

Abstract
Many infectious diseases have emerged and circulated around the world with the development of human civilizations and global commerce. Anthrax, plague and tularemia are three such zoonotic diseases that have been intensely studied through genome characterization and phylogeographic analyses. A few highly fit genotypes within each of the causative species represent the vast majority of observed disease cases. Mutational and selective forces working together create highly adapted pathogens, but this has to be coupled with ecological opportunities for global expansion. This Review describes the distributions of the bacteria that cause anthrax, plague and tularemia and investigates the forces that created a clonal structure in both these species, and specific groups within these species.
Background
Even diseases that were once thought to be quite ancient appear, in fact, to have recently emerged, shaped by evolutionary and ecologic forces in the last ten thousand years. The evidence for their recent emergence invariably comes from molecular genetic analysis of the causative pathogens, which has been greatly aided by the availability of complete genomes sequences. Pathogen genomes are not simple entities but rather can be thought of as a composite of many different types of loci that evolve at very dissimilar rates and are subject to greatly differing selective forces1. As a result, observed mutation rates within a single organism can differ by at least six orders of magnitude. Some short tandemly-repeated sequences may mutate at 10-3 events per generation2, 3 in a genomic background in which the average nucleotide rate is only about 10-10 changes per generation4, 5. Because bacterial populations can be very large and represent a large number of generations, mutations, horizontal transfer, and recombination easily occur at rates fast enough (10-3 to 10-10 per nucleotide per cell division) to generate considerable genetic variation, perhaps even in minor subpopulations.
Selection may enhance locus diversity, suppress it, maintain it, or, in theory, have no effect, which would be considered neutral variation. Mutations almost always become fixed in populations at slower rates than the rates at which they are generated. The fixation or population substitution rate is greatly influenced by selection, pushing molecular evolution towards neutral genetic variation whenever possible. It is true that most variation will have a negative impact and be removed from the population by selection. However, some will remain that is neutral, slightly disadvantageous, or of little advantage in the pathogen's current niche. This genetic variation provides the potential to be highly adaptive in the future given the right opportunity. Thus, pathogen populations contain many individual genotypes possessing genetic adaptations that are possibly important for global expansion into novel niches. For clonal organisms, the lack of recombination means that single mutational changes can influence the entire genome, because if that locus is under selection, the rest of the loci will “hitchhike” along with the selected loci. Currently, molecular variation in all its forms can be precisely measured and modeled using ecological and evolutionary theory to understand past population dynamics of pathogens on multiple spatial scales, local through global.
Genetic diversity within a given pathogen species is rarely evenly distributed across its geographic range. This is because the most-fit genotypes are frequently the most geographically distributed despite being among the most recently derived lineages. The success of a genotype (increase in frequency) can be due to genetic-based adaptive advantages, or merely fortuitous stochastic events. For example, dispersal of recently emerged pathogens may be explosively accelerated by the presence of immunologically naïve and susceptible host species. In contrast, an adaptive radiation may be associated with host shifting facilitated by adaptive fortuitous mutational events or lateral transfer of novel adaptive genes to the pathogen. By definition a highly fit genotype is one that is able to reproduce more successfully than other genotypes. It is evident that highly fit clones become ecologically established in many global locations that contain very different biotic and abiotic environmental factors. As a result, environmental factors combine with dispersal abilities of each pathogen and time dependent evolutionary rates to determine the level of local and overall geographic differentiation. In recently emerged pathogens, phylogenetic patterns at global, regional and local scales show evidence of these forces if the appropriate genomic loci are examined.
Many different pathogenic bacteria are examples of highly fit clonal expansions, and generalization across species is possible to a certain extent. Three of the most highly studied bacterial examples are Bacillus anthracis (anthrax), Yersinia pestis (plague), and Francisella tularensis (tularemia). These zoonotic species are both continuing public health threats and the three most dreaded bacterial biological weapons agents. As with all zoonotic agents, these pathogens evolved, dispersed, and became ecologically established in animal populations in a manner that can be inferred from their present phylogeographic patterns. All three of these bacteria are relatively recently emerged pathogens with even more recently emerged highly fit clonal genotypes nested inside less fit lineages. In this review, we apply what we have learned from phylogeographic studies of B. anthracis, Y. pestis, and F. tularensis to understand the past transmission and phylogeographic patterns of bacterial diseases and how these events are shaped by pathogen ecology and humans. Historical transmission patterns reconstructed from analysis of the phylogeography of these three bacteria will lead to process insights enabling us to better predict the inevitable emergence of novel pathogens.
Bacillus anthracis
Although the geographic origin of anthrax has not been firmly established, it is clear that B. anthracis is derived from a B. cereus ancestor. This ancestor obtained two virulence plasmids and underwent a pleiotropic chromosomal mutation in a plcR transcriptional regulator gene6. The combination of these three evolutionary events in the appropriate genetic background made B. anthracis into a catastrophic animal disease that can be easily distinguished by its distinct pathology, ecology, and evolution from disease caused by B. cereus 6.
Ecologic and evolutionary patterns in B. anthracis are highly dependent upon its ability to form spores. The disease manifestation component of the transmission cycle appears to be strictly dependent on a mammalian host, and is coupled with an environmental reservoir component in which spores persist in soil in a quiescent state for a number of years. There may be cellular replication in the soil but this is controversial and not universally accepted7. Importantly, the stable spore stage allows long-range dispersal of the pathogen while “freezing”, or at the very least greatly slowing, its evolutionary progression. Anthrax is an acute disease, killing most hosts within days or weeks, thereby greatly limiting dispersal of the pathogen via the infected live host. Rather, long distance transmission primarily occurs once the host is dead via human mediated transport of contaminated animal products such as meat, hides, hair, and bones.
B. anthracis isolates from all parts of the globe can be highly similar in their genomic sequences8, which can make subtyping very difficult9, 10. However, once phylogenetically informative characters (e.g., single nucleotide polymorphisms, or SNPs) were identified, they proved to be very stable with almost no homoplasy (i.e., phylogenetic inconsistencies)11. The very low homoplasy level is evidence of a very recently emerged pathogen but also of a highly clonal propagation system. Among multiple whole genome sequences there is no evidence of recombination among lineages or with other species8. Reconstructing the global transmission patterns was achieved12 using rare, but highly stable, phylogenetic SNP characters discovered by comparing multiple whole genome sequences1.
