

Abstract
Progressive augmentation (PA) and ventilatory long-term facilitation (vLTF) of respiratory motor output are forms of respiratory plasticity that are initiated during exposure to intermittent hypoxia. The present study was designed to determine whether PA and vLTF are enhanced in obstructive sleep apnoea (OSA) participants compared to matched healthy controls. The study was also designed to determine whether administration of an antioxidant cocktail mitigates PA and vLTF. Thirteen participants with sleep apnoea and 13 controls completed two trials. During both trials participants were exposed to intermittent hypoxia which included twelve 4-min episodes of hypoxia (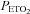 , 50 mmHg;
, 50 mmHg; 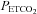 , 4 mmHg above baseline) followed by 30 min of recovery. Prior to exposure to intermittent hypoxia, participants were administered, in a randomized fashion, either an antioxidant or a placebo cocktail. Baseline measures of minute ventilation during the placebo and antioxidant trials were not different between or within groups. During the placebo trial, PA was evident in both groups; however it was enhanced in the OSA group compared to control (last hypoxic episode 36.9 ± 2.8 vs. 27.7 ± 2.2 l min−1; P≤ 0.01). Likewise, vLTF was evident during the recovery period in both groups; on the other hand vLTF was greater in the OSA group compared to control (29.3 ± 2.8 vs. 20.4 ± 1.3 l min−1; P≤ 0.01). PA and vLTF were reduced in the OSA group following antioxidant administration compared to the placebo (PA 30.6 ± 2.0 vs. 36.9 ± 2.8 l min−1, P≤ 0.01; vLTF 23.3 ± 1.4 vs. 29.3 ± 2.8 l min−1, P≤ 0.05). We conclude that PA and vLTF are enhanced in participants with OSA and that these forms of respiratory plasticity are mitigated after treatment with an antioxidant cocktail.
, 4 mmHg above baseline) followed by 30 min of recovery. Prior to exposure to intermittent hypoxia, participants were administered, in a randomized fashion, either an antioxidant or a placebo cocktail. Baseline measures of minute ventilation during the placebo and antioxidant trials were not different between or within groups. During the placebo trial, PA was evident in both groups; however it was enhanced in the OSA group compared to control (last hypoxic episode 36.9 ± 2.8 vs. 27.7 ± 2.2 l min−1; P≤ 0.01). Likewise, vLTF was evident during the recovery period in both groups; on the other hand vLTF was greater in the OSA group compared to control (29.3 ± 2.8 vs. 20.4 ± 1.3 l min−1; P≤ 0.01). PA and vLTF were reduced in the OSA group following antioxidant administration compared to the placebo (PA 30.6 ± 2.0 vs. 36.9 ± 2.8 l min−1, P≤ 0.01; vLTF 23.3 ± 1.4 vs. 29.3 ± 2.8 l min−1, P≤ 0.05). We conclude that PA and vLTF are enhanced in participants with OSA and that these forms of respiratory plasticity are mitigated after treatment with an antioxidant cocktail.
Introduction
Progressive augmentation (PA) and long-term facilitation (LTF) of respiratory motor output are forms of respiratory plasticity that are initiated during exposure to intermittent hypoxia (Powell et al. 1998; Mahamed & Mitchell, 2007; Mateika & Narwani, 2009). Progressive augmentation is characterized by an enhanced respiratory modulated response to hypoxia, which gradually occurs from the initial to the final hypoxic episode throughout exposure to intermittent hypoxia (Powell et al. 1998; Mateika & Narwani, 2009). The increased response to hypoxia may be sustained for minutes to hours after exposure to intermittent hypoxia (Mateika et al. 2004; Fuller, 2005). Progressive augmentation of minute ventilation and phrenic modulated inspiratory discharge has been observed in humans and other animals (Turner & Mitchell, 1997; Mitchell et al. 2001b; Fuller, 2005; Harris et al. 2006; Sokolowska & Pokorski, 2006; Wadhwa et al. 2008). Similarly, PA of hypoglossal or genioglossus muscle modulated inspiratory discharge (Fuller, 2005) and carotid sensory nerve activity (Peng & Prabhakar, 2004; Cummings & Wilson, 2005) has also been reported.
Long-term facilitation is characterized by a progressive increase in respiratory motor output during normoxic periods that separate hypoxic episodes and by a sustained elevation in respiratory activity for up to 90 min after exposure to intermittent hypoxia (Mitchell & Johnson, 2003; Mahamed & Mitchell, 2007). Long-term facilitation of minute ventilation (vLTF) has been observed in healthy humans during wakefulness and sleep (Harris et al. 2006; Wadhwa et al. 2008; Pierchala et al. 2008). Likewise, LTF of minute ventilation, or phrenic, hypoglossal or carotid sinus nerve inspiratory-modulated discharge has been observed in a variety of other animal preparations (Mateika & Narwani, 2009). Long-term facilitation of carotid chemosensory nerve activity differs in part from minute ventilation and phrenic and hypoglossal LTF in that it is induced only following exposure to chronic intermittent hypoxia (e.g. 15 s episodes, 9 episodes per hour, 8 h per day for 10 days) (Peng et al. 2003). In contrast, phrenic and hypoglossal LTF is induced in naive rats not previously exposed to hypoxia (Mitchell et al. 2001a).
Recent studies have indicated that PA and LTF induced by a one time exposure to intermittent hypoxia (i.e. acute intermittent hypoxia) (e.g. six 5-min episodes of hypoxia) in naive rats may be further enhanced following exposure to chronic intermittent hypoxia (Ling et al. 2001; Zabka et al. 2001; McGuire et al. 2003; Peng & Prabhakar, 2003; McGuire et al. 2004; McGuire & Ling, 2005). Given this finding we hypothesized that PA and LTF induced by a one time exposure to intermittent hypoxia might be enhanced in humans chronically exposed to intermittent hypoxia. More specifically, we hypothesized that the magnitude of PA and LTF induced by acute intermittent hypoxia might be greater in individuals with sleep apnoea, who are exposed nightly to intermittent episodes of hypoxia during sleep, compared to healthy individuals. Accordingly, this study was designed in part to test this hypothesis by comparing the magnitude of PA and LTF in participants with sleep apnoea and matched healthy control participants during and following a one time exposure to intermittent hypoxia.
In addition to revealing that the magnitude of PA and LTF might be dependent on prior exposure to hypoxia, recent studies have identified an assortment of mechanisms that impact on the magnitude of PA and LTF, including the accumulation of reactive oxygen species (Macfarlane et al. 2008). These free radicals are known to accumulate as a consequence of exposure to intermittent hypoxia (Macfarlane et al. 2008), and published findings suggest that they impact on the magnitude of the ventilatory response to hypoxia and LTF following exposure to chronic or acute intermittent hypoxia. Peng and colleagues demonstrated that exposure to chronic intermittent hypoxia led to an enhanced in vivo and ex vivo carotid body sensory response to hypoxia (Peng & Prabhakar, 2004) and carotid sensory nerve LTF (Peng et al. 2003). The augmented hypoxic carotid sensory response and carotid sensory nerve LTF was abolished in a group of rats pre-treated with a superoxide dismutase mimetic which is a potent scavenger of superoxide anions (Peng et al. 2003; Peng & Prabhakar, 2004). Likewise, Peng and colleagues also demonstrated that augmentation of phrenic LTF induced by exposure to chronic intermittent hypoxia was abolished following pre-treatment with a potent superoxide anion scavenger (Peng et al. 2003). The latter findings are further supported by results which demonstrated that intravenous administration of 10 mg of a superoxide anion scavenger to anaesthetized, vagotomized and ventilated rats, before exposure to acute intermittent hypoxia, abolished LTF of both phrenic and hypoglossal nerve activity (Wilkerson et al. 2008). Given these findings in animals, we hypothesized that administration of an antioxidant cocktail would reduce the magnitude of the ventilatory response to hypoxia and vLTF during and following exposure to acute intermittent hypoxia in humans. Moreover, we hypothesized that the impact of the antioxidant cocktail on PA and vLTF may be more evident in participants previously exposed to intermittent hypoxia because of the increased likelihood that accumulated reactive oxygen species could not be effectively eliminated by endogenously produced antioxidants. To test this hypothesis, an antioxidant or placebo cocktail was administered, in a randomized blinded fashion, to participants with sleep apnoea and matched healthy participants before exposure to acute intermittent hypoxia. Thereafter the magnitude of the ventilatory response to hypoxia and vLTF was compared.
Methods
Protocol overview
The Human Investigation Committees of Wayne State University and John D. Dingell Veterans Affairs Medical Center approved the experimental protocol. The approved protocol conformed to the standards set by the Declaration of Helsinki. Thirteen healthy males and 13 males with obstructive sleep apnoea (OSA) that were otherwise healthy participated in the study after written informed consent was obtained. The healthy males and males with OSA were matched for height, weight, age and race. Each participant visited the laboratory on four occasions. Before each visit, participants were advised to have a minimum of 7 h of sleep, and to avoid caffeinated beverages and food 4 h prior to the study. For visits 3 and 4 participants arrived at the same time of the day (i.e. 07.00–08.00 h) to account for the influence of circadian rhythms on the measured variables.
During the first visit to the laboratory, participants underwent a physical exam, blood pressure was measured and a 12-lead electrocardiogram (EKG) was completed. Thereafter, participants completed one rebreathing trial followed by exposure to two 4-min episodes of hypoxia to ensure familiarization with the experimental protocol and apparatus. On the second visit to the laboratory, participants completed a sleep study to confirm the absence of sleep apnoea in the case of the healthy participants, and the presence of the disorder in the participants afflicted with sleep apnoea. On the third and fourth visits to the laboratory, participants were exposed to intermittent hypoxia followed by the completion of two rebreathing trials, while in the supine position (see Intermittent hypoxia protocol and Modified rebreathing protocol for further details). Three hours prior to exposure to intermittent hypoxia, 8 ml of venous blood was sampled to obtain a measure of lipid peroxidation, which we used as a biomarker of oxidative stress. Immediately thereafter the oral components of an antioxidant cocktail (see Antioxidant cocktail below for details regarding the antioxidant cocktail) or a placebo were administered. Three hours after the oral components of the antioxidant or placebo cocktail were administered, a second component was administered intravenously immediately prior to exposure to intermittent hypoxia. Following exposure to intermittent hypoxia, and prior to completion of the two rebreathing trials, 8 ml of blood was drawn to obtain an additional measure of oxidative stress. The antioxidant and placebo cocktail were administered randomly. In other words, if the antioxidant cocktail was administered during trial 3 the placebo cocktail was administered during trial 4 and vice versa.
Polysomnography studies
During completion of the screening sleep studies (i.e. visit 2) the monitoring montage included an electroencephalogram (C3/A2, C4/A1, O1/A2, O2/A1), electrooculograms, submental and tibialis anterior electromyogram and a three-lead electrocardiogram. Chest wall and abdominal movements were measured using inductive plethysmography (Respitrace, Ambulatory Monitoring, Inc., Ardsley, NY, USA). Flow and breath timing (inspiratory and expiratory time) were measured using a pneumotachometer (Model RSS100-HR, Hans Rudolph Inc., Kansas, MO, USA) attached to a nasal mask. Additionally, oesophageal pressure was measured using a transducer tipped catheter (MPC-500, Millar, Inc., Houston, TX, USA) to confirm apnoea and ascertain the presence of flow limitation. Oxygen saturation (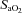 ) was measured using a pulse oximeter (Biox 3700, Ohmeda Corp., Laurel, MD, USA). All physiological variables were analog to digitally converted at a sampling frequency of 100 Hz per channel and input into a microcomputer using a commercially available software package (Gamma v. 4.0, Astro-Med Inc., West Warwick, RI, USA).
