


Abstract
Ribosomal frameshifting on viral RNAs relies on the mechanical properties of structural elements, often pseudoknots and more rarely stem-loops, that are unfolded by the ribosome during translation. In human immunodeficiency virus (HIV)-1 type B a long hairpin containing a three-nucleotide bulge is responsible for efficient frameshifting. This three-nucleotide bulge separates the hairpin in two domains: an unstable lower stem followed by a GC-rich upper stem. Toeprinting and chemical probing assays suggest that a hairpin-like structure is retained when ribosomes, initially bound at the slippery sequence, were allowed multiple EF-G catalyzed translocation cycles. However, while the upper stem remains intact the lower stem readily melts. After the first, and single step of translocation of deacylated tRNA to the 30 S P site, movement of the mRNA stem-loop in the 5′ direction is halted, which is consistent with the notion that the downstream secondary structure resists unfolding. Mechanical stretching of the hairpin using optical tweezers only allows clear identification of unfolding of the upper stem at a force of 12.8 ± 1.0 pN. This suggests that the lower stem is unstable and may indeed readily unfold in the presence of a translocating ribosome.
INTRODUCTION
The ribosome translocates mRNA and bound tRNA molecules accurately in order to maintain the reading frame. This process results in movement of the ribosome along the mRNA by three nucleotides toward the mRNA’s 3′-end. Translocation of mRNA and tRNAs is a property of the ribosome itself (1,2), however binding of elongation factor G (EF-G) and subsequent hydrolysis of GTP strongly catalyzes it (3). Although the ribosome acts as its own helicase, stable folded structures within the coding regions of mRNA affect the rate of translocation, and more seriously, may trigger a change of reading frame (4,5). Such frameshifting mRNA elements play a crucial role in the translational control of viral proteins via –1 programmed ribosomal frameshifting (–1 PRF) where the reading frame has shifted by one base toward the mRNA 5′-end. The –1 PRF requires both the mRNA slippery sequence at the ribosome coding sites as well as a downstream structural element that resists unfolding, representing a physical barrier to the mRNA translocation machinery. While in many cases the downstream barrier constitutes a hairpin (H)-type pseudoknots (6,7), on a rare occasion it can also be a simple stem–loop structure (8–11). It is of interest to note that although these pseudoknots or stem–loops also trigger ribosomal pausing at the slippery site, consistent with notion of them acting as physical barriers, the extent of pausing shows no correlation with frameshift efficiency (12).
The crystal structure of the ribosome in complex with mRNA has revealed that the mRNA is in a single-stranded conformation in the narrow downstream tunnel (13,14). The ribosome therefore has to unwind mRNA secondary structure through its mRNA helicase activity (15,16). A mechanistic basis for mRNA helicase activity has been proposed involving ribosomal proteins S3, S4, S5 at the mRNA entrance and rotational movement of the head of the 30 S subunit (13,15). The 9 Å and the torsional restraint models (17) propose that –1 PRF is dependent on the mechanical tension induced when a pseudoknot resist unfolding by a moving translating ribosome. Possible effects of such tension were directly observed in the cryo-electron microscopic (Cryo-EM) images of eukaryotic ribosomes stalled in the process of –1 frameshifting in complex with eEF2, tRNA and a frameshifting mRNA pseudoknot (18). The opposing actions of translocation catalyzed by eEF2 and resistance to unfolding by the mRNA strand generate strain that deforms the P-site tRNA which may weaken the codon–anticodon interaction and promote the shift by one nucleotide into the 5′ direction.
The notion that mechanical stability of mRNA structural element is crucial for –1 PRF, has triggered mechanical unfolding experiments of individual mRNA pseudoknot, mutants as well as some of the constituent hairpins using optical tweezers hairpins (19–21). Some of these experiments suggest that frameshift efficiencies correlate with unfolding forces rather than the free-energy difference between the folded and unfolded state (21). This indicates that –1 PRF is kinetically controlled, as has been proposed previously (22).
In HIV-1, translational frameshifting leads to synthesis of the Gag–Pol fusion protein which gives rise to the viral protease, reverse transcriptase (RT) and integrase. This HIV-1 RNA frameshifting signal is a potential target for antiviral therapy (23–26). The exact structure of this RNA frameshifting signal has been the subject of debate. Jacks and collaborators initially proposed that it is a stem–loop structure downstream of the slippery sequence that is essential for efficient frameshifting (4). Alternative structures have subsequently been proposed—reviewed by Brierley and Dos Ramos (27)—in which the stimulatory RNA folds as a pseudoknot (28,29); a pseudoknot with an RNA triple helix motif (30) and two-stem helix containing a three-purine bulge (8). Recently however, two independent nuclear magnetic resonance (NMR) studies have shown that, in the absence of the ribosome, the fold is a long hairpin (Figure 1) with an internal three-nucleotide bulge (9,10). A more recent structure–function analysis of the ribosomal –1 FS signal of two human HIV-1 isolates (31) favors the two-stem helix model of Dulude et al. (8). The internal loop of the long stem–loop introduces a distinct bend between the lower and upper helical regions, a structural feature which, remarkably is also often seen with frameshifting pseudoknots. It has been proposed that the lower stem and the bend serve to initiate contacts between the upper stem–loop and the ribosome. Subsequently the lower stem melts allowing the slippery sequence to bind at the decoding site (10,32). Based on the identification of position +11 as the limit of accessibility of an RNA double helix approaching the ribosome (15), we previously proposed that the upper segment of the lower stem and the bulged region could be structured and/or contact the ribosomal surface (9). NMR studies also pointed out that the upper stem, rich in conserved G–C Watson–Crick base pairs, is highly stable whereas the bulge region and the lower stem are much less so, and may readily unfold/melt. We therefore decided to unfold individual HIV-1 hairpins using optical tweezers, which complement existing methods, such as thermal denaturing, chemical probing or NMR spectroscopy, that address local features within the context of the entire global RNA structure. In principle, optical tweezers aid in applying forces locally to a folded RNA molecule in a way that may be more similar to in vivo conditions than, for example, thermal or chemical denaturing. In the case of a hairpin, the mechanical force will act locally on the 5′- and 3′-ends of the RNA unzipping the stem base-pair by base-pair toward the loop. A similar situation may be found in the ribosome where the translocation movement will generate a force pulling the 5′-end of the mRNA inside the ribosome.