There are three deeply rooted lineages within B. anthracis (groups A, B & C; Fig. 1), each with a different global distribution and importance to current disease incidence 8, 12,9, 10. The dominant clonal lineage is group A (∼90% of all known B. anthracis strains), which underwent a recent radiation (Fig. 1) and is now globally distributed12. Subclades within the A lineage are often locally successful but also may be widely distributed. For example, the infamous Ames clade (∼1%) is locally established in south Texas but is also well represented in China13. The Vollum clade (∼5%) also is widely distributed, as it has been commonly found in commodities worldwide, but its source population is uncertain. In strain collections, the most dominant A-subclades are western North American (WNA) and the trans Eurasian (TEA), which combined represent over 37% of all known B. anthracis strains12. Their genetic relationship is very close, with fewer than 100 SNPs separating these two groups14. The wide geographic distributions, prevalence, and short evolutionary separation among these A subclades suggest that they are highly fit clones that could owe their great success to either deterministic adaptive changes in their genomes, or stochastic forces. Although adaptive differences cannot be ruled out, obvious genomic and phenotypic differences among the A, B, and C lineages have not been identified. In contrast, molecular clock estimates associate the timing of the A radiation with major human activities, such as the domestication of animals, suggesting that the stochastic process of human mediated dispersal may explain the great success of this group12; sometimes chance is as important as genetic adaptation to the success of particular bacterial types. Together the B and C lineages account for just a small proportion of globally observed isolates. The B lineage is divided into two main subclades (B1, ∼6%; B2, ∼2%) found primarily in southern Africa 15 and Europe16, respectively, where they appear to be ecologically established. Because spores are still frequently dispersed in animal-derived commodities, identifying atypical outbreaks due to commerce is critical so these events can be distinguished from the overall ecologically established population structure. For example, B2 outbreaks are common in Europe16, 17 arguing that this subclade is ecologically established in this region. In contrast, a single B2 anthrax outbreak near San Jose, CA (USA) in 2001 may not be indicative of a well-established or widespread population and possibly reflects dispersal by human activity because other B2 isolates have not been found in this region. The most basal lineage, C, is very rare (∼0.2% of known isolates) and has only been observed in North America where sampling has been very intense12.
Figure 1.
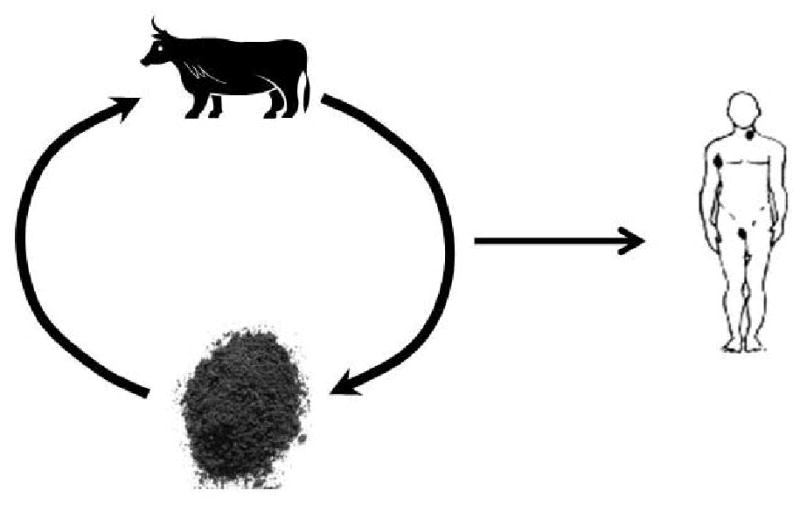
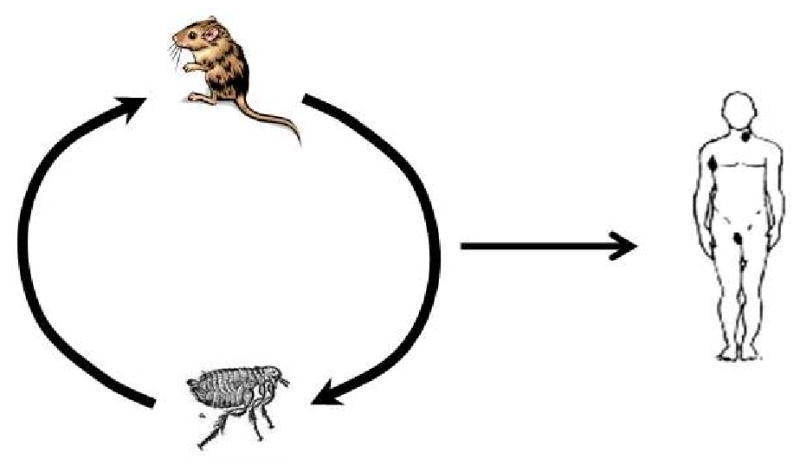
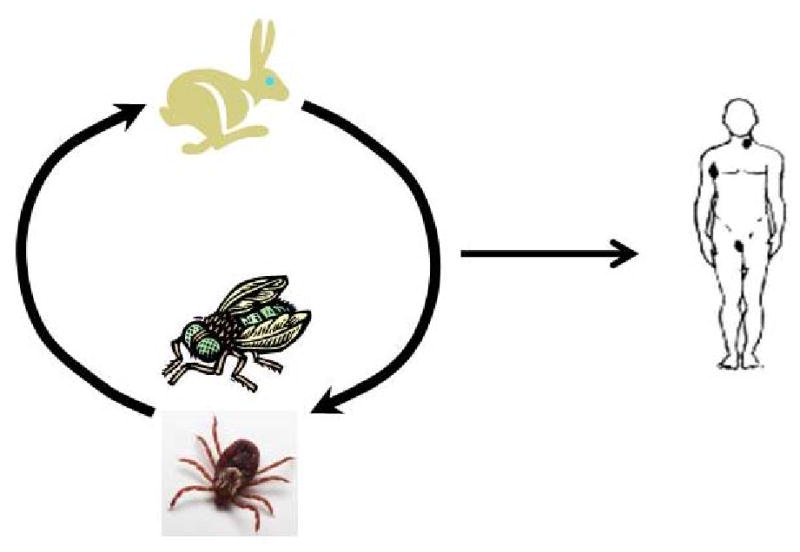
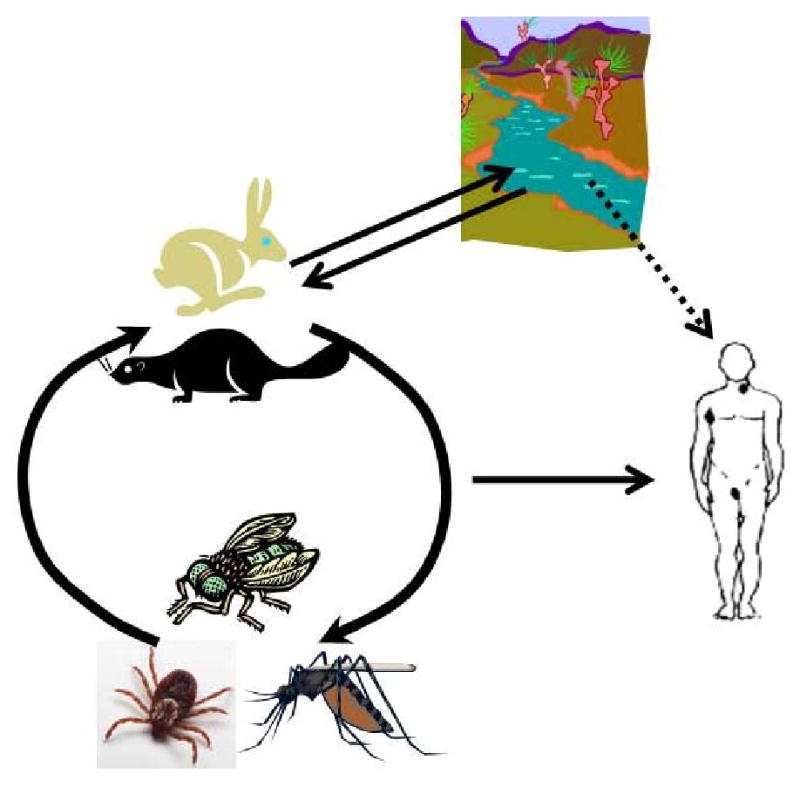
Figure 1a. Infection cycle of Bacillus anthracis. Cattle and other ungulates serve as hosts for Bacillus anthracis, which causes anthrax. They inhale spores from the soil while grazing and, once ingested, these spores germinate. The bacteria then ultimately kill the host. Humans can be exposed to spores from the environment or contaminated animal products, such as meat or skin.