) was measured using a pulse oximeter (Biox 3700, Ohmeda Corp., Laurel, MD, USA). All physiological variables were analog to digitally converted at a sampling frequency of 100 Hz per channel and input into a microcomputer using a commercially available software package (Gamma v. 4.0, Astro-Med Inc., West Warwick, RI, USA).
Intermittent hypoxia protocol
Prior to completion of the IH protocol(s) participants breathed room air for 10 min in order to determine baseline measures of minute ventilation and the partial pressure of end-tidal carbon dioxide (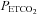 ). Subsequently, carbon dioxide levels were increased 4 mmHg above baseline values and a new baseline was established for 15 min. This level of carbon dioxide was sustained throughout the IH protocol. Carbon dioxide was increased by 4 mmHg because we showed previously that vLTF is evident in humans when carbon dioxide is sustained slightly above the
). Subsequently, carbon dioxide levels were increased 4 mmHg above baseline values and a new baseline was established for 15 min. This level of carbon dioxide was sustained throughout the IH protocol. Carbon dioxide was increased by 4 mmHg because we showed previously that vLTF is evident in humans when carbon dioxide is sustained slightly above the 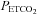 that demarcates the ventilatory threshold but is absent when values are below baseline values (Mateika et al. 2004; Harris et al. 2006). After the new baseline was established, participants were exposed to twelve 4-min episodes of hypoxia with 4 min of normoxia interspersed between episodes. During the hypoxic episodes participants inspired the gas mixture comprising 8% oxygen–balance nitrogen. Supplemental oxygen and carbon dioxide were added to the inspired hypoxic gas mixture to maintain the partial pressure of end-tidal oxygen (
that demarcates the ventilatory threshold but is absent when values are below baseline values (Mateika et al. 2004; Harris et al. 2006). After the new baseline was established, participants were exposed to twelve 4-min episodes of hypoxia with 4 min of normoxia interspersed between episodes. During the hypoxic episodes participants inspired the gas mixture comprising 8% oxygen–balance nitrogen. Supplemental oxygen and carbon dioxide were added to the inspired hypoxic gas mixture to maintain the partial pressure of end-tidal oxygen (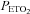 ) at 50 mmHg and to sustain carbon dioxide levels 4 mmHg above resting values. At the completion of each episode, hypoxia was terminated abruptly with a single breath of 100% oxygen to rapidly bring the
) at 50 mmHg and to sustain carbon dioxide levels 4 mmHg above resting values. At the completion of each episode, hypoxia was terminated abruptly with a single breath of 100% oxygen to rapidly bring the 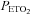 to the normoxic range. Following the last exposure to hypoxia (12th episode), respiration was monitored continuously for 30 min while participants breathed room air with carbon dioxide levels sustained 4 mmHg above baseline.
to the normoxic range. Following the last exposure to hypoxia (12th episode), respiration was monitored continuously for 30 min while participants breathed room air with carbon dioxide levels sustained 4 mmHg above baseline.
During the intermittent hypoxia protocol, participants wore a tight-fitting face mask. The mask was connected to a pneumotachograph (model RSS100-HR, Hans Rudolph, Inc., Shawnee, KS, USA) that was used to monitor breath-by-breath changes in ventilation. The pneumotachograph was attached to a two-way valve. The inspiratory port of the valve was attached to a five-way stopcock. The subject either inspired room air or the contents from one of two bags attached to the stopcock. The bags contained two different gas mixtures. One bag contained 8% oxygen–balance nitrogen to reduce 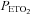 to 50 mmHg at the onset of each hypoxic episode, and the other bag contained 100% oxygen to rapidly induce normoxia at the end of each hypoxic episode. Additionally, the output from a flowmeter was attached to a stopcock port connected to the inspiratory port of the valve. Gas from two cylinders containing 100% oxygen and 100% carbon dioxide were connected to the flowmeter. Thus supplemental oxygen and carbon dioxide could be added to the 8% oxygen–balance nitrogen to maintain desired levels of
to 50 mmHg at the onset of each hypoxic episode, and the other bag contained 100% oxygen to rapidly induce normoxia at the end of each hypoxic episode. Additionally, the output from a flowmeter was attached to a stopcock port connected to the inspiratory port of the valve. Gas from two cylinders containing 100% oxygen and 100% carbon dioxide were connected to the flowmeter. Thus supplemental oxygen and carbon dioxide could be added to the 8% oxygen–balance nitrogen to maintain desired levels of 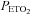 (50 mmHg) and
(50 mmHg) and 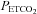 (i.e. 4 mmHg above baseline). End-tidal oxygen and carbon dioxide were sampled from ports on the face mask using oxygen and carbon dioxide analysers (model 17518 and model 17515, Vacumetrics, Inc., Ventura, CA, USA). A three-lead electrocardiogram and oxygen saturation (
(i.e. 4 mmHg above baseline). End-tidal oxygen and carbon dioxide were sampled from ports on the face mask using oxygen and carbon dioxide analysers (model 17518 and model 17515, Vacumetrics, Inc., Ventura, CA, USA). A three-lead electrocardiogram and oxygen saturation (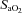 ), via a pulse oximeter (Biox 3700, Ohmeda Corp., Laurel, MD, USA), was monitored. A 16-bit analog-to-digital converter (AT-MIO-16XE-50, National Instruments, Austin, TX, USA) was used to digitize the analog signals for computer analysis using software specifically designed for this purpose. The software calculated minute ventilation,
), via a pulse oximeter (Biox 3700, Ohmeda Corp., Laurel, MD, USA), was monitored. A 16-bit analog-to-digital converter (AT-MIO-16XE-50, National Instruments, Austin, TX, USA) was used to digitize the analog signals for computer analysis using software specifically designed for this purpose. The software calculated minute ventilation, 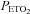 and
and 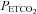 on a breath-by-breath basis.
on a breath-by-breath basis.
Modified rebreathing protocol
A modified rebreathing protocol that has been described in detail in previous studies (Mohan et al. 1999; Mateika et al. 2004; Duffin, 2007) was employed to measure the ventilatory threshold (i.e. the point where ventilation begins to rise with increases in 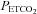 ) and sensitivity (i.e. the slope of ventilation vs.
) and sensitivity (i.e. the slope of ventilation vs.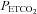 ) to carbon dioxide 30 min after exposure to intermittent hypoxia. Each rebreathing trial was separated by approximately 15 min of rest. During completion of the rebreathing protocol, conditions of quiet wakefulness were maintained, and noise in the laboratory was minimized to reduce any behavioural stimuli that might affect breathing. During each trial, the participants initially breathed room air for 5 min. Thereafter, subjects hyperventilated for 5 min while being coached to maintain
) to carbon dioxide 30 min after exposure to intermittent hypoxia. Each rebreathing trial was separated by approximately 15 min of rest. During completion of the rebreathing protocol, conditions of quiet wakefulness were maintained, and noise in the laboratory was minimized to reduce any behavioural stimuli that might affect breathing. During each trial, the participants initially breathed room air for 5 min. Thereafter, subjects hyperventilated for 5 min while being coached to maintain 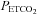 between 20 and 25 mmHg. This period of hyperventilation was employed to lower the stores of the carbon dioxide so that the point where ventilation begins to rise with increases in
between 20 and 25 mmHg. This period of hyperventilation was employed to lower the stores of the carbon dioxide so that the point where ventilation begins to rise with increases in 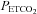 (i.e. the ventilatory threshold) could be delineated. After the desired
(i.e. the ventilatory threshold) could be delineated. After the desired 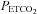 was maintained for 5 min, participants were switched from room air to a rebreathing bag. The
was maintained for 5 min, participants were switched from room air to a rebreathing bag. The 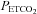 in the bag at the start of the rebreathing experiment was 42 mmHg. Rebreathing began at the end of expiration and was followed by three rapid and deep breaths that produced prompt equilibration of the
in the bag at the start of the rebreathing experiment was 42 mmHg. Rebreathing began at the end of expiration and was followed by three rapid and deep breaths that produced prompt equilibration of the 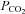 in the bag, lungs and arterial blood to that of mixed venous blood. The presence of a plateau in carbon dioxide during the equilibration was a prerequisite for the continuation of the test. Rebreathing continued until
in the bag, lungs and arterial blood to that of mixed venous blood. The presence of a plateau in carbon dioxide during the equilibration was a prerequisite for the continuation of the test. Rebreathing continued until 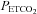 increased 10 mmHg above the ventilatory threshold. The
increased 10 mmHg above the ventilatory threshold. The 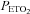 in the rebreathing bag at the start of one rebreathing trial following exposure to intermittent hypoxia was 50 mmHg. This level of
in the rebreathing bag at the start of one rebreathing trial following exposure to intermittent hypoxia was 50 mmHg. This level of 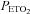 was sustained throughout the trial. The
was sustained throughout the trial. The 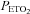 in the rebreathing bag during the second trial was sustained at 150 mmHg.
in the rebreathing bag during the second trial was sustained at 150 mmHg.
During the rebreathing trials, the subjects wore a facemask that was connected to a pneumotachograph (model RSS100-HR, Hans Rudolph) which monitored breath-by-breath changes in ventilation. The pneumotachograph was attached to one side of a three-way valve that allowed us to switch the subjects from room air to the rebreathing bag. The 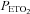 and
and 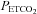 were sampled from the pneumotachograph side of the three-way valve using oxygen and carbon dioxide analysers (model 17518 and model 17515, Vacumetrics, Inc., Ventura, CA, USA). The end-tidal gas that was sampled for monitoring was returned to the bag during rebreathing. The oxygen level in the bag during rebreathing was maintained by a flow of oxygen that was computer controlled. If oxygen decreased below the desired threshold (50 or 150 mmHg), oxygen was immediately bled into the bag. The oxygen saturation (
were sampled from the pneumotachograph side of the three-way valve using oxygen and carbon dioxide analysers (model 17518 and model 17515, Vacumetrics, Inc., Ventura, CA, USA). The end-tidal gas that was sampled for monitoring was returned to the bag during rebreathing. The oxygen level in the bag during rebreathing was maintained by a flow of oxygen that was computer controlled. If oxygen decreased below the desired threshold (50 or 150 mmHg), oxygen was immediately bled into the bag. The oxygen saturation (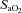 ) was monitored using a pulse oximeter (Biox 3700, Ohmeda Corp., Laurel, MD, USA). A 16-bit analog-to-digital converter (AT-MIO-16XE-50, National Instruments) was used to digitize the analog signals for computer analysis using software specifically designed for this purpose. The software calculated minute ventilation,
) was monitored using a pulse oximeter (Biox 3700, Ohmeda Corp., Laurel, MD, USA). A 16-bit analog-to-digital converter (AT-MIO-16XE-50, National Instruments) was used to digitize the analog signals for computer analysis using software specifically designed for this purpose. The software calculated minute ventilation, 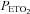 and
and 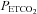 on a breath-by-breath basis.
on a breath-by-breath basis.