Figure 1.

Secondary structure of the mRNAs used in this study. The tow domains of the HIV-1 hairpin are labeled: lower stem (FSL) and upper stem (FSU). The slippery sequence is underlined and the Shine-Dalgarno sequence is boxed. The first nucleotide of codon 1 (position +1) is labeled. Toeprints that are observed when each codon is positioned in the ribosomal P-site are shown on the top. Nucleotides that were mutated are indicated in a box for the mSP-SL-CCC-HIV-1 RNA. The limit of accessibility (position +11) of a RNA double helix at ribosomal surface (15) is highlighted by a triangle. The spacer sequence that is expected to be single-stranded in the mRNA tunnel is indicated by a dashed line.
Questions, however, remain about the structure of the HIV-1 frameshift signal when ribosomes are present and bound at or downstream of the slippery sequence. Here, using Escherichia coli ribosomes, we address this question and probe possible interactions of the HIV-1 frameshift signal with the ribosome by using toeprinting and chemical probing assays. The ability of this eukaryotic mRNA frameshifting signals to promote –1 PRF in the prokaryotic translational machinery has been previously demonstrated (33,34).
MATERIALS AND METHODS
mRNA translocation
The translocation of mRNA was assayed by toeprinting as described (35,36). mRNA (1 µM) was annealed to primer (2 µM) in 50 mM K-Hepes (pH 7.0) and 100 mM KCl by heating to 90°C for 1 min and placing at room temperature until the temperature reached 45°C. To form the complexes, tight-couple ribosomes (2–5 µM) (37) from E. coli MRE600 were added to 0.6 µM of mRNA in 60 mM NH4Cl, 10 mM Tris–Acetate (pH 7.4) and 20 mM MgCl2 and incubated at 37°C for 10 min. A first tRNA (4 µM) was added to fill the P site by incubation at 37°C for 10 min and aliquot (0.6 pmol mRNA) was removed to ice for later extension. A second tRNA (4 µM) was added to fill the A site by incubation at 37°C for 10 min and aliquot (0.6 pmol mRNA) was removed to ice for later extension. EF-G was added in buffer (50 mM Tris–HCl (pH 7.6), 20 mM MgCl2, 100 mM NH4Cl and 1 mM DTT, 1.5 mM GTP) such that the final concentrations of GTP and EF-G were 300 and 1 µM, respectively. Reactions were incubated at 37°C for 10 min, and aliquots (0.6 pmol mRNA) were removed from each reaction lacking (–G) or containing (+G) EF-G. Each of the aliquots was then extended in parallel (38) with (5′-CTTTATCTTCAGAAGAAAAACC-3′) primer, and the product were resolved by 8% denaturing PAGE.
Stepwise translocation
Stepwise translocation experiments were done as previously described (15). Tight-couple 70 S ribosomes (1 µM final concentration) from E. coli MRE600 (39) were incubated with mSP-HIV-1 mRNA (1 µM) in 30 µl binding buffer (10 mM Tris–HCl (pH 7.4), 60 mM NH4Cl, 10 mM Mg(OAc)2, 6 mM β-ME) for 10 min at 37°C, followed by addition of tRNAPhe (1 µM) and a further 10 min incubation to fill the P site. Aliquots (5 µl) of this reaction were then added to separate tubes containing either GTP (600 µM) (F), GTP + tRNALeu (1 µM) (L), GTP + tRNALeu, EF-G (1 µM) (L’), GTP + tRNALeu, EF-G + tRNAGly (1 µM) (G), GTP + tRNALeu, EF-G + tRNAGly + tRNALys (1 µM) (K), GTP + tRNALeu + tRNAGly + tRNALys (K’), in binding buffer (final volume, 10 µl). These tubes were incubated for 10 min at 37°C and then placed on ice for the primer extension in toeprinting analysis (4 µl).