Figure 1b. Infection cycle of Yersinia pestis. Rodents are the hosts of Yersinia pestis, the causative agent of plague, and fleas are the vectors that spread the organism between hosts. Y. pestis continuously cycles between hosts and vectors without ever persisting in the environment. Humans are most commonly infected from the bite of an infected flea but can also become infected through contact with an infected rodent or other host.
Figure 1c. Infection cycle of Francisella tularensis subsp. tularensis. Type A tularaemia, caused by Francisella tularensis subsp. tularensis, is thought to be primarily a disease of lagomorphs (rabbits and hares) and arthropods. Lagomorphs are the hosts for the bacterium, and ticks and flies serve as vectors that transmit the disease between infected and naive hosts and may also serve as long-term reservoirs for the bacteria. Humans are typically infected by the bite of an infected tick or fly or by handling a diseased animal.
Figure 1d. Infection cycle of Francisella tularensis subsp. holarctica. Francisella tularensis subsp. holarctica has a more complex life cycle. A variety of mammals are thought to serve as hosts, including hares, rabbits and beavers. Vectors include blood-feeding mosquitoes, tabanid flies and ticks, and ticks are also possibly long-term reservoirs for the pathogen. Water probably serves as one route of infection for mammals, which may seed aquatic areas. Humans become infected primarily by contact with infected hosts or vectors, but water may also be a source of infection.
Yersinia pestis
The phylogeography of Yersinia pestis has also been shaped by its ecology and human-mediated dispersal. Plague is one of the most notorious diseases in human history due to three major pandemics during which millions of people were killed (see text box) 18. Despite its impacts on human populations, plague is primarily a disease of rodents and their fleas19. Similar to B. anthracis, Y. pestis is a recently emerged clone of a closely-related species, Y. pseudotuberculosis 20. Age estimates based upon molecular clocks suggest that Y. pestis split from Y. pseudotuberculosis within the last 9,000-40,000 years 21. Thus, within a relatively short period of time this species made a drastic niche shift from an enteric pathogen to an obligate blood-borne pathogen that continuously cycles between rodent hosts and flea vectors 19. Genomic comparisons 22, 23 suggest this shift was facilitated by the acquisition of several genomic components not found in Y. pseudotuberculosis, including two additional virulence plasmids; one of these plasmids appears to have a recent common ancestry with a plasmid in Salmonella enterica serovar Typhi 24. With the exception of these two plasmids and a small number of other acquisitions, the rest of the Y. pestis genome has been under decay and is greatly reduced compared to Y. pseudotuberculosis 22, although what remains is quite similar probably because there has been little time for mutations to occur.
Text Box 1. Plasmids.
The acquisition of novel virulence plasmids by the respective ancestors of B. anthracis 1 and Y. pestis 2, 3 were key events leading to the emergence of these two species because genes encoded on the plasmids allowed these organisms to occupy new ecological niches. B. anthracis possesses two virulence plasmids, pXO1 (∼182 kb) and pXO2 (∼95 kb) 4, 5. Important virulence determinants on pXO1 include the genes cya, lef, and pagA 5. Once B. anthracis spores are inhaled by an ungulate host and then convert to vegetative cells, the combined products of these three genes produce the edema and lethal anthrax toxins that eventually kill an infected host, leading to the return of B. anthracis to the soil in spore form and thereby completing the life cycle. The pXO2 plasmid harbors genes (capB, capC, and capA) important for the synthesis of the antiphagocytic poly-d-glutamic acid capsule 5. Most Y. pestis strains typically possess three virulence plasmids 6, 7. Two of these plasmids, pPCP1 (or pPst; 9.5 kb) and pMT1(pFra; 100-110 kb), are only found in Y. pestis, whereas pCD1 (pYV; 70 kb) also is found in Y. pseudotuberculosis and Y. enterocolitica 3. pPCP1 carries the plasminogen activator (pla) gene, which is important for flea-borne transmission because it encodes a surface protease that allows Y. pestis to disseminate away from the site of the flea bite to other areas within the host 7, 8. pMT1 contains a gene (ymt) that enables Y. pestis to colonize the gut of flea vectors 9, as well as the caf gene cluster, which encodes the fraction 1 (F1) capsule 10. pCD1 contains genes that encode both a type III secretion system and the Yersinia outer membrane proteins (Yops) that are secreted into host cells by this system, which together facilitate survival and replication of Yersinia spp. in mammalian lymphoid tissues 11. The genome of F. tularensis contains no plasmids 12.
Y. pestis is very genetically monomorphic and, like B. anthracis and F. tularensis, the limited genetic diversity within this species is not evenly distributed across its global range. There are just three major branches in its phylogenetic tree and eight major molecular groups (Fig. 2). More molecular groups will no doubt be identified and defined once additional isolates from Central Asia are analyzed as this is likely where Y. pestis arose 25. There are specific host associations in plague foci in Central Asia 26, 27 and this local differentiation and adaptation to different hosts and vector species over relatively long periods of time appears to be the main driver of genetic diversity within this species 27 as most of the diverse types are still limited to this region (Fig. 2). Indeed, only two of the eight major groups are found outside of Eurasia: the 1.ANT group is found in central Africa and the highly successful 1.ORI group is found on all continents except Australia and Antarctica (Fig. 2). Despite its global distribution, the 1.ORI group is very monomorphic due to a recent genetic bottleneck. This group was responsible for the 3rd pandemic in the 1800s and 1900s, during which it was introduced to Africa, Australia, North America, and South America18. Limited, but compelling, molecular evidence also suggests that the 1.ORI group was involved in the first two human pandemics28, 29. Although the orientalis biovar correlates well with the 1.ORI group, the other two biovars, mediaevalis and antigua, are not single monophyletic groups 25. As such, Y. pestis biovars provide little help in understanding phylogeography patterns.
Figure 2. Phylogeography of B. anthracis.
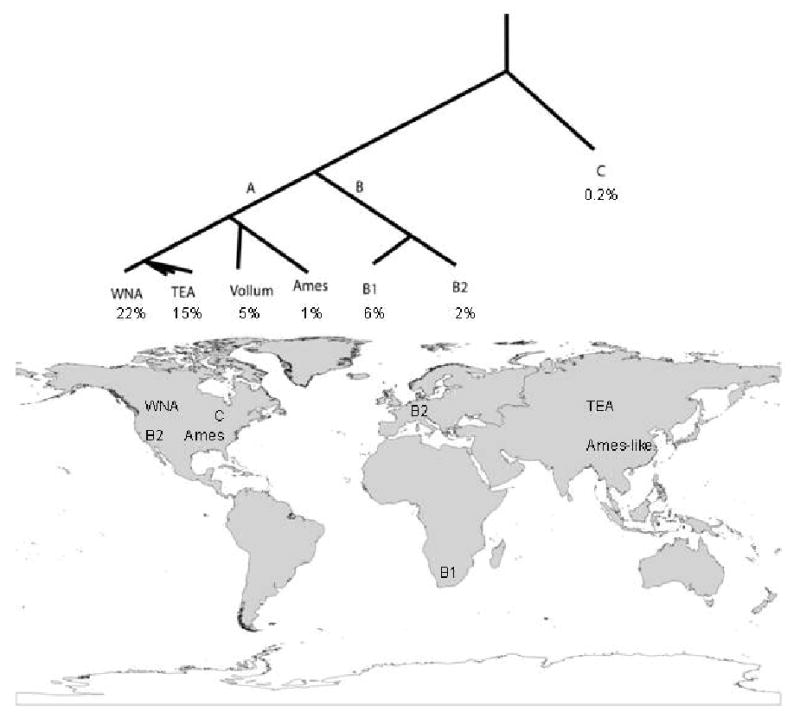
The population structure of B. anthracis revolves around three major groups (A, B, and C). The A group is found in all parts of the world and is very common, whereas the B1, B2, and C groups are relative rare and mostly restricted to subcontinental locations. Highly successful clonal lineage exist even within the A-radiation.
