of 2.573(3) Å and 129(2)°. The new C=N bond is somewhat localized to the phenyl-carrying C atom, having a length 1.310(3) Å, while the other C–N bonds are longer, in the range 1.332(3)–1.399(3) Å. The 24-atom 6-azahemiporphycene core is fairly planar, exhibiting a mean deviation of only 0.047 Å from its mean plane. The five N atoms are even more closely coplanar, with a mean deviation of only 0.021 Å from their best plane. This is in contrast to the t-butyl analogue 12, which has a less planar core, with the additional N atom lying 0.160 Å from the best plane of its four normal corrole N atoms.14
Figure 1.

Molecular structure of 11.
It is interesting to note that the expansion reaction was not successful in the case of corroles 5–8. In the case of 5 and 6, the failure of ring-expansion supports our initial hypothesis that oxidation of the starting corrole ring is a required step for ring-expansion to give the azahemiporphycene macrocycle. When the corrole bears electron-withdrawing substituents at the meso positions, as in the case of 5 and 6, the oxidation is prevented and this leads to a failure of the reaction with the starting material being recovered unaltered. In the case of corroles 7 and 8, the steric hindrance of the 2,6-substituents prevents attack of the triazole to initiate the ring expansion, assuming that the meso-position is the site of the nucleophilic attack as observed when isocorroles are formed by DDQ oxidation of meso-triarylcorroles.20 This hypothesis is confirmed by the failure of the same corroles 5–8 to give isocorrole derivatives by this route.
It should be noted that the formation of 6-azahemiporphycene is always accompanied by a polar blue-green byproduct. Spectroscopic characterization indicated that this product is an open-chain tetrapyrrole formed upon oxidative ring-opening of the starting corrole at the 5 position. We were able to grow single crystals of 20 suitable for X-ray characterization, which allowed the unambiguous characterization of this open chain species (Figure 2).
Figure 2.

Molecular structure of 20.
The molecular structure shows that oxidative ring-opening introduces a benzoyl group and an oxygen atom at the terminal α-pyrrolic positions, producing a biliverdin-like species. Compound 20 adopts a twisted conformation in which the central two pyrrole rings form a porphyrin-like intramolecular hydrogen bond with N···N distance 2.685(2) Å and are approximately coplanar, forming a dihedral angle of 7.7(3)°. The pyrrole carrying the carbonyl oxygen also forms an intramolecular hydrogen bond to the same acceptor, N···N 2.840(2) Å, and is twisted out of the plane of the two central pyrroles, forming a dihedral angle of 33.32(7)° with them. Both the directly linked pyrroles carry N–H hydrogen atoms, and are in an antiperiplanar conformation, with N–C–C–N torsion angle 177.78(13)°. Bonding appears to be more delocalized in the two directly linked pyrroles (C–C range 1.385(2)–1.406(2) Å; C–N range 1.353(2)–1.380(2) Å) than in the other two (C–C range 1.342(2)–1.468(2) Å; C–N range 1.334(2)–1.400(2) Å).
The formation of 20 confirms our previous finding21 that in the case of 5,10,15-triarylcorroles the site where the ring-opening occurs is the 5-position and not the direct pyrrole–pyrrole link, as observed in the case of 10-phenyl-β-alkylcorroles.22
It is worth mentioning that 20 is not an intermediate of the reaction, because any attempt to convert the open chain tetrapyrrole into the corresponding 6-azahemiporphycene species failed; the oxidative ring-opening is competitive with the corrole ring expansion to 6-azahemiporphycene and the total yield of the reaction depends upon the balance among these competing reaction pathways. It is interesting to note that in the case of corroles 5–8, which cannot afford 6-azahemiporphycene, this linear tetrapyrrole species is not observed, further confirming that ring oxidation is the necessary step to initiate the corrole conversion.
To further support the hypothesis that oxidation is a necessary preliminary step for ring expansion, we decided to react the isocorroles 23 and 24 (Figure 3), obtained by DDQ oxidation,20 under the same reaction conditions. It is interesting to note that in both cases the reaction product was the corresponding 6-azahemiporphycene species 10.
Figure 3.

Molecular structures of 23 and 24 isocorroles.
According to these experimental results we can hypothesize a plausible reaction pathway for the 6-azahemiporphycene formation, reported in Scheme 1. The oxidation of corrole to the corresponding radical species is the first step of the reaction: this derivative can be then attacked by triazole to give an isocorrole-like species to give the final 6-azahemiporphycene, or it can be further oxidized leading to the open chain tetrapyrrole. The facile conversion of isocorrole species to corrole has been already reported in the literature,17a and it can explain the results obtained when isocorrole (like 23 or 24) is the starting compound; the conversion to the corrole radical species in the reaction conditions can give the reaction final products. This hypothesis can explain the regio-selectivity of the reaction and the formation of 6-azahemiporphycene as product even when 24 is used as starting reagent.