Footprints
To footprint, binding of mRNAs was performed by incubating 70 S ribosome (in the range of 50–750 pmol according to the mRNA tested) with mRNA (10–50 pmol) in 50 µl reaction buffer A (20 mM MgCl2, 150 mM NH4Cl, 80 mM potassium cacodylate, pH 7.2) at 37°C for 10 min. A first tRNA (320 pmol) was added to fill the P site by incubation at 37°C for 10 min. A second tRNA (320 pmol) was added to fill the A site by incubation at 37°C for 10 min. mRNA–tRNA–70 S complexes were then purified by ultrafiltration (MICROCON YM-100 100 000 Da, Fisher scientific LABOSI). The ternary complex was diluted in 250 µl of buffer A and distributed into 50 µl aliquots (2 pmol mRNA). Chemical probing (38) was performed by addition of 2, 4 or 8 µl dimethyl sulfate (DMS; 1:10 dilution in 95% ethanol), 2, 4 or 8 µl kethoxal (KE; 19 mg/ml in H2O) on 50 µl aliquot, followed by incubation at 37°C for 10 min. All modification reactions were stopped by addition of 150 µl 95% ethanol and 5 µl 3 M sodium acetate followed by rapid mixing. KE-modified samples were adjusted to 25 mM potassium borate (pH 7.0). The pellets were resuspended in 200 µl of 0.3 M sodium acetate, 2.5 mM EDTA and 0.5% SDS (with addition of 25 mM potassium borate for KE samples), extracted three times with phenol, twice with chloroform and resuspended in 10 µl H2O (for DMS samples) or in 10 µl 25 mM potassium borate (for KE samples). Primer extension reactions were performed as described (38).
Purified tRNALys, tRNAfMet and tRNAPhe were purchased from Sigma, tRNAGly and tRNALeu were gracefully donated by Henry Grosjean.
Messenger RNAs were prepared by in vitro transcription. Plasmid pGENE32 is pUC118 containing a region of phage T4 gene 32 (40) from nucleotide position –54 to +84 (where +1 is the translational start) downstream of an engineered T7 promoter sequence. The introduction of slippery sequence in pGENE32 was performed by site-directed mutagenesis kit (Stratagene). Transcripts with stem–loop were obtained by in vitro transcription of synthetic genes flanked upstream by T7 RNA polymerase promoter region and downstream by a BamHI restriction site. The synthetic genes were constructed by shotgun ligation of 10 DNA fragments (24–30-mers) covering both strands and ligated in the KpnI and BamHI sites of pGENE32. All transcripts have been purified by denaturing PAGE.
His-tagged EF-G was purified from pET24b-fusA in E. coli BL21(DE3) as described (41).
Mechanical unfolding using optical tweezers
RNA was synthesized from a template obtained by polymerase chain reaction (PCR) from bases 3821 to 628 of the pBR322 DNA plasmid, where the frameshifting RNA signal from HIV-1 was cloned into the EcoRI and HindIII restriction sites and a T7 promoter was appended to the template in the course of the PCR reaction (42). The DNA components of the handles were prepared by PCR from pBR322. Handle A (pBR322 bases 3821 to 3) was biotinylated, and one of the primers used to amplify handle B (pBR322 bases 30 to 628) was purchased with a 5′ digoxigenin group.
RNA and DNA handles were resuspended in 10 mM sodium phosphate buffer (pH 6.4), and incubated at a ratio of ∼1:1:1 at 90°C for 1 min and transferred to room temperature to cool down gradually, and subsequently diluted to a final concentration (of RNA) of ∼1 μM.
Five microliters of anti-digoxigenin-coated polystyrene beads (0.3 nM; diam. 0.49 μm) were mixed with 1 μl of the DNA–RNA hybrid (∼1 μM) in binding buffer (10 mM Tris buffer [pH 7.0], 250 mM NaCl, 10 mM MgCl2, 0.4% w/v BSA), and incubated at 4°C for overnight on a rotator.
Sample cells were preassembled prior to use. Two thin strip spacers (thickness ∼200 μm) were positioned ∼5 mm apart on the center of a pre-cleaned microscope slide, and epoxy applied at the outer edges of the spacers. A streptavidin functionalized 24 × 40 mm, no. 1.5 coverglass (Xenopore) was then put on the top of the slide. Before introducing the bead and RNA mixture, the sample cell was surface-coated with acetylated BSA by incubation with binding buffer for 30 min at room temperature and washed with 1 ml of the binding buffer, to prevent any sticking of beads to the surface. The bead and RNA mixture was then introduced into the sample cell, and incubated for 30 min at room temperature, and finally washed with 1 ml of binding buffer to remove any unbound beads and RNA.
Molecules were stretched in 10 mM Tris buffer (pH 7.0), 250 mM NaCl, 10 mM MgCl2, or alternatively in 10 mM Tris–acetate (pH 7.4), 60 mM NH4Cl, 6 mM β-mercapto, 20 mM MgCl2. Unfolding/refolding parameters were statistically indistinguishable for both conditions. The spring constant of the optical tweezers was 0.1–0.2 pN nm–1.