Y. pestis is highly monomorphic from a nucleotide perspective 25 and its population structure (Fig. 2) is consistent with a clonally propagating pathogen. Genomic rearrangements, mediated by insertion elements, do appear to be quite common 25, 30 and can be identified even among closely related strains 31. However, it appears to be a strictly clonal pathogen as there is little evidence for genetic recombination among strains, or with other species 25. Exceptions to the latter are the acquisition by Y. pestis of novel plasmids in addition to the three common plasmids 32, which has been reported by several authors 33-36. Most troubling are two novel plasmids, isolated from two separate Y. pestis isolates in Madagascar, that contain multiple antibiotic-resistance genes 35, 36.
Y. pestis occurs in two distinct ecologic situations depending on the type of rodent species involved. The first situation, which is found in most of the ancient foci in Central Asia but also in North America foci and elsewhere, involves Y. pestis cycling within sylvatic, native rodent species that typically are burrow-dwelling or create large nests, which may be important for flea survival. The second situation is found in parts of Asia and Africa, especially in Madagascar, and is the situation associated with most human plague cases today. It involves primarily non-native commensal rodents (such as Rattus spp.) in large populations. These types of rodents and their associated fleas also are important for the dispersal and establishment of Y. pestis in new areas. This likely reflects the presence of commensal rodents, and thereby their fleas, on ships and other forms of transport. Indeed, Rattus spp. were the most important plague hosts during the third pandemic and have been implicated as important hosts during the first two pandemics.
Probably because Y. pestis has the ability to infect a wide variety of rodent species, once introduced to a new region plague can transfer into and become endemic in native rodent species without the further involvement of non-native rodents, as is the case in North America where non-native rodents are no longer involved in plague ecology. If their numbers and geographic distributions are sufficiently large, Y. pestis can be maintained solely by non-native rodents. This is the current situation in Madagascar, where Rattus spp. greatly outnumber native rodents, none of which are involved in plague ecology. Conversely, if Y. pestis is introduced to a new area via non-native commensal rodents but these species are not widespread and there are no native rodents available to support plague, the disease apparently cannot become established. When Y. pestis was introduced in Australia during the third pandemic it briefly caused plague outbreaks among Rattus spp. and humans. But with no native rodents in Australia, coupled with improved hygiene conditions that reduced the number of non-native rodents, plague disappeared from this continent. Thus, the current global distribution of the 1.ORI group of Y. pestis is the result of both human-mediated dispersal and the specific ecologic situation in the invaded regions.
Francisella tularensis
F. tularensis is typically separated into four subspecies that directly correspond to robust molecular phylogenetic groups (Fig. 3), each of which have distinct geographic distributions and disease manifestations37. Two subspecies, F. tularensis subsp. novicida and F. tularensis subsp. mediasiatica, are rarely observed and at best opportunistic pathogens; they occur primarily in North America and Central Asia, respectively. Most human tularemia cases are caused either by F. tularensis subsp. tularensis (Type A), which is generally considered to be the most pathogenic subspecies, or F. tularensis subsp. holarctica (Type B).
Figure 3. Phylogeography of Y. pestis.
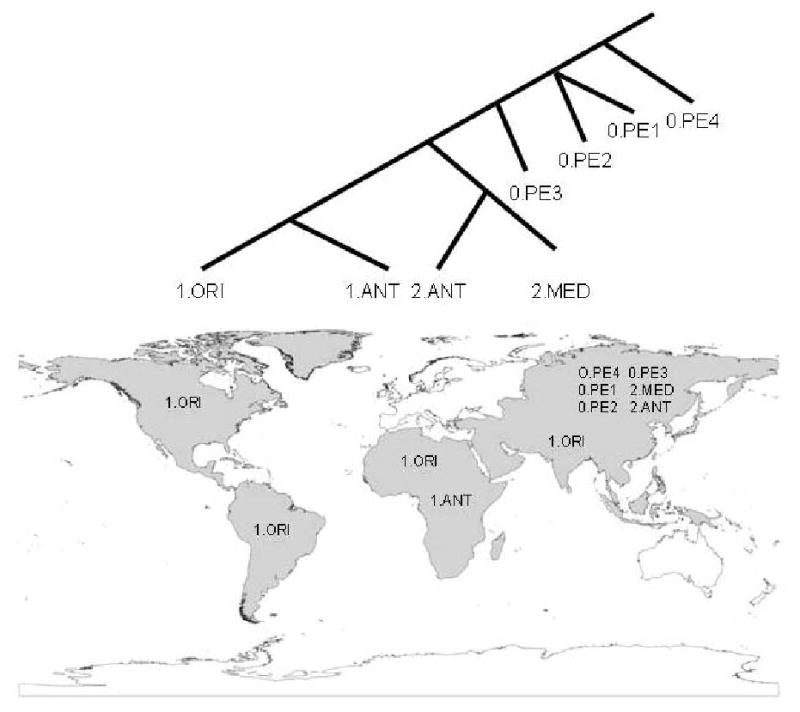
There are currently three major branches within the Y. pestis phylogeny (0, 1, and 2), with eight molecular groups. Six of these groups are only found in Central Asia, where Y. pestis likely evolved from its Y. pseudotuberculosis ancestor. The 1.ANT group is found in just a small region of Africa. The 1.ORI group is widely distributed and currently occurs on all continents except Australia and Antarctic. This group was spread around the world within the last 150 years by humans during the third plague pandemic.
F. tularensis subsp. tularensis is found in North America and recent genetic and genomic analyses have identified two distinct populations within this subspecies, A.I and A.II, that are nearly as genetically distinct as the other subspecies38, 39, 40 Indeed, a recent North American epidemiological analysis suggested the A.II group may be less virulent than even F. tularensis subsp. holarctica41. The A.I and A.II groups have distinct geographic distributions, with the A.II subclade found in the western United States and the A.I subclade primarily found in the eastern US and occasionally California (Fig. 3). These distributions are also highly correlated with specific host and vector species 40. However, it is not clear if these pathogen groups are specifically adapted for transmission by these particular vectors and hosts or if they just share similar geographic distributions and, hence, are just fortuitously associated. Future research needs to move past descriptive science to experimental tests of ecologic differences among the F. tularensis subspecies and groups. This should lead to an initial description of the overall transmission cycles of this pathogen, and may provide insights on ecologic differences that explain the distinct geographic distributions of the subspecies and groups. F. tularensis subsp. tularensis has been identified from the Old World42 but this possibly represents a laboratory escape following human transport, as genotyping39 and genome sequencing43 have established its close identity to the common lab strain SchuS4 (Type A.I).