Scheme 1.

Reaction Pathway for the 6-Azahemiporphycene Formation
Photophysical Characterization
This study was carried out using 12 and the Zn complex 18 as model compounds. The absorption spectrum of 12 in chloroform shows a very intense Soret-like band centered at 416 nm (ε = 71000 M−1 cm−1) and a Q-like set of broader, less intense bands with maximum at 571 nm (ε = 9900 M−1 cm−1) and two shoulders at 533 and 605 nm (Figure 4). The spectrum of 12 is thus less intense, and the energy of the lowest absorption is at higher energies than that of 1 or tetraphenylporphyrin, (TPP)H2, indicating a lower degree of aromaticity for 12, as also suggested by computational results.
Figure 4.

UV—vis spectral variations of 12 upon addition of 1 equiv of triflic acid (solid line). Addition of 1 equiv of tributylamine restores the original spectrum (dashed line).
The fluorescence spectrum at RT shows a moderately weak, broad, unstructured band at 642 nm (Φ = 1.2 × 10−3), presenting an excited state lifetime of 4.4 ns (Figure 4). It should be noted that the radiative rate constant, kr, calculated according to eq 1,14
| (1) |
is for 12 much slower (3 × 105 s−1) than that observed for (TPP)H2 and 1 (8.5 × 106 s−1 and 1.6 × 107 s−1, respectively), suggesting a distorted/non-rigid structure for the excited state, in agreement with the observed band shape. The excitation spectrum is in this case proportional to the absorption spectrum, indicating that the S1 state is also formed with unitary efficiency when S2 and other excited states are initially populated.
Similar results were observed at 77 K. No emissions were observed in the 800–1600 nm region, indicating a negligible efficiency both for phosphorescence (at RT and at 77 K) and for singlet oxygen sensitization (RT) in these experimental conditions. It should be noted that 12 is rather stable under irradiation with visible light while 1, in particular, undergoes rapid degradation upon excitation at 420 nm with a medium pressure Hg lamp. The spectrum of 12 is only slightly affected after 8 h of continuous irradiation.
Interestingly, the addition of 1 equiv of triflic acid led to a small red shift in the spectrum, accompanied by a lowering of the absorption coefficient of the Soret-like band, while the Q-bands underwent noticeable changes (Figure 4). At the same time, a structured band at λmax = 602 nm appears in the emission spectrum, presenting a higher intensity (Φ = 2.0 × 10−3), a smaller Stokes-shift, and a lower excited state lifetime (< 50 ps) (Figure 5). The limit value of kr estimated for the monoprotonated structure is more than 10-fold faster than for the unprotonated compound. All of these results suggest in this case a less distorted structure for the excited state. It is worth noting that the protonation process is perfectly reversed by adding a stoichiometric amount of base such as tributylamine.
Figure 5.

Emission spectrum of 12 before (solid line) and after (dashed line) addition of 1 equiv of triflic acid.
As far as the Zn complex 18 is concerned, it shows generally a more intense spectrum compared with 12, with the Soret-like band centered at 436 nm (ε = 98000 M−1 cm−1, Figure 6). The fluorescence band is in this case structured with a maximum at 626 nm, and with a relatively low quantum yield (3.8 × 10−4) and a short lifetime (0.23 ns). From these data, a kr value of 1.5 × 106 s−1 can be calculated, higher than for 12 (but lower than for the free-base corrole), suggesting for 18 a lower distortion, as also supported by the higher absorption coefficient. As observed also for free-base 6-azahemiporphycene, the excitation spectrum is proportional to the absorption spectrum. In this case, the band typical of singlet oxygen in the NIR region can be observed, although of very low intensity, indicating a higher intersystem crossing efficiency as expected because of the presence of the Zn ion. Also the Zn complex 18 is much more stable than 1 under irradiation with visible light.
Figure 6.

UV-vis absorption (solid line) and emission (dashed line) spectrum of Zn complex 18.
Computational Results
Density functional theory (DFT) calculations were carried with the aim of assessing aromaticity and tautomerism of the basic structure of 6-azahemiporphycene 25, Figure 7 (pyrrole rings A, B, C, D and positions 1 and 6 are indicated in the structure), as well as its relative stability with respect to the 5-aza isomer. All calculations were carried out with the program Gaussian 03.23 The various issues are discussed below under separate headings.
Figure 7.