Antioxidant cocktail
The investigational pharmacist at John D. Dingell, VA Medical Center prepared and randomized the antioxidant and placebo cocktail. Laboratory personnel who administered the intermittent hypoxia and completed the data analysis were blinded to the randomization process. The antioxidant cocktail consisted of 60 mg of coenzyme Q10 (Cell Tech International Inc., Klamath Falls, OR, USA), 400 mg of superoxide dismustase (P. L. Thomas and Co. Inc., Morristown, NJ, USA), 200 IU of vitamin E (Goldline Laboratories Inc., Miami, FL, USA) and vitamin C (2 doses of 1 g in 50 ml of 0.9 NaCl) (Hospira Inc., Rocky Mount, NC, USA). The components of the cocktail were selected because water-soluble ascorbic acid is a potent antioxidant in plasma and the cytosol, while α-tocopherol because of its lipid solubility is an important free radical and lipid peroxide scavenger in membranes. Likewise, it is known that the combined effectiveness of ascorbate and α-tocopherol is synergistic. Moreover, coenzyme Q10 is a potent lipophilic antioxidant and is capable of recycling and regenerating other antioxidants such as α-tocopherol and ascorbate. Superoxide dismutase inactivates superoxide anion, the starting point in the chain production of free radicals, by transforming it into hydrogen peroxide. The components of the cocktail were selected because one or more of the components have been used previously in studies investigating respiratory, or autonomic and vascular responses, in humans (Teppema et al. 2002, 2006; Jablonski et al. 2007) and animals (Peng & Prabhakar, 2004; Durrant et al. 2009).
Coenzyme Q10, superoxide dismustase and vitamin E were ingested orally with 170 g (6 ounces) of plain yogurt 3 h prior to exposure to intermittent hypoxia. Immediately prior to exposure to intermittent hypoxia, participants received an intravenous dose of vitamin C. A peripheral intravenous with 0.9 NaCl infusing at 10 ml h−1 was initiated in the antecubital fossa prior to administering the vitamin C. Subsequently, vitamin C was infused over a 20 min period. Vitamin C was infused immediately before the intermittent hypoxia protocol since peak plasma levels typically occur 90 min after infusion with a half-life of approximately 30 min. The oral and intravenous component of the sham cocktail consisted of cellulose capsules and 0.9% NaCl infusion, respectively. The oral component of the placebo cocktail was ingested along with 170 g (6 ounces) of yogurt. The time frame of administration of the placebo cocktail was identical to that described for the antioxidant cocktail.
Data analysis
Polysomnography studies
Sleep and respiration were scored according to conventional criteria (Rechtschaffen & Kales, 1968). The total number of arousals, apnoeas and hypopnoeas were calculated for the total sleep time. An apnoea was defined as the absence of inspiratory airflow for a minimum of 10 s accompanied by a greater than 3% reduction in oxygen saturation. The apnoea index was defined as the total number of apnoeas per hour of sleep. A hypopnoea was defined as a greater than 50% reduction in flow lasting more than 10 s accompanied by a greater than 3% reduction in oxygen saturation. The hypopnoea index was defined as the total number of hypopnoeas per hour of sleep time. Sleep staging and arousals were performed according to the criteria of the American Academy of Sleep Medicine (1992).
Intermittent hypoxia
During the antioxidant and placebo trials average values for minute ventilation, tidal volume, breathing frequency, 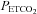 and
and 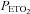 were determined for the last 5 min of the baseline period during which participants breathed room air. Likewise, similar measures were obtained from the last 5 min of the 15 min baseline period recorded immediately before intermittent hypoxia, during which
were determined for the last 5 min of the baseline period during which participants breathed room air. Likewise, similar measures were obtained from the last 5 min of the 15 min baseline period recorded immediately before intermittent hypoxia, during which 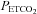 was sustained 4 mmHg above baseline values. Average values were also obtained for the last 2 min of each hypoxic episode and the last 2 min of the normoxic recovery periods that followed each hypoxic episode. The 30 min end recovery period was divided into 5 min intervals and average values were obtained for each interval. For reporting purposes, minute ventilation,
was sustained 4 mmHg above baseline values. Average values were also obtained for the last 2 min of each hypoxic episode and the last 2 min of the normoxic recovery periods that followed each hypoxic episode. The 30 min end recovery period was divided into 5 min intervals and average values were obtained for each interval. For reporting purposes, minute ventilation, 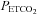 and
and 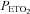 at each of the time points were displayed in graph form (i.e. Figs 1–3). Conversely, tidal volume and breathing frequency measures were reported in tabular form for both baseline periods, for the last hypoxic episode and for the last 5 min interval of the 30 min recovery period. In addition to reporting the average absolute minute ventilation, tidal volume and breathing frequency values, the data measured during the last 5 min of the 30 min end-recovery period were also standardized relative to baseline values, measured when
at each of the time points were displayed in graph form (i.e. Figs 1–3). Conversely, tidal volume and breathing frequency measures were reported in tabular form for both baseline periods, for the last hypoxic episode and for the last 5 min interval of the 30 min recovery period. In addition to reporting the average absolute minute ventilation, tidal volume and breathing frequency values, the data measured during the last 5 min of the 30 min end-recovery period were also standardized relative to baseline values, measured when 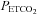 was sustained 4 mmHg above normocapnia, and reported in tabular form.
was sustained 4 mmHg above normocapnia, and reported in tabular form.
Figure 1. Measures of minute ventilation before, during and following intermittent hypoxia in OSA and control participants after administration of the placebo cocktail.

Average values for minute ventilation, end-tidal carbon dioxide partial pressure (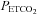 ) and end-tidal oxygen partial pressure (
) and end-tidal oxygen partial pressure (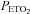 ) recorded from the last 5 min of baseline, the last 2 min of each episode of hypoxia (indicated by division marks on the x-axis), the last 2 min of each normoxic period that separated the hypoxic episodes, and the 30 min post-stimuli recovery period which is divided into six 5-min segments. The data were collected following administration of the placebo cocktail in the OSA (filled circles) and healthy (open circles) participants. Note that a progressive increase in minute ventilation occurred during exposure to intermittent hypoxia and that an elevated level of ventilation was sustained during end-recovery compared to baseline in both groups. However, notice that minute ventilation during the last hypoxic episode and end-recovery was greater in OSA participants compared to control. Lastly, note that the increased minute ventilation during end-recovery compared to baseline in both groups was accompanied by an increase in
) recorded from the last 5 min of baseline, the last 2 min of each episode of hypoxia (indicated by division marks on the x-axis), the last 2 min of each normoxic period that separated the hypoxic episodes, and the 30 min post-stimuli recovery period which is divided into six 5-min segments. The data were collected following administration of the placebo cocktail in the OSA (filled circles) and healthy (open circles) participants. Note that a progressive increase in minute ventilation occurred during exposure to intermittent hypoxia and that an elevated level of ventilation was sustained during end-recovery compared to baseline in both groups. However, notice that minute ventilation during the last hypoxic episode and end-recovery was greater in OSA participants compared to control. Lastly, note that the increased minute ventilation during end-recovery compared to baseline in both groups was accompanied by an increase in 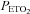 which tended to be greater in the OSA participants compared to control. ‡Significantly different from the initial hypoxic episode; *significantly different from baseline; §significantly different from control; †significantly different from the initial 5 min end-recovery segment.
which tended to be greater in the OSA participants compared to control. ‡Significantly different from the initial hypoxic episode; *significantly different from baseline; §significantly different from control; †significantly different from the initial 5 min end-recovery segment.
Figure 3. Measures of minute ventilation before, during and following intermittent hypoxia in control participants after administration of the placebo and antioxidant cocktail.

Average values for minute ventilation, end-tidal carbon dioxide partial pressure (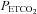 ) and end-tidal oxygen partial pressure (
) and end-tidal oxygen partial pressure (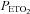 ) recorded from the last 5 min of baseline, the last 2 min of each episode of hypoxia (indicated by division marks on the x-axis), the last 2 min of each normoxic period that separated the hypoxic episodes, and the 30 min post-stimuli recovery period which is divided into six 5-min segments. The data were collected following administration of the placebo (filled circles) and antioxidant (open circles) cocktail in the control participants. Note that a progressive increase in minute ventilation occurred during exposure to intermittent hypoxia following administration of the placebo and antioxidant cocktail. Likewise, notice that an elevated level of ventilation was sustained during end-recovery compared to baseline during both the placebo and antioxidant trials. Lastly, notice that minute ventilation during the last hypoxic episode and end-recovery was similar during the placebo and antioxidant trials. ‡Significantly different from the initial hypoxic episode; *significantly different from baseline.
) recorded from the last 5 min of baseline, the last 2 min of each episode of hypoxia (indicated by division marks on the x-axis), the last 2 min of each normoxic period that separated the hypoxic episodes, and the 30 min post-stimuli recovery period which is divided into six 5-min segments. The data were collected following administration of the placebo (filled circles) and antioxidant (open circles) cocktail in the control participants. Note that a progressive increase in minute ventilation occurred during exposure to intermittent hypoxia following administration of the placebo and antioxidant cocktail. Likewise, notice that an elevated level of ventilation was sustained during end-recovery compared to baseline during both the placebo and antioxidant trials. Lastly, notice that minute ventilation during the last hypoxic episode and end-recovery was similar during the placebo and antioxidant trials. ‡Significantly different from the initial hypoxic episode; *significantly different from baseline.
Modified rebreathing
Before analysis, ventilation was plotted against 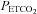 . Subsequently, each plot was fitted with a model made up of the sum of two segments separated by one breakpoint. Model fitting was based on minimizing the sum of least squares for non-linear regressions by use of commercial software (Sigmaplot 7.0, Systat Software Inc., San Jose, CA, USA). The first segment of the response was modelled as an exponential decline to a final value. This value was taken as a measure of ventilation below the recruitment threshold. The exponential decline was chosen to fit any waning of ventilatory post-stimulus potentiation that might have occurred after hyperventilation. However, post-stimulus potentiation was absent so that the time constant of the response was < 1 s.
. Subsequently, each plot was fitted with a model made up of the sum of two segments separated by one breakpoint. Model fitting was based on minimizing the sum of least squares for non-linear regressions by use of commercial software (Sigmaplot 7.0, Systat Software Inc., San Jose, CA, USA). The first segment of the response was modelled as an exponential decline to a final value. This value was taken as a measure of ventilation below the recruitment threshold. The exponential decline was chosen to fit any waning of ventilatory post-stimulus potentiation that might have occurred after hyperventilation. However, post-stimulus potentiation was absent so that the time constant of the response was < 1 s.
The second segment was characterized by a breakpoint followed by a linear increase in minute ventilation that occurred in conjunction with a rise in 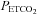 . The first breakpoint was taken as a measure of the recruitment threshold for the minute ventilation response to carbon dioxide. The threshold measured when
. The first breakpoint was taken as a measure of the recruitment threshold for the minute ventilation response to carbon dioxide. The threshold measured when 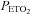 was sustained at 150 mmHg was thought to originate from the central chemoreflex, whereas the threshold measured while
was sustained at 150 mmHg was thought to originate from the central chemoreflex, whereas the threshold measured while 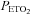 was sustained at 50 mmHg was thought to derive from the sum of the central and peripheral chemoreflex. The slope of the line fitted to ventilation after the breakpoint was taken as a measure of the chemoreflex sensitivity to increases in
was sustained at 50 mmHg was thought to derive from the sum of the central and peripheral chemoreflex. The slope of the line fitted to ventilation after the breakpoint was taken as a measure of the chemoreflex sensitivity to increases in 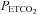 . We assumed that the slope recorded during the rebreathing trial when
. We assumed that the slope recorded during the rebreathing trial when 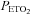 was sustained at 150 mmHg represented central chemoreflex sensitivity, whereas the slope recorded when
was sustained at 150 mmHg represented central chemoreflex sensitivity, whereas the slope recorded when 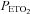 was sustained at 50 mmHg represented the combined peripheral and central chemoreflex sensitivity.
was sustained at 50 mmHg represented the combined peripheral and central chemoreflex sensitivity.