The extension of the unfolded single stranded HIV-1 hairpin, xSS was computed as xSS(F) = Δx + LHP for convenience, with F = (F1+F2)/2 the unfolding force and Δx the increase in extension (Figure S2). The increase in contour length (expressed in number of nucleotides) was subsequently computed using the worm-like chain model for polymer elasticity assuming a stretching modulus of 1000 pN, a persistence length of 1 nm (43,44) and inter-phosphate distance of 0.59 nm. Alternatively, one may choose to do the computation taking into account F1 and F2 explicitly, where it is found that 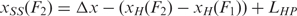 . Then however, one needs to fit the section of the force versus extension curve up to the unfolding event with a worm-like chain model in order to compute the extension of the handles,
. Then however, one needs to fit the section of the force versus extension curve up to the unfolding event with a worm-like chain model in order to compute the extension of the handles, 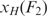 at F2. When performing this more laborious analysis on a subset of our data, we only find a 1–2 nucleotide difference in contour length compared to the analysis that utilizes the applied approximation
at F2. When performing this more laborious analysis on a subset of our data, we only find a 1–2 nucleotide difference in contour length compared to the analysis that utilizes the applied approximation 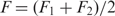 . Standard free-energies at zero force were computed for each trajectory according to
. Standard free-energies at zero force were computed for each trajectory according to 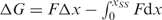 using the worm-like chain model with persistence length of 1 nm and stretching modulus of 1000 pN (43).
using the worm-like chain model with persistence length of 1 nm and stretching modulus of 1000 pN (43).
RESULTS
We used a toeprinting assay to monitor the position and/or structure of the HIV-1 hairpin during the movement of the ribosome along mRNA. This primer-extension inhibition assay has been shown to be a powerful tool for mapping the position of mRNA within 30 S and 70 S ribosomal complexes that contain tRNA (45). When RT encounters the ribosome (a so-called hard-stop), it terminates cDNA synthesis thereby generating a highly specific toeprint. Alternatively, RT may also stall at mRNA structural elements further downstream from the ribosome that prove too hard for RT to unwind, resulting in what is generally called an extended toeprint.
Translocation on the HIV-1 slippery sequence
We first demonstrated that under simple experimental conditions (where tRNA was non-enzymatically delivered at the ribosomal A site), toeprinting allows localization of the ribosome on the mRNA containing the wild-type HIV-1 slippery sequence (mSP-HIV-1) but not the downstream hairpin. The sequences and secondary structures of the mRNA constructs used are derived from T4 gene 32 mRNA in which we introduced the HIV-1 frameshifting signals as shown in Figure 1. In the case of the mSP-HIV-1 construct, two distinct toeprints are observed at +15 and +16 when tRNAPhe is bound at the P site (Figure S1A, lane F), in correspondence with the two possible reading frames created by the slippery sequence. A third toeprint at +17 appears when tRNALeu subsequently binds at the A site (Figure S1A, lane F′). This is thought to be due to a conformational change in the ribosomal complex following A-site binding so that a single position of the tRNA in the A site results in a doublet of bands (at positions +16 and +17) (46,47). It is important to note that the signal at +15 did decrease, indicating that binding of the tRNALeu at the A site assisted in the positioning of the mRNA with tRNAPhe preferentially bound to the u+1u+2u+3 codon adjacent to the leucine codon u+4u+5a+6.
After EF-G catalyzed translocation of tRNALeu to the P site (Figure S1A, lane L), the toeprints at +16, +17 were moved to +20 and +21, corresponding to a 4-nt translocation event. We cannot conclude if a toeprint at +19 exists since this band is also present in the control lane (without ribosome). However, if it exists, this toeprint is very weak. When tRNAGly is subsequently added to the EF-G containing reaction mixture (Figure S1A, lane G), translocation proceeds further giving the expected toeprints at +22 and +23 for tRNAGly bound to P site and paired with the g+7g+8g+9 codon. A weak toeprint at +24 may indicate a low population of tRNAGly bound to P site and paired with the g+8g+9a+10 glycine codon in the +1 frame. The addition of tRNALys (Figure S1A, lane K) triggered translocation leading to a toeprint at +25, which corresponds to a post-translocation complex with tRNALys in the P site bound to the a+10a+11g+12 lysine codon and leaving an empty A site.
Extended toeprints with the downstream frameshifting HIV-1 hairpin
We then applied this assay to a gene 32 message (mSP-SL-HIV-1), which contains the mRNA frameshifting signal (Figure 2A). In order to avoid any unnecessary ambiguity in interpreting the toeprints, we substituted the phenylalanine codon (u+1u+2u+3) for a methionine codon (a+1u+2g+3) (mSP-SL-HIV-1; Figure 1) to avoid complications due to the presence of the slippery site. This allows tRNA binding in only a single unique reading frame.
Figure 2.

Toeprinting analysis of successive EF-G catalyzed translocation steps. (A) mSP-SL-HIV-1; lanes are labeled according to the tRNA species bound to the ribosomal P-site at each step. M, tRNAfMet was bound to the mRNA–ribosome complex followed by stepwise addition of GTP + tRNALeu (M'), GTP + tRNALeu, EF-G (L), GTP + tRNALeu, EF-G + tRNAGly (G), GTP + tRNALeu, EF-G + tRNAGly + tRNALys (K). (B) Toeprints produced by ribosomal complexes with mRNAs mSP-SL-CCC-HIV-1. (C) Toeprints produced by ribosomal complexes with mRNAs mSP-Tyr-SL-HIV-1.