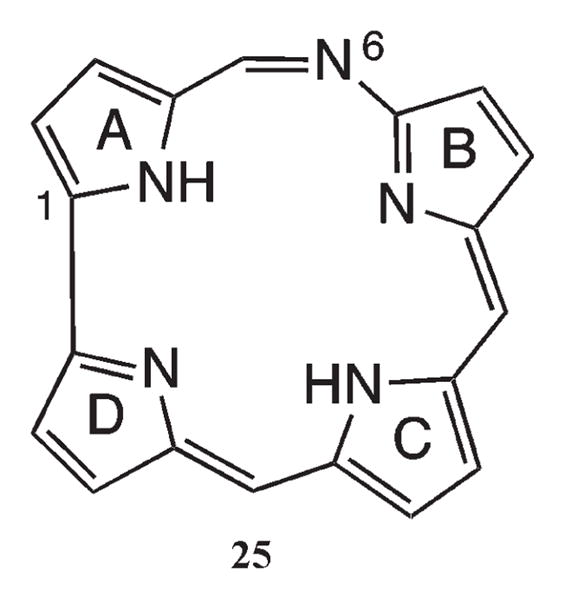
Basic structure of 6-azahemiporphycene 25.
Aromaticity
The Nucleus Independent Chemical Shift (NICS) is presently the most popular index to probe aromaticity because of its simplicity and efficiency.24 It is defined as the negative of the magnetic shielding of a virtual uncharged nucleus placed in selected points in the vicinity of the molecule, typically at ring center, NICS(0), or 1 Å above the ring center, NICS(1). Significantly negative NICS values in interior positions of rings indicate the presence of induced diatropic ring currents or “aromaticity”, whereas positive values at each point denote paratropic ring currents and “antiaromaticity”. The NICS index has been routinely utilized to compare and quantify the aromaticity of porphyrins and porphyrinoids.25 Following the general usage,24,25 we have optimized the geometry of 6-azahemiporphycene, porphyrin, corrole, and hemiporphycene, using the B3LYP/6-311+G(d) method, and computed the NICS(0) and NICS(1) at the same level of theory. The results are shown in Table 2.
Table 2.
NICS(0) and NICS(1), in ppm, at the B3LYP/6-311+G(d) Level of Theory for 6-Azahemiporphycene, Porphyrin, Corrole, and Hemiporphycene
| ring | NICS(0) | NICS(1) |
|---|---|---|
| 6-azahemiporphycene | −12.0 | −11.0 |
| porphyrin | −15.0 | −13.6 |
| corrole | −13.3 | −11.9, −12.7a |
| hemiporphycene | −12.7 | −11.7 |
Owing to the significant deviation from planarity of the corrole ring, NICS(1) and NICS(−1) values are significantly different, thus both have been reported.
Inspection of the data in Table 2 reveals a consistent order of NICS(0) and NICS(1) values suggesting the following order of aromaticity: porphyrin > corrole > hemiporphycene > 6-azahemiporphycene. However, it must be noted that the differences among NICS values are small, indicating that all four compounds possess a comparable aromatic character. The small loss of aromaticity upon passing from hemiporphycene to 6-azahemiporphycene is easily understood as due to the small perturbation in ring current introduced by the more electronegative nitrogen atom.
Tautomerism
The 6-azahemiporphycene basic structure 25 features two hydrogen atoms inside the ring cavity on pyrrole rings A and C, respectively. This is the tautomer that has been detected in the X-ray crystal structures of 12 and 3-nitro-6-aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycene.14 For convenience we indicate this tautomer as 25AC. In addition to 25AC, five other tautomers can be envisaged, depending upon the pair of protonated pyrrole rings, namely, 25BD, 25AB, 25AD, 25BC, and 25CD. Tautomers 25AC and 25BD can be classified as trans the two hydrogens being on opposite rings, whereas the other tautomers with the two hydrogens on adjacent rings can be classified as cis. To evaluate the relative stability of the above tautomers we carried the corresponding geometry optimizations at the B3LYP/6-311++G(d,p) level of theory. The choice of the basis set, with polarization and diffuse functions on the hydrogens beside heavy atoms, was suggested by the role that hydrogen bonding inside the cavity may have on the relative stability of the tautomers. Relative electronic energies are reported in the first column of Table 3.
Table 3.
Relative Electronic Energies (in kcal mol−1) of the Six Tautomers of 6-Azahemiporphycene
| tautomer | B3LYP/6-311++G(d,p) | M05–2X/6-311++G(d,p) |
|---|---|---|
| 25AC | 0.0 | 0.0 |
| 25BD | 0.3 | 1.6 |
| 25AB | 20.2 | 21.4 |
| 25AD | 4.3 | 5.1 |
| 25BC | 4.4 | 5.9 |
| 25CD | 21.0 | 22.3 |
The tautomers can be grouped according to their energies and structural features into three families: the trans tautomers, 25AC and 25BD; the planar cis tautomers, 25AD and 25BC; and the nonplanar cis tautomers, 25AD and 25CD. The trans tautomers are the most stable. The planar cis tautomers, 25AD and 25BC are about 4.5 kcal mol−1 less stable than the corresponding trans isomers probably because of repulsion involving the parallel dipole moments associated with the two N–H bonds. The nonplanar cis tautomers, 25AB and 25CD, are more than 20 kcal mol−1 less stable than the corresponding trans isomers because of transannular steric repulsions between the two hydrogens that cause a significant deviation of the corrole core from planarity.