F. tularensis subsp. holarctica is found throughout much of the Northern Hemisphere and contains little genetic diversity37, 39. This pattern is consistent with a very recent emergence, although there is enough phylogeographic differentiation to suggest the origin of its emergence and global dispersal. The most ancestral lineage is the unusual “japonica” group, which has only been observed in Japan44. Likewise, the second most ancestral lineage has only been observed in California, USA40. Using whole genome analysis, Vogler and colleagues45 identified distinct Old World and New World subclades after the “California ancestor” branch point, which suggests that although F. tularensis subsp. holarctica may have emerged in Asia, the massive radiation within this group commenced in North America. How it has moved among continents is yet to be discerned, although it seems possible that humans may have played a role, similar to the dispersal of the F. tularensis subsp. tularensis A.I., which was transported to the eastern United States via of the transport of infected rabbits 40, 46. F. tularensis subsp. holarctica seems to occupy more diverse ecological niches that provide for enhanced long-term survival in soil and water, perhaps better facilitating long range dispersal.
The ancestry of individual subspecies is unclear as very little is known about the identity of the near neighbors of F. tularensis 37. Like F. tularensis itself, they can be difficult to find, sample, and grow in the laboratory. However, non-culture based sampling of the environment finds evidence for widespread existence of Francisella spp., many of which are probably nonpathogenic but nevertheless could have important evolutionary ties to the pathogenic F. tularensis subspecies47. Indeed, these cryptic near neighbors may represent the ancestral states for each of the more apparent and pathogenic F. tularensis subspecies. Given the phylogenetic structure and many non-culturable examples within Francisella, it seems likely that F. tularensis itself may even contain additional subspecies that are currently unknown.
Despite significant effort, the overall ecology of F. tularensis is not well understood, particularly transmission cycles 37 and ecologic differences among the different subspecies and groups. It has been identified in more than 300 species of mammals, birds, amphibians, and invertebrates 48, which makes identification of specific transmission cycles difficult 37. However, in general, it appears that human cases are most often associated with exposure to lagomorphs, rodents, and blood-feeding arthropods 37. F. tularensis subsp. holarctica infections (Type B) are also associated with rivers, streams, and flooded landscapes37, which may represent persistence of the bacterium within protozoans 37, 49, 50
Synthesis and General Principles
Emergence
B. anthracis, Y. pestis, and F. tularensis are all quite young species, as suggested by their monomorphic population structures (Figs. 1-3). How then did these species arise and spread globally so quickly? Although less clear for F. tularensis, each of these species appears to be the result of a single or few evolutionary changes from their respective ancestors. Many other genetic variants were undoubtedly generated in the ancestral populations but these are the variants that were successful. It seems likely that one of the keys to the success of these clonal species is their increased virulence compared to their ancestors, which was facilitated by lateral gene transfer but also perhaps by genome reductions. This facilitated a shift into much more restricted niches compared to the broad niches of their environmental ancestors. Specialization into restricted niche may help to reinforce their clonality by limiting access to lateral gene donors. Of great importance, this pattern is repeated in highly successful clones found within each of these three species; these successful clones account for most of the global occurrences of each of these pathogens. Changes in virulence also may be related to success of these clones, as differences in disease manifestation and severity are documented among clades of F. tularensis41 and Y. pestis51, though not yet for B. anthracis.
Spread
Many of the phylogeographic patterns observed in these three bacterial pathogens are striking in their similarity, but there are some important differences as well. All three species contain monophyletic and highly clonal clades, representing subspecies or subpopulations that have successfully expanded across multiple continents in the recent past. These events have occurred within an overall population genetic structure in which some clades have remained geographically isolated, while the fit clones have broadly expanded across the globe. Of these three diseases, historical documentation exists only for the spread of plague due to its particularly catastrophic effects on human civilization. The third plague pandemic, caused by the 1.ORI group of Y. pestis, is well documented and the genomic variation and global distribution patterns within this group are highly consistent with 150 years of evolution. Low levels of variation also are observed in F. tularensis subsp. holarctica and in the B. anthracis “A” radiations, suggesting that these two lineages represent comparable recent expansions. The exact time scale is more difficult to discern due to the differential rates of evolution in the spore-forming B. anthracis and the lack of historically documented epidemics in either of these pathogens. However, their phylogeography documents similar clonal expansions and genotype successes. Plague and anthrax expansions have been successful in both the northern and southern hemispheres but, oddly, Francisella has been restricted to North America for F. tularensis subsp. tularensis and to the Northern Hemisphere for F. tularensis subsp. holarctica. The mode of intercontinental transport of F. tularensis subsp. holarctica is unclear and, hence, our understanding of this perceived restriction in the distribution of Francisella is limited.
For recently emerged pathogens, humans themselves are one of the most important parts of a dynamic environment. The third plague pandemic became a global phenomenon because of the advent of efficient steamship transportation that spread infected rats and flea vectors among continents. Anthrax-diseased animals can walk only short distances before succumbing to death. B. anthracis spores, however, have been transported by humans across great distances on the hair, hides, and bones of anthrax killed animals 12. In all cases, the genetic changes that may have been selected for had to match with the dynamic environment, which clearly included human-mediated long distance transport of highly fit clones, in two of these three diseases, to open niches in order to increase genotypic fitness.
Establishment
Why are these species able to establish foci in some regions of the world but not others? Again, F. tularensis is problematic because the overall ecology of this organism is so poorly understood, but several insights can be provided by examining the ecology of B. anthracis and Y. pestis. As described above, the presence of specific types of rodents appear to be required for the dispersal and/or establishment of Y. pestis in new regions, but other factors also may be important. The distribution of Y. pestis includes much of the western United States but stops roughly at the 100th meridian possibly due to climate differences. Recent modeling efforts, based upon climate and other remotely-sensed data for the United States, suggest the predicted ecologic niche of Y. pestis is limited to the western United States 52. For B. anthracis, the presence of ungulates is probably required to maintain foci. This is consistent with patterns from Australia where anthrax now occurs somewhat regularly in domestic cattle. Populations of B. anthracis in Australia, which lacks a native ungulate host, probably were introduced from India around 1850 12, 53, coinciding closely with the introduction of suitable domesticated ungulate hosts. Specific soil properties also may be important to the long-term survival of spores and, hence, establishment and survival of B. anthracis in new disease foci 7. Together, these patterns suggest that both abiotic and biotic factors are important for the survival and establishment of these pathogens in new geographic regions. Understanding these complex requirements and using them to predict other regions of suitability for these pathogens is challenging, but can be accomplished using approaches such as ecological niche modeling 52, 54-56, which also can be used to examine how the distribution of these species may change under predicted global climate change scenarios 52.
Adaptation
Highly fit pathogen clones result from an interaction between the pathogen and its environment, including hosts. The acquisition of a genotypic advantage can be in the form of a plasmid, phage, gene, or even a single SNP that then dramatically increases the fitness of a bacterium. A mutational change alone, however, is not sufficient if the environmental conditions do not allow that genetic change to confer a selective advantage.