When tRNAfMet was bound at the P site, a doublet of toeprints at positions +U16/+G17 was detected (Figures S2 and 2A). We note the existence of a stop at position +17 in the control lane (in the absence of ribosomes). However, the signal in presence of ribosome is substantially stronger and therefore is interpreted as a ribosomal toeprinting signal. tRNALeu4 was subsequently bound to the A site (Figures S2, lane 3, and 2A, lane M′) which resulted in the disappearance of the +16 toeprint. The +16 and +17 toeprints are hard stops, independent of the reverse transcription activity indicating that the mRNA secondary structure unfolds during cDNA synthesis as previously seen for a stem–loop (48) and pseudoknots (49). However, in addition to these hard stops downstream extended toeprints can also exist indicating that mRNA interactions with the ribosome can extend beyond the usual 15 nucleotides buried in the ribosome-mRNA track (48,50). Unlike hard stops, extended toeprints are generally dependent upon change of temperature, RT concentration, or the source of RT. Therefore, we varied the temperature and also tested different RTs to look for extended toeprints in the case of mSP-SL-HIV-1. Two additional reverse transcription stops were identified within the 3′ region of the stem–loop at positions +47 (very weak) and +43 (Figures 2A and 3A). These extended toeprints are weak in comparison to the hard stop at +17 but are absent in the control lane (Figure 2A, lane C, in the absence of the ribosome). The +43 and +47 toeprints indicate that a fraction of the RT molecules halted when the enzyme approached or encountered bulge region in the HIV-1 hairpin.
Figure 3.

Toeprint results of the m SP-SL-HIV-1 RNA in pre- (A) and post-translocation complexes (B). The relative position of the mRNA hairpin with respect to the ribosome is indicated. The positions of the observed toeprints are indicated by arrows.
In order to test the contribution of the bulge region to the extended toeprint, we changed the GGA-bulge by a CCC-bulge (mSP-SL-CCC-HIV-1, Figure 1). The intensities of both toeprint signals (at +43 and +47) decreased (Figure 2B) and were reproducibly of lower level than the RT stops found in the control lane –70 S. Interestingly, the substitution of the three purines in the bulge by pyrimidines also decreases frameshifting efficiency (8,32).
Translocation with the downstream frameshifting HIV-1 stem–loop
We subsequently monitored the position of mSP-SL-HIV-1 within the ribosome allowing a single or multiple rounds of EF-G catalyzed translocation. The signal at positions +43 and +47 disappeared to give a new toeprint at +39 (Figure 2, lane L, and 3B), indicating that the lower stem readily gave way upon just a single translocation step. An identical result was observed in the mSP-SL-HIV-1 RNA which has a wild-type slippery sequence (first codon is u+1u+2u+3) indicating that codon substitution to a methionine codon did not affect the observed toeprints except for the +47 signal that is ambiguous (Figure S1B). Furthermore upon a single translocation, the +16/+17 toeprint signals disappeared without, however, any occurrence of a new toeprint at position +19 (lane L). The disappearance of the mSP-SL-HIV-1 +16/+17 toeprints upon addition of EF-G is intriguing. We also observed the same phenomenon with the construct containing the wild-type slippery sequence (Figure S1B), and interestingly also with an mRNA containing a pseudoknot (BWYV) bound to ribosome (51). This phenomenon is characteristic to mRNAs containing downstream structural elements, however its cause still need to be elucidated. Ribosomes are known to change conformation upon EF-G binding (52–55). One may speculate that this either blocks RT access directly or stabilizes the mRNA structure.
Any subsequent addition of tRNAGly (lane G) and tRNALys (lane K) in presence of EF-G did not further affect the position of the extended toeprint, which remained at +39, indicating that further movement of the mRNA through the ribosome was impaired.
We next tested an mRNA with an additional codon between the AUG and the start of the lower stem (mSP-Tyr-SL-HIV-1 RNA) extending the spacer, which should at least allow for one round of translocation to be visualized before the ribosome stalls. The toeprints at the new positions A+16/U+17 were unambiguously identified (Figure S3), which places the boundary region contacting the ribosome upstream from the bulge region. These bases are in the upper stem for the mSP-SL-HIV-1 RNA. In Figure 2C, where experimental conditions were tuned as to specifically detect the extended toeprints, the toeprint signals in the region +17 prove somewhat weaker than in Figure S3, but are always present. As expected, with the longer spacer, a band at +19 appeared upon addition of EF-G (Figure 2C, lane L). Interestingly, the extended toeprints did not change and appeared at the exact same positions at A + 46 and U + 50 (corresponding to the A + 43 and U + 47 in mSP-SL-HIV-1 RNA) (Figure 2C). Subsequent addition of EF-G and tRNATyr and tRNAGly produced the same changes in the toeprint pattern as for the mSP-SL-HIV-1 RNA (a shift of 4-nt from +46 to +42). We note that in this case the +19 toeprint remains the strongest toeprint in the upper region of the gel (by comparison with the +17 toeprint) (Figure 2C, lanes Y and G) indicating that translocation, as before, seems to be impaired with the downstream upper stem.