We were somewhat surprised to find that the two trans isomers have approximately the same energy, especially because the X-ray structures show the exclusive existence of the tautomer 25AC in the solid state. Since Zhao and Truhlar’s recently proposed M05–2X functional has been demonstrated to outperform the popular B3LYP functional in the energetic description of organic systems,26b we carried out geometry optimizations of the six tautomers also at the M05–2X/6-311++G(d,p) level of theory. The results reported in the second column of Table 3 showed the same pattern of the B3LYP results, although now the experimentally found tautomer appears 1.6 kcal mol−1 more stable than the other trans tautomer. In contrast with the computed planar structure of the tautomer 25AC, the X-ray crystal structures of the aryl substituted 6-azahemiporphycenes14 show that the corrole core is significantly warped in the solid state. We suspected that the aryl groups could be the cause of such warp. To put this hypothesis to the test we calculated the energy of the two trans tautomers of 6-aza-5,11,16-triphenylhemiporphycene at the M05–2X/6-311++G-(d,p)//M05-2X/6-31G(d) level of theory. The lowest energy conformations of both the tautomers with respect to rotations around the phenyl-corrole axes were found by a systematic conformational search at M05–2X/6-31G(d) level. The presence of the phenyl rings causes significant deviations from planarity in both the structures. The AC tautomer is now 2.9 kcal mol−1 more stable than the corresponding BD tautomer thus justifying the fact that only the AC tautomer has been found in the solid state. The origin of this increased relative stability can be understood by comparing the geometries of the phenyl substituted structures with the corresponding unsubstituted ones: the warping caused by the phenyl groups increases the distance between the central hydrogens in the case of the AC tautomer thus decreasing their steric repulsion, whereas the opposite occurs in the case of the BD tautomer where the same distance is reduced.
Interestingly, the most stable conformer of the AC tautomer corresponds to the X-ray structure conformation; thus computational results are in perfect agreement with crystallography both for the position of the central protons and for the dihedral angle around the phenyl-corrole bonds.
Stability of the 5-Aza Isomer
To rationalize the regio-selectivity observed in the synthesis of aryl substituted 6-azahemiporphycenes14 (i.e., the finding that the nitrogen atom is exclusively introduced in position 6), it is instructive to compare the energy of 5-azahemiporphycene with that of the 6-aza isomer. Of course a careful theoretical study of the regioselectivity would require a comparison of the energies of the corresponding transition states, but owing to the well-known difficulties to locate and characterize transition states and since transition states must resemble to some extent the final products, we thought that a rational for the regioselectivity can be simply gained by comparing the energies of the final products. Geometry optimizations of unsubstituted 5-azahemiporphycene at both the B3LYP/6-311++G(d,p) and the M05-2X/6-311++G(d,p) levels of theory were carried out. Relative energy of 5-azahemiporphycene with respect to the 6-aza isomer was 0.2 kcal mol−1 with the B3LYP functional and 1.3 kcal mol−1 with M05–2X functional. Although the relative energy calculated with the M05–2X functional should be more reliable,26 it is not large enough to justify the observed regioselectivity. To investigate a possible role of the phenyl rings, a comparison between the energies of the AC tautomer of 5-aza-6,11,16-triphenylhemiporphycene and 6-aza-5,11,16-triphenylhemiporphycene was performed at the M05–2X/6-311++G(d,p)//M05–2X/6-31G(d) levels of theory. The most stable conformation of the 5-aza isomer was found to be 4.7 kcal mol−1 higher in energy than the 6-aza isomer. Thus, computational results reasonably indicate that the observed regioselectivity has to be ascribed to a significantly greater stability of the aryl substituted 6-azahemiporphycene. Inspection of the geometry of the 5-aza isomer suggests that steric effects between the phenyl rings and the corrole structure play a major role in increasing its energy.
Metal Complexes
The metal complexes 13–18 were prepared to investigate both the coordination abilities of the macrocycle 12 and the behavior of the resulting metal derivatives. We adopted synthetic procedures usually exploited for the preparation of the analogous metalloporphyrinates,27 obtaining in all cases good yields of the metal complexes of 12. These results indicate that the strong NH–N intramolecular hydrogen bond present in the macrocyclic inner core does not hinder the coordination properties of the 6-azahemiporphycene, which is able to stabilize both divalent metal ions such as Co(II), Ni(II), Cu(II), and Zn(II) and trivalent metals such as Mn(III) and Fe(III). These last two complexes are particularly interesting because the presence of the chloride axial ligand leads to a mixture of enantiomers due to the lower symmetry of the 6-azahemiporphycene structure compared with a porphyrin. Spectroscopic characterizations of these complexes show features expected for porphyrinoid metal derivatives. Considering that in the case of porphycene metal complexes, electrochemical behavior showed notable peculiarities, we decided to study in detail the electrochemistry of the 6-azahemiporphycene metal derivatives. These results are described below.