Measures of oxidative stress
We used measures of lipid peroxidation as a biomarker of oxidative stress, since previous studies have revealed that this measure is altered in participants with OSA (Carpagnano et al. 2003; Lavie et al. 2004). The measure of lipid peroxidation was determined from plasma samples obtained from EDTA treated peripheral venous blood centrifuged at 1000 g for 10 min at 4°C. Following centrifugation the plasma was stored at −80°C until lipid peroxidation was measured using a standard assay (Cayman Chemical Co., Ann Arbor, MI, USA). The assay is designed to measure the concentration of isoprostane which is produced by the random oxidation of tissue phospholipids by oxygen radicals. The assay is based on the competition between 8-isoprostane and an 8-isoprostane–acetylchoinesterase conjugate (8-isoprostane tracer) for a limited number of 8-isoprostane-specific rabbit antiserum binding sites. Plates are initially coated with rabbit IgG monoclonal antibody. The tracer, antiserum and sample are then added to the plate. The rabbit antiserum-8-isoprostane complex binds to the rabbit IgG monoclonal antibody attached to the well. The concentration of the 8-isoprostane tracer is held constant while the concentration of the 8-isoprostane in the sample varies. The amount of 8-isoprostane tracer that is able to bind to the rabbit antiserum is inversely proportional to the concentration of 8-isoprostane. The plate is washed to remove any unbound reagents and then Ellman's reagent is added to the well. The product of this enzymatic reaction has a distinct yellow colour and absorbs strongly at 412 nm. The intensity of the colour is proportional to the amount of 8-isoprostane tracer bound to the well, which is inversely proportional to the amount of free 8-isoprostane that was present in the well during incubation.
Statistical analysis
Student's t test for unpaired data was used to determine whether age, height, weight, body mass index and apnoea index were different between the groups. A two-way analysis of variance in conjunction with the Holm–Sidak post hoc test was used to determine whether minute ventilation, tidal volume, breathing frequency, 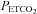 and
and 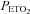 (hereinafter referred to as the physiological variables) were different between the OSA and control groups during the baseline period in which participants were breathing room air. The following comparisons were also made at different time points of the intermittent hypoxia protocol using the same statistical analysis. (a) The physiological variables measured during the final hypoxic episode were compared to measures obtained during the initial hypoxic episode in the OSA and control groups following administration of the placebo cocktail. Likewise the variables measured during the hypoxic episodes were compared between groups. The two factors in the design were group (i.e. OSA versus control) and period (initial hypoxic episode vs. 12th hypoxic episode). (b) The physiological variables (both absolute and standardized values) measured during the end recovery period following administration of the placebo cocktail were compared to baseline within and between each group. The two factors in the design were group (i.e. OSA versus control) and period (i.e. baseline and end-recovery). (c) The comparisons (i.e. baseline vs. end-recovery; initial vs. final hypoxic episode) outlined in (a) and (b) were made within and between the OSA and control groups following administration of the antioxidant cocktail. (d) The physiological variables were also compared across trials within each group. The two factors in the design were cocktail (i.e. antioxidant vs. placebo) and period (i.e. baseline vs. end-recovery or baseline vs. initial vs. final hypoxic episode). (e) The central chemoreflex threshold and sensitivity, or peripheral & central chemoreflex threshold and sensitivity, were compared between groups (i.e. OSA vs. placebo) or trials (placebo vs. antioxidant) within a group. (f) Measures of 8-isoprostane before and after intermittent hypoxia during the placebo trial both within (before vs. after intermittent hypoxia) and between groups (OSA vs. control) were compared. (g) Measures of 8-isoprostane before and after intermittent hypoxia were compared between trials (placebo vs. antioxidant) within each group. For this final comparison the data were standardized relative to baseline values by dividing the 8-isoprostane measures obtained following intermittent hypoxia by the concomitant baseline values measured during either the antioxidant or placebo trial. The data were standardized for two reasons. First, baseline values obtained during the antioxidant and placebo trials were significantly different in a few of the participants. Second, although the antioxidant cocktail reduced oxidative stress overall relative to placebo in the OSA participants, the response was variable, increasing relative to baseline in some cases (although the increase was less than that observed under placebo conditions) and decreasing relative to baseline in other cases. Consequently, standardization of the data relative to baseline was necessary to compare measures between the placebo and antioxidant trials. Given that the data were not normally distributed, a non-parametric signed rank test was used to compare the standardized data between trials for each group. All results were presented as means ±s.e.m. A value of P≤ 0.05 was considered significant. In addition P values ≤ 0.2 were reported in the results. For clarity, P values > 0.2 were not reported.
(hereinafter referred to as the physiological variables) were different between the OSA and control groups during the baseline period in which participants were breathing room air. The following comparisons were also made at different time points of the intermittent hypoxia protocol using the same statistical analysis. (a) The physiological variables measured during the final hypoxic episode were compared to measures obtained during the initial hypoxic episode in the OSA and control groups following administration of the placebo cocktail. Likewise the variables measured during the hypoxic episodes were compared between groups. The two factors in the design were group (i.e. OSA versus control) and period (initial hypoxic episode vs. 12th hypoxic episode). (b) The physiological variables (both absolute and standardized values) measured during the end recovery period following administration of the placebo cocktail were compared to baseline within and between each group. The two factors in the design were group (i.e. OSA versus control) and period (i.e. baseline and end-recovery). (c) The comparisons (i.e. baseline vs. end-recovery; initial vs. final hypoxic episode) outlined in (a) and (b) were made within and between the OSA and control groups following administration of the antioxidant cocktail. (d) The physiological variables were also compared across trials within each group. The two factors in the design were cocktail (i.e. antioxidant vs. placebo) and period (i.e. baseline vs. end-recovery or baseline vs. initial vs. final hypoxic episode). (e) The central chemoreflex threshold and sensitivity, or peripheral & central chemoreflex threshold and sensitivity, were compared between groups (i.e. OSA vs. placebo) or trials (placebo vs. antioxidant) within a group. (f) Measures of 8-isoprostane before and after intermittent hypoxia during the placebo trial both within (before vs. after intermittent hypoxia) and between groups (OSA vs. control) were compared. (g) Measures of 8-isoprostane before and after intermittent hypoxia were compared between trials (placebo vs. antioxidant) within each group. For this final comparison the data were standardized relative to baseline values by dividing the 8-isoprostane measures obtained following intermittent hypoxia by the concomitant baseline values measured during either the antioxidant or placebo trial. The data were standardized for two reasons. First, baseline values obtained during the antioxidant and placebo trials were significantly different in a few of the participants. Second, although the antioxidant cocktail reduced oxidative stress overall relative to placebo in the OSA participants, the response was variable, increasing relative to baseline in some cases (although the increase was less than that observed under placebo conditions) and decreasing relative to baseline in other cases. Consequently, standardization of the data relative to baseline was necessary to compare measures between the placebo and antioxidant trials. Given that the data were not normally distributed, a non-parametric signed rank test was used to compare the standardized data between trials for each group. All results were presented as means ±s.e.m. A value of P≤ 0.05 was considered significant. In addition P values ≤ 0.2 were reported in the results. For clarity, P values > 0.2 were not reported.
Results
As expected age, height, weight, body mass index and race were similar between the OSA and control groups since these variables were used to match the groups (Table 1). Table 2 shows that minute ventilation (P= 0.07), tidal volume, breathing frequency, 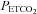 and
and 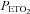 during the initial baseline period (i.e. while participants were breathing room air) were not significantly different between the OSA and control participants following administration of either the antioxidant or the placebo cocktail. Likewise, there were no differences in the measured variables during the baseline period of the antioxidant and placebo trials within each group. Minute ventilation (P= 0.13), breathing frequency,
during the initial baseline period (i.e. while participants were breathing room air) were not significantly different between the OSA and control participants following administration of either the antioxidant or the placebo cocktail. Likewise, there were no differences in the measured variables during the baseline period of the antioxidant and placebo trials within each group. Minute ventilation (P= 0.13), breathing frequency, 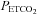 and
and 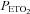 were similar between groups throughout the second baseline period when carbon dioxide was sustained 4 mmHg above baseline (Figs 1–3 and Table 3). However, tidal volume during the placebo trial was significantly greater in the OSA group compared to control (P= 0.04) (Table 3). The measured variables did not differ between the antioxidant and placebo trials during the second baseline period within each group (Figs 1–3 and Table 3).
were similar between groups throughout the second baseline period when carbon dioxide was sustained 4 mmHg above baseline (Figs 1–3 and Table 3). However, tidal volume during the placebo trial was significantly greater in the OSA group compared to control (P= 0.04) (Table 3). The measured variables did not differ between the antioxidant and placebo trials during the second baseline period within each group (Figs 1–3 and Table 3).
Table 1.
Anthropometric and apnea severity indices
| OSA | Control | |
|---|---|---|
| Age (years) | 28.2 ± 1.4 | 27.8 ± 1.4 |
| Height (cm) | 177.5 ± 1.9 | 178.2 ± 1.7 |
| Weight (kg) | 86.9 ± 2.7 | 82.8 ± 3.0 |
| Body mass index (kg m−2) | 27.3 ± 0.7 | 26.3 ± 0.5 |
| Apnoea index (events h−1) | 28.8 ± 6.7* | 0.5 ± 0.2 |
| Race | 3 Caucasian, 3 Asian, 6 African American, 1 Hispanic | 3 Caucasian, 3 Asian, 6 African American, 1 Hispanic |
P≤ 0.0001
Table 2.
Baseline measures of respiratory parameters
| OSA |
Control |
|||
|---|---|---|---|---|
| Antioxidant | Placebo | Antioxidant | Placebo | |
| Minute ventilation (l min−1) | 11.8 ± 0.5* | 13.0 ± 1.5* | 10.7 ± 0.3 | 11.2 ± 0.4 |
| Tidal volume (ml) | 890.6 ± 67.4 | 954.2 ± 93.7 | 801.7 ± 59.3 | 836.5 ± 72.0 |
| Frequency (breaths min−1) | 14.1 ± 0.9 | 14.3 ± 0.8 | 14.3 ± 0.9 | 14.5 ± 0.9 |
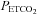 (mmHg) (mmHg) |
40.4 ± 0.8 | 40.0 ± 0.7 | 39.8 ± 0.6 | 39.7 ± 0.7 |
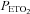 (mmHg) (mmHg) |
98.2 ± 1.1 | 98.1 ± 1.1 | 98.8 ± 0.9 | 97.9 ± 1.0 |
P= 0.07
Table 3.