Chemical probing of mRNA with the downstream frameshifting HIV-1 stem-loop
Toeprinting assays fail to provide information on more detailed structural changes that the mRNA might undergo upon binding to the ribosome. Therefore, a footprint assay was performed to obtain structural information upon the interaction of mSP-SL-HIV-1 mRNA with 70 S ribosome–tRNA complexes. In these complexes the ribosome is positioned with the slippery sequence at the decoding site. Results of the chemical probing with KE, 1-cyclohexyl-2-morpholino-carbo-diimide-bmetho-p-toluene sulfonate (CMCT) and DMS are shown in Figure 4. For free mSP-SL-HIV-1 mRNA the chemical modification patterns and levels of reactivities are identical to those previously published (9). Nucleotides in the apical ACAA teraloop as well as A+43 and A+44 from the bulge are found to be reactive to DMS (Figure 4), while nucleotides G+41 and G+42 were reactive to KE. In the lower stem most of the guanine, adenine and uracil bases are accessible to the chemical probes indicating poor stability (9).
Figure 4.

Chemical probing of mSP-SL-HIV-1 mRNA complexed with 70 S, tRNAMet and tRNALeu analyzed by primer extension. (A) Left panel: DMS and CMCT (the control lane without DMS or CMCT but in presence of 70 S particles is on the left; right panel: KE. Nucleotides with modified reactivity are highlighted with an arrow. (B) Summary of the chemical modifications in the structure of the frameshift stimulatory signal. The footprint protections observed upon binding of the ribosome are shaded. The nucleotides with increased reactivity are highlighted with a square. Observed toeprints in the pre-translocation complex are indicated by arrows.
Subsequently, mSP-SL-HIV-1 mRNA was probed in complex with the ribosome and tRNA. For each chemical probe tested, bands that were present in a control lane of unmodified mRNA incubated with ribosome were not taken into account. In the complex, the characteristic strong protections at the guanine nucleotides in the Shine–Dalgarno sequence (G–11, G–10) are clearly seen. Base A+1, of the methionine codon is protected from chemical modifications demonstrating, as previously described (56), Watson–Crick pairing with the anticodon of tRNAfMet. Interestingly, nucleotide A+6 experienced an increase in reactivity similar to what has previously been seen for a mRNA containing a hairpin (selenocysteine incorporation sequence SECIS) in complex with 30 S subunit and tRNAfMet (57). In the spacer and hairpin regions, most of the changes in chemical reactivities were detected in the lower stem that is supposed to be close to the ribosomal surface. Bases G+7, G+8 and G+12 were protected from modification by KE (Figure 4). The reactivity of G+9 toward KE slightly increased in a way similar as A+10 toward DMS.
Mechanical unfolding of the HIV-1 RNA hairpin
Since toeprinting experiments suggested reduced structural stability of the lower stem, we investigated the mechanical stability of the HIV-1 hairpins. Individual hairpins, sandwiched between two differentially end-labeled hybrid DNA–RNA handles were unfolded using optical tweezers (42). Molecules were tethered between a streptavidin-coated glass cover slip and anti-digoxygenin coated polystyrene beads (diameter 1 µm), and stretched by moving the piezo-actuated microscope stage while holding the bead with optical tweezers. Force versus extension curves were computed taking into account this experimental geometry (58). Stage velocities were 67 nm/s, so that the system remained at or close to thermodynamic equilibrium as confirmed by the overlap of the stretching and relaxation force versus extension curves (Figure 5B). In addition, repeated unfolding and folding can be observed within single force versus extension curves (Figure 5B), a further indication that the loading rate is sufficiently low as to assure thermodynamic equilibrium. The increase in contour length (expressed in number of nucleotides) upon unfolding was subsequently computed using the worm-like chain model for polymer elasticity assuming a stretching modulus of 1000 pN, a persistence length of 1 nm (43,59) and a inter-phosphate distance of 0.59 nm (see ‘Materials and Methods’ section). Results are summarized in Figure 5C, yielding a mean increase of 25.3 ± 3.4 nm (mean ± SD) nucleotides, consistent with the contour length of the upper stem of the HIV-1 hairpin. The mean unfolding force is 12.8 ± 1.0 pN (mean ± SD), whereas the standard free-energy change at zero force was found as ΔG = 10 ± 2 kcal/mol (mean ± SD). Refolding statistics are summarized in the Figures S4 and S5 and yield a decrease of contour length of 26.0 ± 4.7 nm (mean ± SD) at an average refolding force of 13.1 ± 1.1 pN (mean ± SD), and ΔG = 11 ± 3 kcal/mol (mean ± SD), essentially unchanged from the unfolding statistics, as one would expect when at thermal equilibrium.
Figure 5.

Mechanical unfolding of the HIV-1 hairpin. (A) HIV-1 hairpin sequence and structure. (B) Typical force versus extension curves upon stretching and relaxation of a single molecule using optical tweezers. The relaxation curve has been offset by 50 nm for clarity, since both curves would overlap otherwise. (C) Histogram of contour lengths of unfolded structures. (D) Histogram of unfolding forces.