Electrochemistry
Compounds 12–18 were examined as to their electrochemical and spectroelectrochemical behavior in PhCN and CH2Cl2 containing 0.1 M TBAP. An example of the cyclic voltammograms obtained in PhCN are displayed in Figure 8, and a summary of the half-wave or peak potential for each redox reaction is given in Table 4.
Figure 8.

Cyclic voltammograms of M-6-aza-5,11,16-tris(4-terf-butyl-phenyl)hemiporphycene in PhCN containing 0.1 M TBAP where M and the compound number are indicated in the figure.
Table 4.
Half-Wave or Peak Potentials (V vs SCE) of 6-Azahemiporphycene Derivatives in PhCN and CH2Cl2 Containing 0.1 M TBAP
| cpda | solvent | M | oxidation | MII/III | reduction | ||||
|---|---|---|---|---|---|---|---|---|---|
| 12 | PhCN | 2H | 1.54b | 1.17b | −0.89 | −1.27 | |||
| 13 | MnCl | c | −0.21 | −1.21 | −1.52 | ||||
| 14 | FeCl | 1.60b | 1.24b | 0.95b | −0.28b | −1.31d | |||
| 15 | Co | 1.63b | 1.26b | 1.02b | 0.56b | −0.90 | −1.43 | ||
| 16 | Ni | 1.82b | 1.48b | 1.34b | 1.02b | −0.93 | −1.34 | ||
| 17 | Cu | 1.94b | 1.50b | 1.30b | 1.09b | −0.97 | −1.37 | ||
| 18 | Zn | 1.86b | 1.35b | 1.13b | 0.90b | −1.05 | −1.38 | ||
| 12 | CH2Cl2f | 2H | 1.75b | 1.56b | 1.06b | −0.92 | −1.26 | ||
| 13 | MnCle | 1.38b | 1.21b | −0.51b | −1.18 | −1.72 | |||
| 14 | FeCl | 1.68b | 1.22b | 0.82b | −0.26 | −0.92 | −1.32 | ||
| 15 | Co | 1.67b | 1.20b | 0.98b | 0.72b | −0.86 | −1.43 | ||
| 16 | Ni | 1.86b | 1.52b | 1.37b | 1.00b | −0.91 | −1.35 | ||
| 17 | Cu | 1.47b | 1.08b | −0.96 | −1.37 | ||||
| 18 | Zn | 1.32b | 1.09b | 0.85b | −1.10 | −1.39 | |||
As shown in Figure 8, the free-base and metal(II) derivatives of 6-azahemiporphycenes all undergo two well-defined reversible reductions. Each one-electron addition occurs on the macrocycle as confirmed by UV–visible spectroelecrochemistry in a thin-layer cell that indicates a loss of intensity in the Soret band and produces a typical π-anion radical type spectrum as the final electroreduction product (see examples of spectral changes in Figures 9b and S1–S2). The first reductions of the MnIIICl and FeIIICl derivatives (13 and 14) are located at El/2 = −0.21 V (MnIII/MnII) or EPC = −0.28 V (FeIII/FeII) and are easier than for reduction of the free-base or metal(II) complexes (12 and 15–18). Further reversible reductions are obtained at E1/2 = −1.21 and −1.52 V (compound 13, MnIIICl) or El/2 = −1.31 V (compound 14, FeIIICl).
Figure 9.

UV-visible spectral changes during the first reduction of (a) MnIIICl and (b) CuII azahemiporphycenes in PhCN, 0.1 M TBAP.
The ring-centered reductions of each 6-azahemiporphycenes are about 250 mV easier than those for the ring-centered reductions of (TPP)M complexes28 and not so different from E1/2 values for tetrapropylporphycenes29 containing the same central metal ion.
The oxidations of compounds 12–18 are not as well-defined as the reductions and the current–voltage curves indicate coupled chemical reactions after electron abstraction in each case. A CoII/CoIII process is observed for 15 at Epa. = 0.56 V for a scan rate of 0.1 V/s while most oxidations of the other derivatives are also irreversible. The current–voltage curve for oxidation of the MnIII compound 13 is not only ill-defined but also has larger than expected currents for a "simple" electron transfer process in PhCN. However, at −70 °C in CH2Cl2, a reversible peak is observed at E1/2 = 1.21 V (Supporting Information, Figure S3), and this suggests a catalytic reaction involving the singly oxidized species at RT in PhCN. This was not further investigated in the present study.