Adaptation for dispersal regardless of virulence could be the key fitness factor in clone success. It is commonly assumed that recently emerged pathogens are able to expand into a novel niche due to some newly acquired genetic adaptation. Although research frequently focuses on virulence, many other pathogen phenotypes are important. Spore stability in B. anthracis, for example, is a highly regulated trait that affects long-range transmissibility. Spore characteristics could be adaptive traits affecting dispersal and transmission rates. These long-range dispersal events, coupled with adaptations for dispersal, provide pathogens with access to naïve host populations, which is a great fitness advantage for the fortuitous clones. As these new areas may contain similar ecologic conditions to the ancestral regions, pathogens invading these areas are not necessarily occupying completely novel niches but rather occupying the same or similar niches to those found in the ancestral regions. These areas can be thought of as open niches, as the pathogens of interest did not previously have access to these locations and they were unoccupied by other pathogens. In the case of B. anthracis, Y. pestis, and F. tularensis this capacity to invade new areas is facilitated, at least in part, by their ability to infect a wide variety of hosts (i.e., wide niche breadth). Doubtlessly, open niches, access to naïve hosts, rapid global transportation, and the dynamic human-nature interface will create new emerging infectious diseases in the coming years. Anthrax, plague, and tularemia are models for other pathogen population dynamics in our future.
Potentially genetically fit pathogens waiting for an opportunity to reach the global stage can be thought of as “Hopeful monsters”57, 58. These fit pathogen clones have emerged and dispersed widely across the globe in pandemics associated with many different diseases. The three diseases discussed here are no exception: B. anthracis has the A-clade radiation; Y. pestis has the global spread of 1.ORI clade during the third pandemic; and F. tularensis has the F. tularensis subsp. holarctica expansion. The supposition is that these clones have genetically-based attributes that makes them more fit and, therefore, better suited for a global expansion. (This supposition is supported in some examples, but unknown and unproven in most.) If they are genetically superior, why didn't they emerge previously or later? We believe that humans with our ancient migratory patterns and more recent global transportation networks are a paradigm-changing development that dramatically alters the fitness of pre-existing genotypes. Humans provide an opportunity for pathogens to both shift niches, using their genetic adaptations in novel environmental contexts, and obtain access to open niches. Just as macro-evolutionary biologists have been surprised by apparently rapid evolutionary change in their study species, these highly fit clones appear on the global stage as if from nowhere but in reality have developed in isolated foci and just stood ready for emergence.
Text Box 2. Plague Pandemics.
It is widely accepted that there have been three major plague pandemics during the course of human history that may have resulted in as many as 200 million deaths. The first pandemic, known as the Justinian Plague, occurred from approximately 541 to 750 A.D. and affected Arabia, central and southern Asia, Europe, and north Africa. The second pandemic, which included the infamous epidemic known as the Black Death (1347-1351 A.D.), started around 1347 A.D. and lasted into the 17th century. The third pandemic likely started in the Chinese province of Yunnan in 1855 and last well into the 20th century 18. Some researchers question whether Y. pestis was the causative agent of the first two pandemics (see discussion and citations in 59), but compelling molecular data suggest that plague was the cause of at least some of mortality during these pandemics 28, 29, 60, 61. Y. pestis was definitely the causative agent of the third pandemic, specifically strains from the 1.ORI group 25.
Figure 4. Phylogeography of F. tularensis.
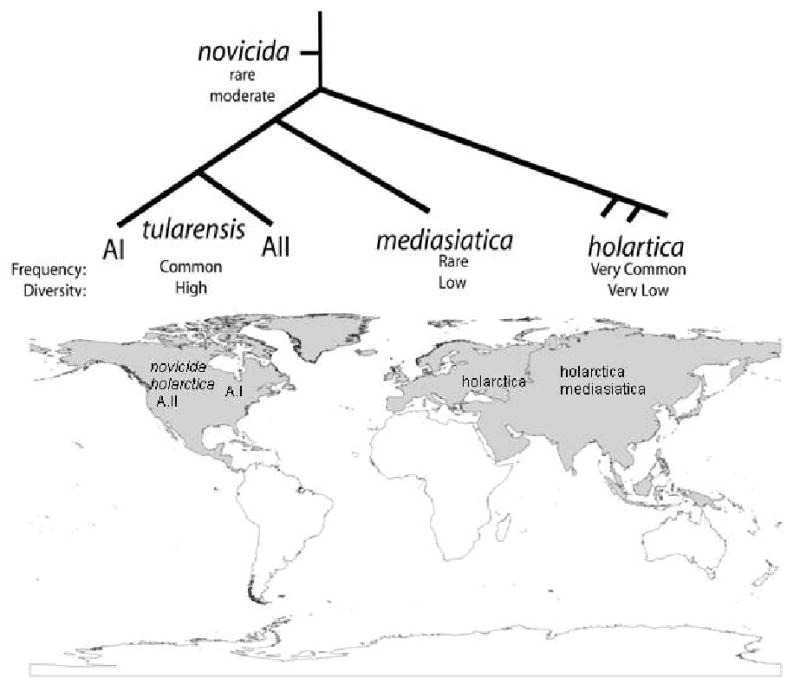
The subspecies of F. tularensis are clonal, with differential success on a global scale. F. t. subsp. tularensis is differentiated into two subpopulations locally restricted to parts of North America. The subspecies novicida and mediasiatica are the rarest and restricted mostly to North America and Central Asia, respectively. F.t. subsp. holarctica appears to be highly fit and highly mobile with a circumpolar distribution in the northern hemisphere.
Acknowledgments
We are grateful to Jeff Foster, Richard Okinaka, Talima Pearson, and several anonymous reviewers for their comments. This work was supported by the National Institutes of Health (GM060795, AI070183), the Pacific-Southwest Region Center of Excellence (AI065359), and the Department of Homeland Security Science and Technology Directorate (NBCH2070001 and HSHQDC-08-C00158).
Glossary of Terms
- Clonal organisms
Organisms that only reproduce by the division of somatic cells without gene transfer or recombination. In this situation, genetic diversity is generated by mutation alone, and not by gene transfer or recombination.
- Fixation
This is a genetic phenomenon in which a previously polymorphic feature takes on the same state in every individual in a population and, hence, is no longer polymorphic.
- Horizontal gene transfer
The movement of DNA from one organism's genome into another's. Three primary mechanisms are documented: 1) transformation involves uptake of naked DNA from the environment by a bacterium, 2) phage transduction can facilitate the transfer of DNA, and 3) some bacteria have mating mechanisms for transfer of DNA via conjugation. This word usage is frequently synonymous with lateral gene transfer or recombination.
- Homoplasy
The occurrence of same character states in two organisms that is not due to shared descent but rather to other forces, such as a reverse mutation or recombination.
- Host shifting
A phenomenon where pathogens acquire the capacity to infect a new host species and perhaps become adapted specifically to that new host.
- Mutation rates
Frequency that nucleotide changes occur in a genome, usually standardized by events per generation.
- Neutral genetic variation
Polymorphic genetic features that have no impact on the survival or reproductive success of the organism.
- Obligate pathogen
An organisms that is only able to survive in a host and/or vector and cannot persist directly in the environment.
- Opportunistic pathogens
Organism that are normally benign but given the right situation (e.g., an immuno-compromised host) can cause disease. Often humans, or other hosts that are occasionally infected by these organisms, are not important to the overall lifestyle of these pathogens but rather are dead-end hosts.