DISCUSSION
Extended toeprints (albeit weak ones) at +43 and +47 in the pre-translocation state indicate that a fraction of RTs was incapable of unwinding part of the lower stem and bulge region when encountering the HIV-1 hairpin bound at the ribosome. Since such toeprints do not occur with free hairpins, contacts with the ribosome (34) may be responsible for these signals. Extending the spacer sequence by an extra codon yields an identical toeprint; not surprising as it is hard to imagine that a longer spacer would stand in the way of forming ribosomal contacts. This will be discussed further below. Furthermore, we showed that these toeprints are dependent on the structure of the bulged region of the frameshifting signal. The subsequent replacement of the GGA-bulge with a CCC-bulge, known to reduce –1 PRF efficiency (8,32), practically erases the +43 and +47 toeprints, indicating such a mutation affects either contacts with the ribosomes or stability of the hairpin itself. We tend to favor the former possibility as mechanical unfolding of free hairpins indicates the lower stem is fairly weak to begin with. Certainly it remains possible that contact with the ribosome can stabilize part of the lower stem. If such stabilization was to occur, it does not prevent further translocation as a single cycle of EF-G catalyzed translocation moves the toeprint to +39 providing direct evidence for melting of the lower stem. Interestingly, the extended toeprint at +39 remains even under conditions where further translocation is allowed. This suggests that the upper stem is capable of inhibiting translocation of a sizeable fraction of ribosomes. Our mechanical unfolding study supports the notion of a weak lower stem since no transition in the force versus extension curves indicative of unfolding of solely the lower stem have been observed. Although on occasion we suspect such a transition (by visual inspection of the force versus extension data, typically at forces ∼6 pN and lower), the increase in extension in those cases should be ∼4–5 nm (by theory), too small to reliably distinguish from thermal fluctuations at those force levels. We note that were these transitions to occur in the steeper part of the force versus extension curve (F > 6 pN), we should be able to detect a shift of the curve toward the right consistent with unfolding of the lower stem. However, the increases in contour lengths (Figure 5C) observed are consistent with unfolding of solely the upper stem. Not on any occasions have we observed increases in contour lengths consistent with simultaneous unfolding of the lower and upper stems. Since unfolding is hierarchical, the lower stem has to unfold before the upper stem does. We conclude that the lower stem therefore has much reduced mechanical stability compared to the upper stem. Quantitative assessment of the stability of the lower stem is currently not possible in the existing experimental geometry, and requires higher resolution experimental designs (60), if at all possible. The question naturally arises how these findings accord with the observed +43 and +47 toeprint and the dependence of frameshifting upon the lower stem and bulge regions. It seems unlikely that tensions as low as a mere 6 pN and below, can trigger a frameshift (61). Therefore, it is appealing to consider that unfolding of the hairpin in the presence of ribosomes will differ significantly from that when ribosomes are absent. When adhering to a tension-dependent mechanism of –1 frameshifting this suggest that contacts with the ribosome’s exterior surface may indeed stabilize the part of the lower stem of the hairpin. Single-molecule mechanical unfolding experiments in the presence of ribosomes are considered, but beyond the scope of this article.
Our chemical probing experiments provide further insight into the structure of the HIV-1 frameshift signal when bound to the ribosome. The chemical reactivity pattern indicates that almost the entire structure of the hairpin is maintained. Nucleotides from the ACAA tetraloop and the GGA bulge region remained reactive to chemical probes whereas nucleotides in the upper stem were unreactive. As expected, most of the changes in chemical reactivity of the bases are concentrated in the lower stem. The changes observed in the spacer at positions +7, +8 are consistent with this segment of the RNA being engaged in the 30 S mRNA tunnel (13). Further downstream in the spacer sequence, nucleotides G+9, A+10 and G+12 that are expected to be located in the vicinity or at the ribosomal helicase center indeed experienced a change in reactivity. In this segment of the RNA, the DMS reactivity of nucleotide A+11 remained mostly unaffected by the presence of the ribosome. Unfortunately, our chemical probing experiments could not provide useful information on the 3′ strand of the lower stem. In summary, the data demonstrate that the 5′ region of the lower stem experiences structural changes when contacted by the ribosome.
A number of other observations, although of no direct consequence for the interpretation of the extended toeprints, are of interest. First is the observation of the occurrence of an apparent 4-nt translocation step during transfer of tRNALeu from A site to P site. The disappearance and appearance of respectively the +43 and +39 toeprints indicate that the addition of EF-G triggered a movement of the mSP-SL-HIV-1 RNA inside the ribosome of four nucleotides. However, we note that this 4-nt shift is detected on the 3′ strand of the mRNA hairpin that is located outside the ribosome and therefore may not necessarily present a 1:1 reflection of what happened at the coding site. On the other hand, single translocation cycles with only the slippery sequence (mSP-HIV-1) also show a 4-nt step. In this experiment we analyzed the toeprint signals that result from A site, P site tRNA binding and subsequent EF-G catalyzed translocation. The mSP-HIV-1 toeprint at +15 corresponds to tRNAPhe that recognizes the overlapping codon, one nucleotide upstream (u–1u+1u+2). Binding of tRNAPhe to the u–1u+1u+2 codon results in an optimal spacing of seven bases between the SD sequence and the start codon (62). The toeprint at +16 corresponds to tRNAPhe paired to codon 1 (u+1u+2u+3). Interestingly, overlapping codons u+2u+3u+4 and u+3u+4u+5 are not pairing with tRNAPhe because no toeprints were observed at +17 and +18. This is most likely due to unfavorable spacing between the P-site codon and the Shine-Dalgarno sequence (47,63). After P-site filling and the first round of translocation, the toeprint at +19 is the expected signal for a post-translocation complex with tRNALeu in the P site bound at the u+4u+5a+6 codon. However, the major toeprint signal is at position +20 suggesting that in the post-translocation complex the P site-bound tRNALeu is interacting with the u+5a+6g+7 codon. This corresponds to a 4-nt translocation event. Subsequent translocation cycles seem to force the toeprints back into register. It remains unclear what gives cause to this behavior. We note that a 4-nt translocation has been observed for punctuated mRNA with an extra, unpaired nucleotide between codons (64). Interestingly, Leger and collaborators proposed that slippage and repairing of P-site-bound tRNAPhe in the –1 frame would leave an unpaired nucleotide between the Phe and Leu codons (32). But this is unlikely at equilibrium in our assay because A-site binding of tRNALeu mostly repositioned tRNAPhe into the canonical frame removing any unpaired nucleotide between Phe and Leu codons.