UV–visible spectral changes during the first controlled potential reduction of the MnIIICl 13 and CuII 17 derivatives are shown in Figure 9. Upon applying a potential of −0.60 V, a new intense Soret band at 448 nm is seen for 13 which indicates a metal-centered reaction (Figure 9a). This contrasts with what is observed during the first reduction of 17 (Figure 9b) where the Soret band decreases in intensity as new near-IR bands grow in at 706 and 751 nm. These latter spectral changes are indicative of a ring-centered reduction. The second reduction of 13 at −1.21 V leads to [13]2– whose spectrum is shown in Supporting Information, Figure S4.
The first oxidation of 15 involves a Co(II)/Co(III) process. The electrogenerated Co(III) form of the compound has a Soret-like band at 439 nm after controlled potential oxidation at 0.70 V (Figure l0a). Controlled potential reduction of the same compound may generate either a Co(II) π-anion radical or a Co(I) compound. The spectrum of the singly reduced product (Figure l0b) lacks the intense radical "marker bands" seen for the electro-generated CuII17 anion radical (Figure 9b), thus suggesting the formation of a Co(I) azahemiporphycene with an uncharged macrocycle.
Figure 10.
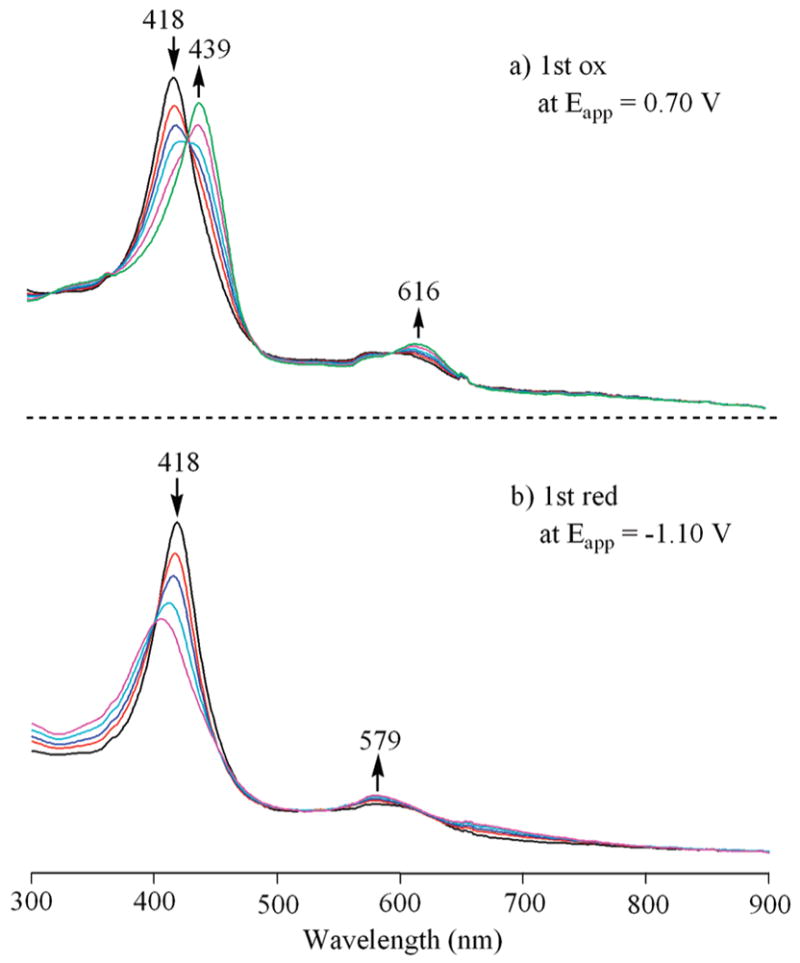
UV–visible spectral changes of CoII azahemiporphycene 15 during (a) the first oxidation at 0.70 V and (b) the first reduction at −1.10 V inPhCN,0.1M TBAP.
Conclusions
6-Azahemiporphycene represents a further example of corrole ring "breathing", with the macrocycle expansion occurring regioselectively at the 6-position. This approach offers a facile way to prepare a novel member of the porphyrinoid family, considering the significant developments recently achieved by the synthetic chemistry of corrole. Theoretical and spectroscopic characterization of 6-azahemiporphycene offer important information on both the formation pathway and behavior of such macrocycles. Different metal complexes of 6-azahemiporphycene have been prepared, and the reductive electrochemistry of compounds 12–18 resembles that of both porphyrins and porphycenes with similar metal ions. The singly and doubly reduced products are stable but this is not the case for the oxidations where coupled chemical reactions occur to give unidentified new products. These features, together with the photophysical behavior showed by 12 and 18, make these compounds of interest for applications in catalysis or as chemical sensors, where porphyrinoids have already shown interesting potential. This research is currently ongoing in our laboratories.