Measures of respiratory parameters before, during and after exposure to intermittent hypoxia
| OSA |
Control |
|||
|---|---|---|---|---|
| Antioxidant | Placebo | Antioxidant | Placebo | |
| Ventilation | ||||
| Baseline (l min−1) | 16.8 ± 0.9 | 18.8 ± 2.1 | 14.9 ± 2.5 | 14.8 ± 0.5 |
| Initial hypoxic episode (l min−1) | 29.0 ± 1.7 | 31.6 ± 3.0§ | 24.3 ± 1.0 | 24.5 ± 0.9 |
| Final hypoxic episode (l min−1) | 30.6 ± 2.0 | 36.9 ± 2.8‡§† | 26.9 ± 1.3‡ | 27.7 ± 2.2‡ |
| End-recovery (l min−1) | 23.3 ± 1.4* | 29.3 ± 2.8*§† | 20.2 ± 1.3* | 20.4 ± 1.3* |
| End-recovery (fraction of baseline) | 1.40 ± 0.07* | 1.59 ± 0.07*§† | 1.36 ± 0.06* | 1.39 ± 0.09* |
| Tidal volume | ||||
| Baseline (ml) | 1136.4 ± 77.6 | 1298.5 ± 114.4§ | 1006.6 ± 52.6 | 1030.8 ± 52.1 |
| Initial hypoxic episode (ml) | 1849.7 ±136.4 | 2089.6 ± 200.6§ | 1600.8 ± 78.9 | 1676.7 ± 102.9 |
| Final hypoxic episode (ml) | 1858.5 ± 135.0 | 2138.1 ± 182.0§ | 1708.3 ± 97.8‡ | 1626.9 ± 71.0 |
| End-recovery (ml) | 1452.3 ± 107.8* | 1672.7 ± 152.1*§ | 1360.8 ± 98.1* | 1339.7 ± 80.2* |
| End-recovery (fraction of baseline) | 1.30 ± 0.07* | 1.30 ± 0.05* | 1.34 ± 0.06* | 1.31 ± 0.07* |
| Breathing frequency | ||||
| Baseline (breaths min−1) | 15.3 ± 0.7 | 15.0 ± 0.9 | 15.4 ± 0.8 | 15.4 ± 0.8 |
| Initial hypoxic episode (breaths min−1) | 16.3 ± 0.8 | 15.7 ± 0.8 | 15.8 ± 1.0 | 15.7 ± 1.2 |
| Final hypoxic episode (breaths min−1) | 17.1 ± 1.1 | 18.5 ± 1.3‡† | 16.6 ± 1.4‡ | 18.2 ± 2.3‡ |
| End-recovery (breaths min−1) | 17.0 ± 1.1* | 18.3 ± 1.1*† | 15.5 ± 0.9 | 16.2 ± 1.1 |
| End-recovery (fraction of baseline) | 1.10 ± 0.05(P= 0.07)* | 1.24 ± 0.06*§† | 1.02 ± 0.04 | 1.05 ± 0.03 |
Significantly different from baseline;
significantly different from control;
significantly different from antioxidant trial;
significantly different from initial hypoxic episode.
Progressive augmentation of minute ventilation
Following administration of the placebo cocktail minute ventilation progressively increased from the initial to the final hypoxic episode in both the OSA (P < 0.0001) and control participants (P < 0.0001), which is characteristic of the PA phenomenon (Fig. 1 and Table 3). The progressive increase in minute ventilation was primarily the consequence of a gradual increase in breathing frequency, which was greater during the last compared to the initial hypoxic episode in both the OSA and control groups (P= 0.04) (Table 3). Minute ventilation during the last hypoxic episode was significantly greater in the OSA participants compared to control (P= 0.004) (Fig. 1 and Table 3). This difference was due primarily to tidal volume, which was greater in the OSA group compared to control during the last hypoxic episode (P= 0.02). The difference in minute ventilation observed between groups was not the result of a variation in stimulus intensity since 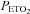 was maintained at a similar value during each hypoxic episode (Fig. 1). Likewise, the differences in minute ventilation observed between groups during the final episode existed even though
was maintained at a similar value during each hypoxic episode (Fig. 1). Likewise, the differences in minute ventilation observed between groups during the final episode existed even though 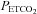 values measured within and between groups did not vary throughout the intermittent hypoxia protocol (Fig. 1).
values measured within and between groups did not vary throughout the intermittent hypoxia protocol (Fig. 1).
In contrast to the findings obtained during the placebo trial, minute ventilation during the last hypoxic episode was similar to values measured during the initial hypoxic episode following administration of the antioxidant cocktail in the OSA group (P= 0.13) (Fig. 2 and Table 3). Consequently, minute ventilation during the final hypoxic episode was significantly less during the antioxidant trial compared to placebo in the OSA group (P= 0.04) (Fig. 2 and Table 3). The reduction in minute ventilation was replicated in the breathing frequency measure, which was significantly less during the antioxidant trial compared to the placebo trial in the OSA group (P= 0.05) (Table 3). Differences in minute ventilation following antioxidant and placebo administration were independent of stimulus intensity since 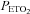 and
and 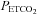 were similar during each hypoxic episode (Fig. 2). Progressive augmentation of minute ventilation was similar during the antioxidant and placebo trials in the control group (Fig. 3). Consequently, minute ventilation during the last hypoxic episode was equivalent between trials (Fig. 3). Likewise, Table 3 shows that minute ventilation (P= 0.11) and its components (i.e. tidal volume and breathing frequency) following antioxidant administration were similar between the OSA and control group during the last hypoxic episode.
were similar during each hypoxic episode (Fig. 2). Progressive augmentation of minute ventilation was similar during the antioxidant and placebo trials in the control group (Fig. 3). Consequently, minute ventilation during the last hypoxic episode was equivalent between trials (Fig. 3). Likewise, Table 3 shows that minute ventilation (P= 0.11) and its components (i.e. tidal volume and breathing frequency) following antioxidant administration were similar between the OSA and control group during the last hypoxic episode.
Figure 2. Measures of minute ventilation before, during and following intermittent hypoxia in OSA participants after administration of the placebo and antioxidant cocktail.

Average values for minute ventilation, end-tidal carbon dioxide partial pressure (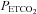 ) and end-tidal oxygen partial pressure (
) and end-tidal oxygen partial pressure (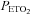 ) recorded from the last 5 min of baseline, the last 2 min of each episode of hypoxia (indicated by division marks on the x-axis), the last 2 min of each normoxic period that separated the hypoxic episodes, and the 30 min post-stimuli recovery period which is divided into six 5-min segments. The data were collected following administration of the placebo (filled circles) and antioxidant (open circles) cocktail in the OSA participants. Note that a progressive increase in minute ventilation occurred during exposure to intermittent hypoxia following administration of the placebo cocktail. Likewise, notice that an elevated level of ventilation was sustained during end-recovery compared to baseline during both the placebo and antioxidant trials. Lastly, notice that minute ventilation during the last hypoxic episode and end-recovery was greater during the placebo compared to the antioxidant trial. ‡Significantly different from the initial hypoxic episode; *significantly different from baseline; §significantly different from antioxidant trial; †significantly different from the initial 5 min end-recovery segment.
) recorded from the last 5 min of baseline, the last 2 min of each episode of hypoxia (indicated by division marks on the x-axis), the last 2 min of each normoxic period that separated the hypoxic episodes, and the 30 min post-stimuli recovery period which is divided into six 5-min segments. The data were collected following administration of the placebo (filled circles) and antioxidant (open circles) cocktail in the OSA participants. Note that a progressive increase in minute ventilation occurred during exposure to intermittent hypoxia following administration of the placebo cocktail. Likewise, notice that an elevated level of ventilation was sustained during end-recovery compared to baseline during both the placebo and antioxidant trials. Lastly, notice that minute ventilation during the last hypoxic episode and end-recovery was greater during the placebo compared to the antioxidant trial. ‡Significantly different from the initial hypoxic episode; *significantly different from baseline; §significantly different from antioxidant trial; †significantly different from the initial 5 min end-recovery segment.
The central chemoreflex and central and peripheral chemoreflex sensitivities measured using the modified rebreathing method after exposure to intermittent hypoxia are shown in Fig. 4 for the OSA and control groups. The data show that the ventilatory sensitivity to hypercapnia in the presence of sustained hyperoxia, which we assumed was a measure of central chemoreflex sensitivity, was not different between the OSA and control groups during the placebo trial (Fig. 4A). Likewise, central chemoreflex sensitivity following exposure to intermittent hypoxia was similar during the placebo and antioxidant trials for both the OSA and control participants (Fig. 4A). In contrast, the ventilatory sensitivity to hypercapnia in the presence of sustained hypoxia, which we assumed was a measure of central and peripheral chemoreflex sensitivity, was significantly greater in the OSA participants compared to control for the placebo trial (P= 0.01) (Fig. 4B). Following administration of the antioxidant cocktail, central and peripheral chemoreflex sensitivity was reduced compared to placebo in the OSA group (P= 0.02) (Fig. 4B). Conversely, central and peripheral chemoreflex sensitivity was similar during the placebo and antioxidant trials in the control group. Similarly, no differences in central chemoreflex or central and peripheral chemoreflex threshold were observed between the OSA and control groups, or between trials (placebo vs. antioxidant) within each group.
Figure 4. Chemoreflex sensitivity measures following intermittent hypoxia in OSA and control participants after administration of the placebo and antioxidant cocktail.

Bar graphs showing the average chemoreflex sensitivity obtained from the rebreathing trials completed while oxygen was maintained at 150 mmHg (central chemoreflex, A) or 50 mmHg (central and peripheral chemoreflex, B) after exposure to intermittent hypoxia during the placebo (filled bars) and antioxidant trials (open bars) in the OSA and control participants. Note that central and peripheral chemoreflex sensitivity during the placebo trial was significantly greater in the OSA participants compared to control. In addition, note that central and peripheral chemoreflex sensitivity following antioxidant administration was significantly reduced compared to placebo. A raw example of the reduction in central and peripheral chemoreflex sensitivity (i.e. a reduction in the slope of the minute ventilation response to carbon dioxide in the presence of hypoxia) in one OSA participant following antioxidant administration is shown in the inset figure. *Significantly different from placebo trial; §significantly different from control.
Ventilatory long-term facilitation
Figure 1 shows that minute ventilation gradually increased during the recovery periods that were interspersed between the hypoxic episodes and remained sustained above baseline levels during the end-recovery period in both the OSA (P < 0.0001) and control groups (P= 0.001), which is characteristic of vLTF. Table 3 indicates that the increase during end-recovery was evident in both the absolute and standardized measures of minute ventilation (P < 0.0001) and tidal volume (P < 0.0001) in both groups. Conversely, relative to baseline measures, increases in the absolute and standardized measures of breathing frequency during end-recovery were evident only in the OSA group (P < 0.0001). Although vLTF was evident in both groups, the absolute (P= 0.002) (Fig. 1) and standardized measures of ventilation (P= 0.02) (Table 3) were greater in the OSA participants compared to control (P= 0.002) (Fig. 1). The difference observed between groups was primarily a consequence of increases in breathing frequency (P < 0.0001) as indicated by the standardized values shown in Table 3. In addition to differences in magnitude between the OSA and control groups, the appearance of vLTF also differed during the end-recovery period. Minute ventilation progressively increased in the OSA group (Fig. 1); consequently, ventilation recorded during the last 5 min of the end-recovery period was greater than the values measured during the initial 5 min of recovery (P= 0.001). In contrast, the progressive increase in minute ventilation observed in the OSA participants during the end-recovery period was absent in the control group. The increase in minute ventilation during end-recovery was accompanied by a sustained increase in 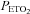 in both groups (P= 0.001). Moreover, the
in both groups (P= 0.001). Moreover, the 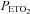 in the OSA group tended to be greater compared to control although this difference was not statistically significant (P= 0.07) (Fig. 1). The presence of vLTF in both groups and the enhanced magnitude of vLTF in the OSA group during end-recovery were independent of
in the OSA group tended to be greater compared to control although this difference was not statistically significant (P= 0.07) (Fig. 1). The presence of vLTF in both groups and the enhanced magnitude of vLTF in the OSA group during end-recovery were independent of 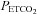 , which by design was sustained at a constant level throughout the intermittent hypoxia protocol in both the OSA and control groups (Fig. 1).