- Phylogenetically informative characters
These are polymorphic features (e.g., a locus possessing multiple alleles) that have a character state (e.g., an allele) shared by two or more members (e.g., isolates, strains) of the study group (e.g., a population or a species) in a fashion that leads to insights into the evolutionary relationships among members. An example of a non-informative character would a character state that is found only in a single member of the study group. In this case, one knows that that member is unique but this character provides no information about how it is related to other members of the study group.
- Phylogeny
A model of the evolutionary history of a set of individuals, species, or other taxonomic units. Phylogenies should be thought of as hypotheses that are based upon analysis of phylogenetically informative characters that arose as the organisms evolved. Phylogenies are traditionally used to understand the evolution of species and other taxonomic units, but are applicable to clonally propagating populations as well.
- Phylogeography
The combination of two disciplines, phylogenetics and geography, to understand the geographic distribution of evolutionary patterns in a given organism.
- Pleiotropic mutation
A single genetic change that affects multiple phenotypes. For example, a regulatory mutation may impact many different genes and thereby the multiple phenotypes associated with those different genes.
- Recently emerged pathogen
A pathogen species, subspecies, population, or genotype with a short evolutionary history. They emerge from another species or population possibly, but not always, due to a genetic change (mutation) that increases their fitness leading to an increase in their frequency and distribution. The recent emergence of such pathogens is often inferred based upon a relative lack of genetic diversity among individual isolates.
- Substitution rate
The speed at which a previously polymorphic feature becomes fixed or identical in all members of a population or species.
- Sylvatic
Having to do with wild animals, as opposed to commensal animals, which are associated with humans.
References
- 1.Pearson T, et al. Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. PNAS. 2004;101:13536–13541. doi: 10.1073/pnas.0403844101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Girard JM, et al. Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. Proc Natl Acad Sci U S A. 2004;101:8408–13. doi: 10.1073/pnas.0401561101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogler AJ, et al. Effect of repeat copy number on variable-number tandem repeat mutations in Escherichia coli O157:H7. J Bacteriol. 2006;188:4253–63. doi: 10.1128/JB.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price LB, et al. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob Agents Chemother. 2003;47:2362–5. doi: 10.1128/AAC.47.7.2362-2365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogler AJ, et al. Molecular analysis of rifampin resistance in Bacillus anthracis and Bacillus cereus. Antimicrob Agents Chemother. 2002;46:511–3. doi: 10.1128/AAC.46.2.511-513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okinaka R, Pearson T, Keim P. Anthrax, but Not Bacillus anthracis? PLoS Pathogens. 2006;2:e122. doi: 10.1371/journal.ppat.0020122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dragon DC, Rennie RP. The ecology of anthrax spores: tough but not invincible. The Canadian Veterinary Journal a Revue Veterinaire Canadienne. 1995;36:295–301. [PMC free article] [PubMed] [Google Scholar]
- 8.Pearson T, et al. Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proc Natl Acad Sci U S A. 2004;101:13536–41. doi: 10.1073/pnas.0403844101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keim P, et al. Molecular evolution and diversity in Bacillus anthracis as detected by amplified fragment length polymorphism markers. J Bacteriol. 1997;179:818–24. doi: 10.1128/jb.179.3.818-824.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keim P, et al. Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J Bacteriol. 2000;182:2928–36. doi: 10.1128/jb.182.10.2928-2936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keim P, et al. Anthrax molecular epidemiology and forensics: using the appropriate marker for different evolutionary scales. Infection, Genetics and Evolution. 2004;4:205–213. doi: 10.1016/j.meegid.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Van Ert MN, et al. Global genetic population structure of Bacillus anthracis. PLoS ONE. 2007;2:e461. doi: 10.1371/journal.pone.0000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonson TS, Okinaka Richard, Wang Bingxiang, Van Ert Matthew N, Easterday Ryan W, Huynh Lynn, U'Ren Jana M, Dukerich M, Zanecki S, Kenefic Leo, Beaudry Jodi, Schupp James, Pearson T, Wagner David M, Hoffmaster Alex, Ravel J, Keim P. Bacillus anthracis in China its Relationship to World-wide Lineages. BMC Microbiology. 2009 doi: 10.1186/1471-2180-9-71. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenefic LJ, Pearson Talima, Okinaka Richard T, Schupp James M, Wagner David M, Ravel Jacques, Hoffmaster Alex R, Trim Carla P, Chung Wai-Kwan, Beaudry Jodi A, Foster Jeffrey T, Mead James I, Keim Paul. Pre-Columbian Origins for North American Anthrax. PLoS ONE. 2009 doi: 10.1371/journal.pone.0004813. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith KL, et al. Bacillus anthracis diversity in Kruger National Park. J Clin Microbiol. 2000;38:3780–4. doi: 10.1128/jcm.38.10.3780-3784.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fouet A, et al. Diversity among French Bacillus anthracis isolates. J Clin Microbiol. 2002;40:4732–4. doi: 10.1128/JCM.40.12.4732-4734.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gierczynski R, et al. Intriguing diversity of Bacillus anthracis in eastern Poland--the molecular echoes of the past outbreaks. FEMS Microbiol Lett. 2004;239:235–40. doi: 10.1016/j.femsle.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 18.Perry RD, Fetherston JD. Yersinia pestis - Etiologic agent of plague. Clinical Microbiology Reviews. 1997;10:35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gage KL, Kosoy MY. Natural history of plague: perspectives from more than a century of research. Annual Review Of Entomology. 2005;50:505–528. doi: 10.1146/annurev.ento.50.071803.130337. [DOI] [PubMed] [Google Scholar]
- 20.Achtman M, et al. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1999;96:14043–14048. doi: 10.1073/pnas.96.24.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Achtman M. In: Yersinia: Molecular and Cellular Biology. Carniel E, Hinnebusch BJ, editors. Horizon Bioscience; Norwich, UK: 2004. p. 432. [Google Scholar]