Supplementary Material
Acknowledgments
Financial support from Fondazione Banca del Monte (L.P.), MIUR-PRIN project n. 2007C8RW53 (L.P., R.P.), the U.S. National Institutes of Health (K.M.S., Grant CA132861), and the Robert A. Welch Foundation (K.M.K., Grant E-680) are gratefully acknowledged.
Footnotes
Supporting Information Available: CIF files of 11 and 20, optimized geometries, energies of all computed structures, and UV–visible spectroelectrochemistry. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. 1–10 Academic Press; San Diego, CA: 2000. [Google Scholar]; (b) Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. 11–20 Academic Press; San Diego, CA: 2003. [Google Scholar]
- 2.Drain CM, Hupp JT, Suslick KS, Wasielewski MR, Chen XJ. Porphyrins Phthalocyanines. 2002;6:243–258. [Google Scholar]
- 3.(a) Sessler JL, Weghorn SJ. Expanded, Contracted and Isomeric Porphyrins. Pergamon; Oxford: 1997. [Google Scholar]; (b) Jasat A, Dolphin D. Chem Rev. 1997;97:2267–2340. doi: 10.1021/cr950078b. [DOI] [PubMed] [Google Scholar]; (c) Sessler JL, Gebauer A, Vogel E. In: The Porphyrin Handbook. Kadish KM, Smith K, Guilard R, editors. Vol. 2. Academic; San Diego, CA: 2000. pp. 1–54. [Google Scholar]; (d) Sessler JL, Gebauer A, Weghorn SJ. In: The Porphyrin Handbook. Kadish KM, Smith K, Guilard R, editors. Vol. 2. Academic; San Diego, CA: 2000. pp. 55–124. [Google Scholar]; (e) Sessler JL, Seidel D. Angew Chem, Int Ed. 2003;42:5134–5175. doi: 10.1002/anie.200200561. [DOI] [PubMed] [Google Scholar]; (f) Misra R, Chandrashekar TK. Acc Chem Res. 2008;41:265–279. doi: 10.1021/ar700091k. [DOI] [PubMed] [Google Scholar]; (g) Callot HJ, Rohrer A, Tschamber Th. New J Chem. 1995;19:155–159. [Google Scholar]
- 4.(a) Horn S, Dahms K, Senge MO. J Porphyrins Phthalocyanines. 2008;12:1053–1077. [Google Scholar]; (b) Jux N. Angew Chem, Int Ed. 2009;48:4284–4286. doi: 10.1002/anie.200901195. [DOI] [PubMed] [Google Scholar]
- 5.(a) Callot H. Dalton Trans. 2008:6346–6357. doi: 10.1039/b807066k. [DOI] [PubMed] [Google Scholar]; (b) Senge MO, Sergeeva NN. Angew Chem, Int Ed. 2006;45:7492–7495. doi: 10.1002/anie.200603249. [DOI] [PubMed] [Google Scholar]
- 6.Grigg R, Johnson AW, Richardson K, Smith MJJ. Chem Soc C. 1970:1289–1295. [Google Scholar]
- 7.(a) Callot HJ, Tschamber T. Tetrahedron Lett. 1974;15:3155–3158. [Google Scholar]; (b) Callot HJ, Tschamber TJ. Am Chem Soc. 1975;97:6175–6178. [Google Scholar]
- 8.(a) Nardis S, Monti D, Paolesse R. Mini-Rev Org Chem. 2005;2:546–564. [Google Scholar]; (b) Paolesse R. Synlett. 2008:2215–2230. [Google Scholar]; (c) Gryko DT. J Porphyrins Phthalocyanines. 2008;12:906–917. [Google Scholar]
- 9.(a) Aviv I, Gross Z. Chem Commun. 2007:1987–1999. doi: 10.1039/b618482k. [DOI] [PubMed] [Google Scholar]; (b) Aviv-Harel I, Gross Z. Chem–Eur J. 2009;15:8382–8394. doi: 10.1002/chem.200900920. [DOI] [PubMed] [Google Scholar]; (c) Flamigni L, Gryko DT. Chem Soc Rev. 2009;38:1635–1646. doi: 10.1039/b805230c. [DOI] [PubMed] [Google Scholar]; (d) Lvova L, Di Natale C, D’Amico A, Paolesse RJ. Porphyrins Phthalocyanines. 2009 in press. [Google Scholar]
- 10.Simkhovich L, Mahammed A, Goldberg I, Gross Z. Chem– Eur J. 2001;7:1041–1055. doi: 10.1002/1521-3765(20010302)7:5<1041::aid-chem1041>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 11.Gros CP, Barbe JM, Espinosa E, Guilard R. Angew Chem, Int Ed. 2006;45:5642–5645. doi: 10.1002/anie.200601580. [DOI] [PubMed] [Google Scholar]