, which by design was sustained at a constant level throughout the intermittent hypoxia protocol in both the OSA and control groups (Fig. 1).
Similar to observations made during the placebo trial, minute ventilation during the end-recovery period remained elevated compared to baseline in the OSA participants following antioxidant administration (P= 0.001) (Fig. 2 and Table 3). However, the magnitude of the increase was less than that observed during the placebo trial. Consequently, minute ventilation during end-recovery was less following administration of the antioxidant cocktail compared to placebo in the OSA participants (P= 0.05) (Fig. 2 and Table 3). The reduction in minute ventilation during the antioxidant trial was due primarily to a diminution in the absolute (P= 0.01) and standardized measures (P= 0.0001) of breathing frequency (Table 3). The reduction in minute ventilation following antioxidant administration did not manifest itself in a concomitant reduction in 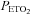 . Moreover, the reduction in minute ventilation occurred even though
. Moreover, the reduction in minute ventilation occurred even though 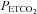 values were similar to measures obtained following administration of the placebo cocktail (Fig. 2). Figure 3 and Table 3 show that the sustained increase in minute ventilation and tidal volume during end-recovery compared to baseline was similar during the antioxidant and placebo trials in the control participants. Likewise, minute ventilation, tidal volume and breathing frequency following antioxidant administration were similar between the OSA and control group during end-recovery.
values were similar to measures obtained following administration of the placebo cocktail (Fig. 2). Figure 3 and Table 3 show that the sustained increase in minute ventilation and tidal volume during end-recovery compared to baseline was similar during the antioxidant and placebo trials in the control participants. Likewise, minute ventilation, tidal volume and breathing frequency following antioxidant administration were similar between the OSA and control group during end-recovery.
Measures of oxidative stress (8-isoprostane)
Measures of 8-isoprostane in the OSA participants were significantly greater compared to control before exposure to intermittent hypoxia during the placebo trial (P= 0.01) (Fig. 5). After exposure to intermittent hypoxia measures of 8-isoprostane increased significantly in the OSA group compared to baseline (P= 0.01) (Fig. 5). In contrast, measures of 8-isoprostane did not increase significantly in the control participants following intermittent hypoxia (Fig. 5). 8-Isoprostane measures were reduced during the antioxidant compared to the placebo trial in the OSA (1.04 ± 0.18 vs. 1.39 ± 0.12 fraction of baseline, P≤ 0.04) but not in the control participants (1.08 ± 0.14 vs. 1.19 ± 0.15 fraction of baseline) when the data were expressed as a fraction of baseline but not when the data were expressed in absolute values (OSA antioxidant trial: before intermittent hypoxia 48.4 ± 15.5 pg ml−1vs. after 33.9 ± 5.1; control antioxidant trial: before intermittent hypoxia 26.2 ± 4.5 pg ml−1vs. after 22.5 ± 2.9; see Fig. 5 for OSA and control placebo data).
Figure 5. Measures of 8-isoprostane obtained from OSA and control participants before and after intermittent hypoxia during the placebo trial.

Bar graphs showing average measures of 8-isoprostane obtained from the OSA and control participants before and after intermittent hypoxia during the placebo trial. Note that 8-isoprostane measures increased in the OSA participants but not in the control participants after (open bars) compared to before (filled bars) intermittent hypoxia. Additionally, note that the measures of 8-isoprostane were greater in the OSA compared to the control participants both before and after intermittent hypoxia. *Significantly different from before intermittent hypoxia; §significantly different from control.
Discussion
The novel findings of our study are threefold. First, we confirmed that PA of minute ventilation and vLTF can be induced consistently in participants with sleep apnoea during wakefulness. We also showed that the minute ventilation response during and following exposure to intermittent hypoxia is greater in sleep apnoea participants compared to control. Lastly, we demonstrated that the magnitude of the increase in minute ventilation during and following exposure to intermittent hypoxia was diminished after administration of an antioxidant cocktail.
Progressive augmentation and ventilatory LTF (placebo trial)
During the placebo trial, PA and vLTF were evident in both the sleep apnoea and control participants. In both groups, the increase in minute ventilation during the final hypoxic episode compared to the initial episode was evidence of the presence of PA, while the sustained increase in ventilation during the end-recovery period compared to baseline was a reflection of the existence of vLTF. Progressive augmentation and vLTF has been observed previously in healthy males and females during wakefulness (Harris et al. 2006; Wadhwa et al. 2008). On the other hand, this is the first time to our knowledge that both PA and vLTF have been observed during exposure to intermittent hypoxia in individuals with sleep apnoea during wakefulness, despite previous attempts on our part to elicit the phenomenon (Khodadadeh et al. 2006). The absence of PA and vLTF previously was likely to have been the consequence of the carbon dioxide level sustained during and following exposure to intermittent hypoxia. We and others have shown that PA and vLTF are not evident in the presence of hypo- or isocapnia (McEvoy et al. 1996; Jordan et al. 2002; Mateika et al. 2004; Morelli et al. 2004; Khodadadeh et al. 2006) but is clearly evident when 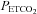 is elevated slightly above resting values in healthy adults (Harris et al. 2006; Wadhwa et al. 2008). Thus, sustaining
is elevated slightly above resting values in healthy adults (Harris et al. 2006; Wadhwa et al. 2008). Thus, sustaining 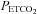 above baseline in the present study ensured the expression of PA and vLTF in the sleep apnoea participants, which was lacking when
above baseline in the present study ensured the expression of PA and vLTF in the sleep apnoea participants, which was lacking when 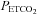 was sustained previously at hypocapnic levels (Khodadadeh et al. 2006). The expression of PA and vLTF in both healthy and apnoeic humans during wakefulness adds to an extensive list of species, including goats (Turner & Mitchell, 1997), rats (McGuire et al. 2002) and ducks (Mitchell et al. 2001b), in which PA (Turner & Mitchell, 1997; Mitchell et al. 2001b) and vLTF have been observed during wakefulness (Turner & Mitchell, 1997; Mitchell et al. 2001b; McGuire et al. 2002; Rey et al. 2004). Likewise, PA and LTF of minute ventilation (Sokolowska & Pokorski, 2006), phrenic nerve activity (Fuller, 2005), hypoglossal nerve or genioglossus muscle activity (Fuller, 2005) and carotid sensory nerve activity (Peng et al. 2003; Peng & Prabhakar, 2003; Peng & Prabhakar, 2004) have also been observed in anaesthetized preparations.
was sustained previously at hypocapnic levels (Khodadadeh et al. 2006). The expression of PA and vLTF in both healthy and apnoeic humans during wakefulness adds to an extensive list of species, including goats (Turner & Mitchell, 1997), rats (McGuire et al. 2002) and ducks (Mitchell et al. 2001b), in which PA (Turner & Mitchell, 1997; Mitchell et al. 2001b) and vLTF have been observed during wakefulness (Turner & Mitchell, 1997; Mitchell et al. 2001b; McGuire et al. 2002; Rey et al. 2004). Likewise, PA and LTF of minute ventilation (Sokolowska & Pokorski, 2006), phrenic nerve activity (Fuller, 2005), hypoglossal nerve or genioglossus muscle activity (Fuller, 2005) and carotid sensory nerve activity (Peng et al. 2003; Peng & Prabhakar, 2003; Peng & Prabhakar, 2004) have also been observed in anaesthetized preparations.
Although PA and vLTF were evident in both groups, the magnitude of PA and vLTF was greater in the sleep apnoea participants compared to control. As a consequence of the enhanced responses (i.e. PA and vLTF), minute ventilation was greater during the final hypoxic episode and during the end-recovery period in the sleep apnoea participants compared to control. The prominent PA observed in the sleep apnoea participants during the intermittent hypoxia protocol was also evident in the results obtained from the rebreathing studies completed 30 min following exposure. The ventilatory sensitivity to hypercapnia in the presence of hypoxia was increased in the sleep apnoea participants compared to control. Conversely, the ventilatory sensitivity to hypercapnia in the presence of hyperoxia was not significantly different between the groups.
A number of factors might account for the increased level of PA and vLTF observed in the sleep apnoea participants. However, both groups were closely matched for a variety of anthropometric variables, and the chemical stimuli (i.e. 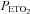 and
and 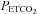 ) were similar within and between each group throughout the intermittent hypoxia protocols. Thus, we propose that chronic nightly exposure to intermittent hypoxia, a hallmark of sleep apnoea, might be responsible for the enhanced PA and vLTF observed in the sleep apnoea participants. This suggestion is supported by a variety of studies which have shown that chronic exposure to intermittent hypoxia enhances the magnitude of the ventilatory response to hypoxia in awake cats (Rey et al. 2004) and vLTF in awake rats (McGuire et al. 2003, 2004; McGuire & Ling, 2005), as well as, phrenic nerve LTF in anaesthetized rats (Ling et al. 2001; Peng & Prabhakar, 2003). Likewise, Peng and colleagues (Peng et al. 2003; Peng & Prabhakar, 2004) showed that a one-time exposure to intermittent hypoxia did not elicit carotid sensory nerve PA or LTF in rats; however the same stimulus induced PA (Peng & Prabhakar, 2004) and LTF (Peng et al. 2003) following chronic exposure to intermittent hypoxia. Studies completed in healthy humans (Serebrovskaya et al. 1999; Foster et al. 2005; Lusina et al. 2006; Koehle et al. 2007), in addition to findings obtained from sleep apnoea participants published in abstract form (Gerst et al. 2009), indicate that exposure to intermittent hypoxia ranging from 1 to 2 weeks enhances PA (Serebrovskaya et al. 1999; Lusina et al. 2006; Koehle et al. 2007; Gerst et al. 2009) and vLTF (Foster et al. 2005; Gerst et al. 2009). Thus, the available evidence indicates that the difference in the magnitude of PA and vLTF between the sleep apnoea and control participants we observed may be a consequence long-term nightly exposure to intermittent hypoxia typically experienced by those individuals with sleep apnoea.
) were similar within and between each group throughout the intermittent hypoxia protocols. Thus, we propose that chronic nightly exposure to intermittent hypoxia, a hallmark of sleep apnoea, might be responsible for the enhanced PA and vLTF observed in the sleep apnoea participants. This suggestion is supported by a variety of studies which have shown that chronic exposure to intermittent hypoxia enhances the magnitude of the ventilatory response to hypoxia in awake cats (Rey et al. 2004) and vLTF in awake rats (McGuire et al. 2003, 2004; McGuire & Ling, 2005), as well as, phrenic nerve LTF in anaesthetized rats (Ling et al. 2001; Peng & Prabhakar, 2003). Likewise, Peng and colleagues (Peng et al. 2003; Peng & Prabhakar, 2004) showed that a one-time exposure to intermittent hypoxia did not elicit carotid sensory nerve PA or LTF in rats; however the same stimulus induced PA (Peng & Prabhakar, 2004) and LTF (Peng et al. 2003) following chronic exposure to intermittent hypoxia. Studies completed in healthy humans (Serebrovskaya et al. 1999; Foster et al. 2005; Lusina et al. 2006; Koehle et al. 2007), in addition to findings obtained from sleep apnoea participants published in abstract form (Gerst et al. 2009), indicate that exposure to intermittent hypoxia ranging from 1 to 2 weeks enhances PA (Serebrovskaya et al. 1999; Lusina et al. 2006; Koehle et al. 2007; Gerst et al. 2009) and vLTF (Foster et al. 2005; Gerst et al. 2009). Thus, the available evidence indicates that the difference in the magnitude of PA and vLTF between the sleep apnoea and control participants we observed may be a consequence long-term nightly exposure to intermittent hypoxia typically experienced by those individuals with sleep apnoea.