- 22.Chain PSG, et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. PNAS. 2004;101:13826–13831. doi: 10.1073/pnas.0404012101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkhill J, et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- 24.Prentice MB, et al. Yersinia pestis pFra Shows Biovar-Specific Differences and Recent Common Ancestry with a Salmonella enterica Serovar Typhi Plasmid. J Bacteriol. 2001;183:2586–2594. doi: 10.1128/JB.183.8.2586-2594.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Achtman M, et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17837–17842. doi: 10.1073/pnas.0408026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anisimov AP, Lindler LE, Pier GB. Intraspecific Diversity of Yersinia pestis. Clin Microbiol Rev. 2004;17:434–464. doi: 10.1128/CMR.17.2.434-464.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou D, et al. DNA Microarray Analysis of Genome Dynamics in Yersinia pestis: Insights into Bacterial Genome Microevolution and Niche Adaptation. J Bacteriol. 2004;186:5138–5146. doi: 10.1128/JB.186.15.5138-5146.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drancourt M, et al. Genotyping, Orientalis-like Yersinia pestis, and plague pandemics. Emerging Infectious Diseases. 2004;10:1585–1592. doi: 10.3201/eid1009.030933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drancourt M, et al. Yersinia pestis orientalis in remains of ancient plague patients. Emerging Infectious Diseases. 2007;13:332–333. doi: 10.3201/eid1302.060197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng W, et al. Genome Sequence of Yersinia pestis KIM. J Bacteriol. 2002;184:4601–4611. doi: 10.1128/JB.184.16.4601-4611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auerbach RK, et al. Yersinia pestis evolution on a small timescale: comparison of whole genome sequences from North America. PLoS ONE. 2007;2:e770. doi: 10.1371/journal.pone.0000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu P, et al. Structural Organization of Virulence-Associated Plasmids of Yersinia pestis. J Bacteriol. 1998;180:5192–5202. doi: 10.1128/jb.180.19.5192-5202.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong X, Ye F, Peng H. Geographic distribution and feature of Yersinia pestis plasmid isolated from Yunnan province. Zhonghua Liu Xing Bing Xue Za Zhi = Zhonghua Liuxingbingxue Zazhi. 2001;22:344. [PubMed] [Google Scholar]
- 34.Filippov AA, Solodovnikov NS, Kookleva LM, Protsenko OA. Plasmid content in Yersinia pestis strains of different origin. FEMS Microbiology Letters. 1990;67:45–48. doi: 10.1016/0378-1097(90)90165-m. [DOI] [PubMed] [Google Scholar]
- 35.Galimand M, Carniel E, Courvalin P. Resistance of Yersinia pestis to Antimicrobial Agents. Antimicrob Agents Chemother. 2006;50:3233–3236. doi: 10.1128/AAC.00306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guiyoule A, et al. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerging Infectious Diseases. 2001;7:43–48. doi: 10.3201/eid0701.010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keim P, Johansson A, Wagner DM. Molecular epidemiology, evolution, and ecology of Francisella. Annals of the New York Academy Of Sciences. 2007;1105:30–66. doi: 10.1196/annals.1409.011. [DOI] [PubMed] [Google Scholar]
- 38.Farlow J, et al. Francisella tularensis strain typing using multiple-locus, variable-number tandem repeat analysis. J Clin Microbiol. 2001;39:3186–92. doi: 10.1128/JCM.39.9.3186-3192.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johansson A, et al. Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. J Bacteriol. 2004;186:5808–18. doi: 10.1128/JB.186.17.5808-5818.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farlow J, et al. Francisella tularensis in the United States. Emerg Infect Dis. 2005;11:1835–41. doi: 10.3201/eid1112.050728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Staples JE, Kubota KA, Chalcraft LG, Mead PS, Petersen JM. Epidemiologic and molecular analysis of human tularemia, United States, 1964-2004. Emerg Infect Dis. 2006;12:1113–8. doi: 10.3201/eid1207.051504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gurycova D. First isolation of Francisella tularensis subsp. tularensis in Europe. Eur J Epidemiol. 1998;14:797–802. doi: 10.1023/a:1007537405242. [DOI] [PubMed] [Google Scholar]
- 43.Chaudhuri RR, et al. Genome sequencing shows that European isolates of Francisella tularensis subspecies tularensis are almost identical to US laboratory strain Schu S4. PLoS ONE. 2007;2:e352. doi: 10.1371/journal.pone.0000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogler AJ, Birdsell Dawn, Price Lance B, Bowers Jolene, Beckstrom-Sternberg Stephen, Auerbach Raymond K, Beckstrom-Sternberg James S, Johansson Anders, Clare Ashley, Buchhagen Jordan, Petersen Jeannine M, Pearson Talima, Vaissaire Josée, Dempsey Michael P, Foxall Paul, Engelthaler David M, Wagner David M, Keim Paul. Phylogeography of Francisella tularensis: Whole Genome Sequences and SNP Analysis. J Bacteriol. 2009 doi: 10.1128/JB.01786-08. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogler AJ, et al. Phylogeography of Francisella tularensis: global expansion of a highly fit clone. J Bacteriol. 2009;191:2474–84. doi: 10.1128/JB.01786-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belding DL, Merrill B. Tularemia in imported rabbits in Massachusetts. New England Journal of Medicine. 1941;224:1085–1087. [Google Scholar]
- 47.Barns SM, Grow CC, Okinaka RT, Keim P, Kuske CR. Detection of diverse new Francisella-like bacteria in environmental samples. Appl Environ Microbiol. 2005;71:5494–500. doi: 10.1128/AEM.71.9.5494-5500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mörner T, Addison E. In: Infectious diseases of wild animals. Williams ES, Barker IK, editors. Iowa State University; Ames, Iowa: 2001. pp. 303–312. [Google Scholar]
- 49.Abd H, Johansson T, Golovliov I, Sandstrom G, Forsman M. Survival and growth of Francisella tularensis in Acanthamoeba castellanii. Appl Environ Microbiol. 2003;69:600–6. doi: 10.1128/AEM.69.1.600-606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berdal BP, Mehl R, Meidell NK, Lorentzen-Styr AM, Scheel O. Field investigations of tularemia in Norway. FEMS Immunol Med Microbiol. 1996;13:191–5. doi: 10.1111/j.1574-695X.1996.tb00235.x. [DOI] [PubMed] [Google Scholar]
- 51.Zhou D, Han Y, Song Y, Huang P, Yang R. Comparative and evolutionary genomics of Yersinia pestis. Microbes and Infection. 2004;6:1226–1234. doi: 10.1016/j.micinf.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Nakazawa Y, et al. Climate Change Effects on Plague and Tularemia in the United States. Vector-Borne and Zoonotic Diseases. 2007;7:529–540. doi: 10.1089/vbz.2007.0125. [DOI] [PubMed] [Google Scholar]
- 53.Freedman ML, Thorpe ME. Anthrax: a case report and a short review of Anthrax in Australia. The Medical Journal Of Australia. 1969;1:154. doi: 10.5694/j.1326-5377.1969.tb92078.x. [DOI] [PubMed] [Google Scholar]
- 54.Blackburn JK, McNyset KM, Curtis A, Hugh-Jones ME. Modeling the geographic distribution of Bacillus anthracis, the causative agent of anthrax disease, for the contiguous United States using predictive ecologic niche modeling. Am J Trop Med Hyg. 2007;77:1103–1110. [PubMed] [Google Scholar]
- 55.Neerinckx S, Peterson A, Gulinck H, Deckers J, Leirs H. Geographic distribution and ecological niche of plague in sub-Saharan Africa. International Journal of Health Geographics. 2008;7:54. doi: 10.1186/1476-072X-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peterson A. Biogeography of diseases: a framework for analysis. Naturwissenschaften. 2008;95:483–491. doi: 10.1007/s00114-008-0352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldschmidt R. The Material Basis of Evolution. Yale University Press; 1940. [Google Scholar]
- 58.Gould S. The Return of Hopeful Monsters. Natural History. 1977;86:22–30. [Google Scholar]
- 59.Papagrigorakis MJ, Yapijakis C, Synodinos PN, Baziotopoulou-Valavani E. DNA examination of ancient dental pulp incriminates typhoid fever as a probable cause of the Plague of Athens. International Journal of Infectious Diseases. 2006;10:206–214. doi: 10.1016/j.ijid.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 60.Drancourt M, Raoult D. Molecular insights into the history of plague. Microbes and Infection. 2002;4:105–109. doi: 10.1016/s1286-4579(01)01515-5. [DOI] [PubMed] [Google Scholar]
- 61.Wiechmann I, Grupe G. Detection of Yersinia pestis DNA in two early medieval skeletal finds from Aschheim (Upper Bavaria, 6th century A.D.) American Journal Of Physical Anthropology. 2005;126:48–55. doi: 10.1002/ajpa.10276. [DOI] [PubMed] [Google Scholar]