- 12.Kin Tse M, Zhang Z, Mak TCW, Shing Chan K. Chem Commun. 1998:1199–1200. [Google Scholar]
- 13.Paolesse R, Nardis S, Stefanelli M, Fronczek FR, Vicente MGH. Angew Chem, Int Ed. 2005;44:3047–3050. doi: 10.1002/anie.200500481. [DOI] [PubMed] [Google Scholar]
- 14.Mandoj F, Stefanelli M, Nardis S, Mastroianni M, Fronczek FR, Smith KM, Paolesse R. Chem Commun. 2009:1580–1582. doi: 10.1039/b821390a. [DOI] [PubMed] [Google Scholar]
- 15.Montalti M, Credi A, Prodi L, Gandolfi MT. Handbook of Photochemistry. 3. CRC Taylor & Francis; Boca Raton, FL: 2006. [Google Scholar]
- 16.Lin XQ, Kadish KM. Anal Chem. 1985;57:1498–1501. doi: 10.1021/ac00284a079. [DOI] [PubMed] [Google Scholar]
- 17.(a) Paolesse R, Nardis S, Sagone F, Khoury RGJ. Org Chem. 2001;66:550–556. doi: 10.1021/jo005661t. [DOI] [PubMed] [Google Scholar]; (b) Paolesse R, Marini A, Nardis S, Froiio A, Mandoj F, Nurco DJ, Prodi L, Montalti M, Smith KM. J Porphyrins Phthalocyanines. 2003;7:25–36. [Google Scholar]
- 18.Richeter S, Hadj-Aïssa A, Lee A, Leclercq D. Chem Commun. 2007:2148–2150. doi: 10.1039/b704681b. [DOI] [PubMed] [Google Scholar]
- 19.Stefanelli M, Shen J, Zhu W, Mastroianni M, Mandoj F, Nardis S, Ou Z, Kadish KM, Fronczek FR, Smith KM, Paolesse R. Inorg Chem. 2009;48:6879–6887. doi: 10.1021/ic900859a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nardis S, Pomarico G, Fronczek FR, Vicente MGH, Paolesse R. Tetrahedron Lett. 2007;48:8643–8646. [Google Scholar]
- 21.Nardis S, Mandoj F, Paolesse R, Fronczek FR, Smith KM, Prodi L, Montalti M, Battistini G. Eur J Inorg Chem. 2007:2345–2354. [Google Scholar]
- 22.(a) Tardieux C, Gros C, Guilard R. J Heterocycl Chem. 1998;55:965–970. [Google Scholar]; (b) Paolesse R, Froiio A, Nardis S, Mastroianni M, Russo M, Nurco DJ, Smith KM. J Porphyrins Phthalocyanines. 2003;7:585–592. [Google Scholar]
- 23.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Jr, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian, Inc; Wallingford, CT: 2004. Revision E.01. [Google Scholar]
- 24.Chen Z, Wannere CS, Corminboeuf C, Puchta R, von Ragué Schleyer P. Chem Rev. 2005;105:3842–3888. doi: 10.1021/cr030088+. [DOI] [PubMed] [Google Scholar]
- 25.(a) Cyrañski MK, Krygowski TM, Wisiorowski M, van Eikema Hommes NJR, von Ragué Schleyer P. Angew Chem, Int Ed. 1998;37:177–180. [Google Scholar]; (b) Furuta H, Maeda H, Osuka A. J Org Chem. 2001;66:8563–8572. doi: 10.1021/jo016051b. [DOI] [PubMed] [Google Scholar]; (c) Yoon ZS, Noh SB, Cho DG, Sessler JL, Kim D. Chem Commun. 2007:2378–2380. doi: 10.1039/b618674b. [DOI] [PubMed] [Google Scholar]; (d) Stepień M, Latos-Grażyński L. Top Heterocycl Chem. 2009;19:83–153. [Google Scholar]
- 26.(a) Wodrich MD, Corminboeuf C, Schreiner PR, Fokin AA, Schleyer PvR. Org Lett. 2007;9:1851–1854. doi: 10.1021/ol070354w. [DOI] [PubMed] [Google Scholar]; (b) Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 27.Buchler JW. In: Porphyrins and Metalloporphyrins. Smith KM, editor. Elsevier; Amsterdam: 1975. pp. 157–231. [Google Scholar]
- 28.Kadish KM, Royal G, Van Caemelbecke E, Gueletti L. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 9. Academic Press; San Diego, CA: 2003. pp. 1–220. [Google Scholar]
- 29.Gisselbrecht JP, Gross M, Koecher M, Lausmann M, Vogel E. J Am Chem Soc. 1990;112:8618–8620. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.