If our hypothesis is correct, enhanced measures of resting minute ventilation, indicative of the presence of vLTF, and/or enhanced measures of the ventilatory response to a brief hypoxic exposure (e.g. 3 min) might be evident consistently in individuals with sleep apnoea. However, studies which have reported resting minute ventilation indicate that this measure is similar between OSA and matched controls (Narkiewicz et al. 1999; Osanai et al. 1999). Moreover, some studies have indicated that the ventilatory response to a brief hypoxic exposure is enhanced in OSA participants (Narkiewicz et al. 1999; Tun et al. 2000), while others have reported the opposite finding (i.e. the hypoxic response is depressed) (Lin, 1994; Osanai et al. 1999). Reasons for the divergent findings have not been fully elucidated. However, we have hypothesized previously (Mateika & Narwani, 2009) that chronic exposure to mild hypoxia might enhance vLTF and the acute hypoxic response, while chronic exposure to severe hypoxia might blunt the response, in a manner observed in individuals exposed to hypoxia at high altitude for short (i.e. days) or long periods of time (i.e. years) (Weil, 1986). Thus, factors such as age (i.e. duration of exposure to hypoxia), apnoea/hypopnoea index (i.e. frequency of hypoxic exposure) and degree of oxygen desaturation at night (i.e. intensity of hypoxaemia) might impact on the acute response measured in individuals with OSA. If this is the case, enhancement of vLTF and/or ventilatory sensitivity to hypoxia might be particularly evident in young individuals who experience mild-to-moderate forms of apnoea. Conversely, exposure to CIH might blunt these responses in individuals who are older and experience severe hypoxaemia throughout a given night.
Potential mechanisms responsible for progressive augmentation and ventilatory long-term facilitation
It is tempting to speculate that the site of origin of PA and vLTF observed in our participants was the carotid bodies, since these receptors are the primary detectors of hypoxia (Duffin, 2007) and the ventilatory response to hypoxia both during and after (i.e. rebreathing trials) exposure to intermittent hypoxia was enhanced. Moreover, PA and LTF of carotid sensory nerve activity have been recorded in rats. Nevertheless, further investigation is required to substantiate the site of origin of PA and vLTF in humans.
Independent of the site of origin, we hypothesize that the accumulation of reactive oxygen species might be the mechanistic link between chronic intermittent hypoxia and the enhanced PA and vLTF observed in the sleep apnoea participants. This postulation is supported by findings which showed that chronic exposure to intermittent hypoxia led to an enhanced in vivo and ex vivo carotid body sensory response to hypoxia (Peng & Prabhakar, 2004), as well as carotid sensory nerve LTF (Peng et al. 2003). The augmented hypoxic carotid sensory response and carotid sensory LTF were abolished in a group of rats pre-treated with a potent scavenger of superoxide anions (Peng et al. 2003; Peng & Prabhakar, 2004). Peng and colleagues (Peng & Prabhakar, 2003) also demonstrated that augmentation of phrenic LTF induced by exposure to chronic intermittent hypoxia was abolished following pre-treatment with a superoxide anion scavenger. At present it is not established whether a reactive oxygen species has a role in initiating PA in naive animals not previously exposed to intermittent hypoxia. However, Wilkerson et al. (2008) demonstrated that intravenous administration of 10 mg of a superoxide anion scavenger to anaesthetized, vagotomized and ventilated rats, before exposure to acute intermittent hypoxia, abolished LTF of both phrenic and hypoglossal nerve activity.
One of the major sources of reactive oxygen species might be NADPH oxidase, since recent findings have shown that NADPH oxidase is necessary for chronic intermittent hypoxia induced carotid sensory LTF (Peng et al. 2006, 2009) and acute intermittent hypoxia induced phrenic LTF (Macfarlane et al. 2009). Additionally, the link between reactive oxygen species generated by NADPH oxidase and the manifestation of LTF may be via the suppression of protein phosphatases (Wilkerson et al. 2007; Macfarlane et al. 2008). Indeed, it has been recently demonstrated that the restraint of phosphatases via the administration of okadaic acid induces phrenic LTF following exposure to sustained hypoxia, a stimulus that typically does not initiate LTF (Wilkerson et al. 2008).
In the present study we did not attain measures of oxygen radicals, and thus we do not have direct evidence that levels of reactive oxygen species were altered in response to intermittent hypoxia or antioxidant administration in the OSA participants. However, we did obtain measures of 8-isoprostane, an indicator of oxidative stress. Whether this biomarker was the most appropriate to detect the presence of oxidative stress remains to be determined. We selected the biomarker because previous studies (Carpagnano et al. 2003; Lavie et al. 2004) reported that 8-isoprostane levels are elevated in OSA patients compared to healthy controls. Although our findings using this measure add support to our ventilation data, we did observe some variability in the measure, particularly in the baseline measures of two OSA participants in which 8-isoprostane measures were significantly higher during the antioxidant trial compared to the placebo trial. We have no reason to believe the variability is due to experimental error. Although the reason for the variability was not established, diet or the degree of exposure to hypoxia on the night(s) prior to each trial, which we did not control, might account for it.
Nevertheless, we demonstrated that 8-isoprostane, an indicator of oxidative stress, was elevated in the sleep apnoea group compared to control prior to exposure to intermittent hypoxia during the placebo trial. This result is similar to previous findings (Carpagnano et al. 2003; Lavie et al. 2004). Likewise, the levels of 8-isoprostane increased following exposure to the intermittent hypoxia protocol. We speculate that the increase in oxidative stress measured under baseline conditions was a consequence of nightly exposure to intermittent hypoxia naturally experienced by the sleep apnoea participants, while additional increases in oxidative stress were a consequence of the experimentally induced intermittent hypoxia. Our results also showed that 8-isoprostane levels were reduced following administration of the antioxidant cocktail in the OSA participants. In conjunction with the decrease in 8-isoprostane we observed a concurrent decrease in PA and vLTF in the sleep apnoea participants. Consequently, minute ventilation measured from the sleep apnoea participants during the final hypoxic episode and end-recovery period of the antioxidant trial was less than the response measured during the placebo trial. On the other hand the values obtained from the sleep apnoea participants during the antioxidant trial were similar to those obtained from the control participants during the antioxidant and placebo trials.
In contrast to the sleep apnoea participants, the level of oxidative stress did not increase significantly following acute intermittent hypoxia in the control population during the placebo trial. Thus, it is possible that the protocol that we employed only induced significant oxidative stress in those individuals exposed previously to intermittent hypoxia. We speculate that the absence of oxidative stress in the control group may have been due to the presence of an adequate level of endogenous antioxidants. If this speculation is correct, the administration of exogenous antioxidants might not be expected to impact significantly on PA or vLTF in individuals that are exposed to an intermittent hypoxia protocol of moderate intensity and short duration. This appears to be the case for our control participants since the level of ventilation during the final hypoxic episodes and throughout end-recovery was similar in both the placebo and antioxidant trials.
The presence of PA and vLTF in the control participants, the magnitude of which was seemingly unaffected by administration of the antioxidant cocktail, might indicate that other cellular mechanisms have a role in initiating and maintaining PA and vLTF independent of the accumulation of reactive oxygen species. It is well established that serotonin has a role in initiating PA of carotid sensory nerve activity (Peng et al. 2006), as well as phrenic nerve LTF (Ling, 2008). Moreover, there is evidence that this neuromodulator activates a series of cellular events that are responsible for the maintenance of LTF. For example, brain derived neurotrophic factor has a role in maintaining LTF (Baker-Herman et al. 2004) likely via activation of tyrosine kinase receptors and downstream signalling molecules including MAP kinases such as extracellular signal regulated kinases (ERK) 1 and 2 (Wilkerson & Mitchell, 2009). Although it is not established whether the maintenance of vLTF can occur independently of the suppression of protein phosphatases via reactive oxygen species, it is reasonable to hypothesize that the activation of MAP kinases may lead to the manifestation of LTF independent of the accumulation of these oxygen radicals. Thus, given that the mechanisms responsible for the manifestation of PA and LTF may be multifactorial it is not surprising that PA and vLTF were evident, albeit to a lesser degree, in the control participants despite evidence indicating the lack of accumulation of reactive oxygen species. As suggested the increased magnitude of PA and vLTF in the sleep apnoea group might reflect the impact that chronic intermittent hypoxia and reactive oxygen species has on PA and vLTF, while the presence of the two phenomena in the control group represents the role of other mechanisms outlined above in the initiation and manifestation of PA and vLTF.
Physiological significance
Our findings suggest that chronic exposure to intermittent hypoxia may enhance PA of the ventilatory response to hypoxia, as well as vLTF in humans. Moreover, we have provided evidence that oxidative stress might be the mechanistic link between exposure to chronic intermittent hypoxia and enhancement of PA and vLTF in humans. Lastly, our findings denote that administration of an antioxidant cocktail might prove to be effective in diminishing oxidative stress induced by exposure to intermittent hypoxia, thereby diminishing the magnitude of PA and vLTF. Given the findings of our study, it is possible that PA and vLTF are typically enhanced in individuals suffering from sleep apnoea, given that this condition is associated with exposure to chronic intermittent periods of hypoxia. Likewise, because alterations in the control of breathing are known to impact on the severity of sleep apnoea (Mateika & Narwani, 2009), it is likely that enhancement of these phenomena have a role in the overall degree of breathing (in)stability experienced during the night in individuals with OSA. Whether the contributions of these phenomena ultimately serve to promote or mitigate apnoea remains to be determined (Mateika & Narwani, 2009). Nevertheless, given that we have shown that administration of an antioxidant cocktail mitigates PA and vLTF, it is possible that such a cocktail might serve as adjunctive treatment for this condition, particularly if one or both of these phenomena contribute to the exacerbation of apnoea.
Acknowledgments
This work was supported by the National Heart, Lung and Blood Institute (R01HL085537). We thank Anita D'Souza, Magalie Anthouard and Gunjan Narwani for their contributions to data collection and analysis. We thank Timothy J. Hadden PhD for his advice and guidance on measuring oxidative stress.
Glossary
Abbreviations
- OSA
obstructive sleep apnoea
- PA
progressive augmentation
- vLTF
ventilatory long-term facilitation
Author contributions
D.S.L. contributed to the design of the study, analysis and interpretation of the data and drafting of the article. M.S.B. contributed to the conception and design of the study and revision of the article. J.H.M. contributed to the conception and design of the study, analysis and interpretation of data, drafting of the article and revising it critically for important intellectual content. All authors gave final approval of the version to be published.
References
- American Academy of Sleep Medicine. EEG arousals: scoring rules and examples: a preliminary report from the Sleep Disorders Atlas Task Force of the American Sleep Disorders Association. Sleep. 1992;15:173–184. [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ. 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest. 2003;124:1386–1392. doi: 10.1378/chest.124.4.1386. [DOI] [PubMed] [Google Scholar]
- Cummings KJ, Wilson RJ. Time-dependent modulation of carotid body afferent activity during and after intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1571–R1580. doi: 10.1152/ajpregu.00788.2004. [DOI] [PubMed] [Google Scholar]
- Duffin J. Measuring the ventilatory response to hypoxia. J Physiol. 2007;584:285–293. doi: 10.1113/jphysiol.2007.138883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant JR, Seals DR, Connell ML, Russell MJ, Lawson BR, Folian BJ, Donato AJ, Lesniewski LA. Voluntary wheel running restores endothelial function in conduit arteries of old mice: direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J Physiol. 2009;587:3271–3285. doi: 10.1113/jphysiol.2009.169771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster GE, McKenzie DC, Milsom WK, Sheel AW. Effects of two protocols of intermittent hypoxia on human ventilatory, cardiovascular and cerebral responses to hypoxia. J Physiol. 2005;567:689–699. doi: 10.1113/jphysiol.2005.091462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD. Episodic hypoxia induces long-term facilitation of neural drive to tongue protrudor and retractor muscles. J Appl Physiol. 2005;98:1761–1767. doi: 10.1152/japplphysiol.01142.2004. [DOI] [PubMed] [Google Scholar]
- Gerst DG, Lee DS, Badr MS, Qureshi T, Mateika JH. Impact of chronic intermittent hypoxia on ventilatory long term facilitation and the hypoxic ventilatory response in participants with sleep apnea. FASEB J. 2009;23:621.17. Meeting Abstract. [Google Scholar]
- Harris DP, Balasubramanian A, Badr MS, Mateika JH. Long-term facilitation of ventilation and genioglossus muscle activity is evident in the presence of elevated levels of carbon dioxide in awake humans. Am J Physiol Regul Integr Comp Physiol. 2006;29:R1111–R1119. doi: 10.1152/ajpregu.00896.2005. [DOI] [PubMed] [Google Scholar]
- Jablonski KL, Seals DR, Eskurza I, Monahan KD, Donato AJ. High-dose ascorbic acid infusion abolishes chronic vasoconstriction and restores resting leg blood flow in healthy older men. J Appl Physiol. 2007;103:1715–1721. doi: 10.1152/japplphysiol.00533.2007. [DOI] [PubMed] [Google Scholar]
- Jordan AS, Catcheside PG, O’Donoghue FJ, McEvoy RD. Long-term facilitation of ventilation is not present during wakefulness in healthy men or women. J Appl Physiol. 2002;93:2129–2136. doi: 10.1152/japplphysiol.00135.2002. [DOI] [PubMed] [Google Scholar]
- Khodadadeh B, Badr MS, Mateika JH. The ventilatory response to carbon dioxide and sustained hypoxia is enhanced after episodic hypoxia in OSA patients. Respir Physiol Neurobiol. 2006;150:122–134. doi: 10.1016/j.resp.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Koehle MS, Sheel AW, Milsom WK, McKenzie DC. Two patterns of daily hypoxic exposure and their effects on measures of chemosensitivity in humans. J Appl Physiol. 2007;103:1973–1978. doi: 10.1152/japplphysiol.00545.2007. [DOI] [PubMed] [Google Scholar]
- Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep. 2004;27:123–128. [PubMed] [Google Scholar]
- Lin CC. Effect of nasal CPAP on ventilatory drive in normocapnic and hypercapnic patients with obstructive sleep apnoea syndrome. Eur Respir J. 1994;7:2005–2010. [PubMed] [Google Scholar]
- Ling L. Serotonin and NMDA receptors in respiratory long-term facilitation. Respir Physiol Neurobiol. 2008;164:233–241. doi: 10.1016/j.resp.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusina SJ, Kennedy PM, Inglis JT, McKenzie DC, Ayas NT, Sheel AW. Long-term intermittent hypoxia increases sympathetic activity and chemosensitivity during acute hypoxia in humans. J Physiol. 2006;575:961–970. doi: 10.1113/jphysiol.2006.114660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane PM, Satriotomo I, Windelborn JA, Mitchell GS. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J Physiol. 2009;587:1931–1942. doi: 10.1113/jphysiol.2008.165597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane PM, Wilkerson JE, Lovett-Barr MR, Mitchell GS. Reactive oxygen species and respiratory plasticity following intermittent hypoxia. Respir Physiol Neurobiol. 2008;164:263–271. doi: 10.1016/j.resp.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Mateika JH, Mendello C, Obeid D, Badr MS. Peripheral chemoreflex responsiveness is increased at elevated levels of carbon dioxide after episodic hypoxia in awake humans. J Appl Physiol. 2004;96:1197–1205. doi: 10.1152/japplphysiol.00573.2003. [DOI] [PubMed] [Google Scholar]
- Mateika JH, Narwani G. Intermittent hypoxia and respiratory plasticity in humans and other animals: does exposure to intermittent hypoxia promote or mitigate sleep apnoea? Exp Physiol. 2009;94:279–296. doi: 10.1113/expphysiol.2008.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy RD, Popovic RM, Saunders NA, White DP. Effects of sustained and repetitive isocapnic hypoxia on ventilation and genioglossal and diaphragmatic EMGs. J Appl Physiol. 1996;81:866–875. doi: 10.1152/jappl.1996.81.2.866. [DOI] [PubMed] [Google Scholar]
- McGuire M, Ling L. Ventilatory long-term facilitation is greater in 1- vs. 2-mo-old awake rats. J Appl Physiol. 2005;98:1195–1201. doi: 10.1152/japplphysiol.00996.2004. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Effect of hypoxic episode number and severity on ventilatory long-term facilitation in awake rats. J Appl Physiol. 2002;93:2155–2161. doi: 10.1152/japplphysiol.00405.2002. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Chronic intermittent hypoxia enhances ventilatory long-term facilitation in awake rats. J Appl Physiol. 2003;95:1499–1508. doi: 10.1152/japplphysiol.00044.2003. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Serotonin receptor subtypes required for ventilatory long-term facilitation and its enhancement after chronic intermittent hypoxia in awake rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R334–R341. doi: 10.1152/ajpregu.00463.2003. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001a;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Powell FL, Hopkins SR, Milsom WK. Time domains of the hypoxic ventilatory response in awake ducks: episodic and continuous hypoxia. Respir Physiol. 2001b;124:117–128. doi: 10.1016/s0034-5687(00)00197-3. [DOI] [PubMed] [Google Scholar]
- Mohan RM, Amara CE, Cunningham DA, Duffin J. Measuring central-chemoreflex sensitivity in man: rebreathing and steady-state methods compared. Respir Physiol. 1999;115:23–33. doi: 10.1016/s0034-5687(99)00003-1. [DOI] [PubMed] [Google Scholar]
- Morelli C, Badr MS, Mateika JH. Ventilatory responses to carbon dioxide at low and high levels of oxygen are elevated after episodic hypoxia in men compared with women. J Appl Physiol. 2004;97:1673–1680. doi: 10.1152/japplphysiol.00541.2004. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Osanai S, Akiba Y, Fujiuchi S, Nakano H, Matsumoto H, Ohsaki Y, Kikuchi K. Depression of peripheral chemosensitivity by a dopaminergic mechanism in patients with obstructive sleep apnoea syndrome. Eur Respir J. 1999;13:418–423. doi: 10.1183/09031936.99.13241899. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci. 2009;29:4903–4910. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behaviour elicited by chronic intermittent hypoxia. J Appl Physiol. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol. 2006;576:289–295. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierchala LA, Mohammed AS, Grullon K, Mateika JH, Badr MS. Ventilatory long-term facilitation in non-snoring subjects during NREM sleep. Respir Physiol Neurobiol. 2008;160:259–266. doi: 10.1016/j.resp.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Kales AA. A Manual of Standardized Terminology, Techniques and Scoring System for Sleep Stages of Human Subjects. Washington, DC: National Institutes of Health; 1968. [Google Scholar]
- Rey S, Del RR, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebrovskaya TV, Swanson RJ, Karaban IN, Serebrovskaya ZA, Kolesnikova EE. Intermittent hypoxia alters hypoxic ventilatory responses. Fiziol Zh. 1999;45:9–18. [PubMed] [Google Scholar]
- Sokolowska B, Pokorski M. Ventilatory augmentation by acute intermittent hypoxia in the rabbit. J Physiol Pharmacol. 2006;57(Suppl 4):341–347. [PubMed] [Google Scholar]
- Teppema LJ, Bijl H, Romberg RR, Dahan A. Antioxidants reverse depression of the hypoxic ventilatory response by acetazolamide in man. J Physiol. 2006;572:849–856. doi: 10.1113/jphysiol.2005.104174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppema LJ, Nieuwenhuijs D, Sarton E, Romberg R, Olievier CN, Ward DS, Dahan A. Antioxidants prevent depression of the acute hypoxic ventilatory response by subanaesthetic halothane in men. J Physiol. 2002;544:931–938. doi: 10.1113/jphysiol.2002.025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tun Y, Hida W, Okabe S, Kikuchi Y, Kurosawa H, Tabata M, Shirato K. Effects of nasal continuous positive airway pressure on awake ventilatory responses to hypoxia and hypercapnia in patients with obstructive sleep apnea. Tohoku J Exp Med. 2000;190:157–168. doi: 10.1620/tjem.190.157. [DOI] [PubMed] [Google Scholar]
- Turner DL, Mitchell GS. Long-term facilitation of ventilation following repeated hypoxic episodes in awake goats. J Physiol. 1997;499:543–550. doi: 10.1113/jphysiol.1997.sp021947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa H, Gradinaru C, Gates GJ, Badr MS, Mateika JH. Impact of intermittent hypoxia on long-term facilitation of minute ventilation and heart rate variability in men and women: do sex differences exist? J Appl Physiol. 2008;104:1625–1633. doi: 10.1152/japplphysiol.01273.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil JV. Ventilatory control at high altitude. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, section 3, The Respiratory System, vol II, Control of Breathing, part 2. Bethesda, MD: American Physiological Society; 1986. pp. 703–728. [Google Scholar]
- Wilkerson JE, Macfarlane PM, Hoffman MS, Mitchell GS. Respiratory plasticity following intermittent hypoxia: roles of protein phosphatases and reactive oxygen species. Biochem Soc Trans. 2007;35:1269–1272. doi: 10.1042/BST0351269. [DOI] [PubMed] [Google Scholar]
- Wilkerson JE, Mitchell GS. Daily intermittent hypoxia augments spinal BDNF levels, ERK phosphorylation and respiratory long-term facilitation. Exp Neurol. 2009;217:116–123. doi: 10.1016/j.expneurol.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JE, Satriotomo I, Baker-Herman TL, Watters JJ, Mitchell GS. Okadaic acid-sensitive protein phosphatases constrain phrenic long-term facilitation after sustained hypoxia. J Neurosci. 2008;28:2949–2958. doi: 10.1523/JNEUROSCI.5539-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabka AG, Behan M, Mitchell GS. Selected contribution: Time-dependent hypoxic respiratory responses in female rats are influenced by age and by the estrus cycle. J Appl Physiol. 2001;91:2831–2838. doi: 10.1152/jappl.2001.91.6.2831. [DOI] [PubMed] [Google Scholar]