The P-site tRNA binding triggered a major RT stop at position +17 while only a weak signal at position +16 (the ‘classical toeprint’). Previous toeprinting assays under identical conditions but with different phage T4 mRNAs showed a variety of toeprints signals confined to the +14 to +17 range (45). Thus toeprinting assays are very sensitive to the type of mRNA tested and we note that the toeprint signals with the mSP-HIV-1 and mSP-SL-HIV-1 mRNAs respectively at +16 and +17 fall into this range. Could the observed difference between the two mRNAs be attributed to the hairpin? With unstructured mRNA, we expect a pulling force on the spacer mRNA out of the ribosome purely based on entropic arguments. Such a force presumably exposes base +16 giving rise to the classical toeprint. In the case of the HIV-1 hairpin, physical interactions of the ribosome with the hairpin may provide some sort of strain relieve allowing +16 to slightly relax back into the ribosome. On the other hand, how could such a mechanism be consistent with +16/+17 toeprint obtained when an additional codon was inserted in the spacer, between the AUG codon and the hairpin? If the hairpin provides strain relieve would one not expect the mRNA to recede further back into ribosome, not only protect +16 but also perhaps +17. However, lacking any quantitative information about tension in the spacer, such extra movement may be too small to also protect +17. These are intriguing possibilities and raise questions that require the design of new experiments, well beyond the scope of this work, for answering.
Site-directed mutagenesis and amino-acid sequencing localized the site of frameshifting to the UUA codon of the HIV-1 slippery sequence (65). The presence of the GC-rich upper stem strongly enhances frameshifting efficiencies. This level of mRNA slippage is enhanced further by the bulge and lower stem. We show here that when the slippery sequence is bound at the decoding site, the ribosome directly influences the spacer sequence in the lower stem that very likely enters the mRNA track. In addition, extended toeprints localized in the upper segment of the lower stem support a physical interaction between this region and the ribosome. These results are in agreement with the importance of the lower stem and bulge regions for the –1 frameshift (8,32,66). It is conceivable that this interaction enhances –1 frameshifting at the particularly slippery UUUUUUA sequence (67).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Young Investigator Award from the Human Frontier Science Program (HFSP) to D.F. and K.V. Funding for open access charge: CNRS
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Kevin Wilson for the kind gift of the pET24b-fusA vector for His-tagged EF-G.
REFERENCES
- 1.Gavrilova LP, Kostiashkina OE, Koteliansky VE, Rutkevitch NM, Spirin AS. Factor-free (“non-enzymic”) and factor-dependent systems of translation of polyuridylic acid by Escherichia coli ribosomes. J. Mol. Biol. 1976;101:537–552. doi: 10.1016/0022-2836(76)90243-6. [DOI] [PubMed] [Google Scholar]
- 2.Fredrick K, Noller HF. Catalysis of ribosomal translocation by sparsomycin. Science. 2003;300:1159–1162. doi: 10.1126/science.1084571. [DOI] [PubMed] [Google Scholar]
- 3.Rodnina MV, Savelsbergh A, Katunin VI, Wintermeyer W. Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature. 1997;385:37–41. doi: 10.1038/385037a0. [DOI] [PubMed] [Google Scholar]
- 4.Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag–pol expression. Nature. 1988;331:280–283. doi: 10.1038/331280a0. [DOI] [PubMed] [Google Scholar]
- 5.Brierley I, Digard P, Inglis SC. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell. 1989;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baranov PV, Gesteland RF, Atkins JF. Recoding: translational bifurcations in gene expression. Gene. 2002;286:187–201. doi: 10.1016/s0378-1119(02)00423-7. [DOI] [PubMed] [Google Scholar]
- 7.Giedroc DP, Cornish PV. Frameshifting RNA pseudoknots: structure and mechanism. Virus research. 2009;139:193–208. doi: 10.1016/j.virusres.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dulude D, Baril M, Brakier-Gingras L. Characterization of the frameshift stimulatory signal controlling a programmed –1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res. 2002;30:5094–5102. doi: 10.1093/nar/gkf657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaudin C, Mazauric MH, Traikia M, Guittet E, Yoshizawa S, Fourmy D. Structure of the RNA signal essential for translational frameshifting in HIV-1. J. Mol. Biol. 2005;349:1024–1035. doi: 10.1016/j.jmb.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 10.Staple DW, Butcher SE. Solution structure and thermodynamic investigation of the HIV-1 frameshift inducing element. J. Mol. Biol. 2005;349:1011–1023. doi: 10.1016/j.jmb.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 11.Marcheschi RJ, Staple DW, Butcher SE. Programmed ribosomal frameshifting in SIV is induced by a highly structured RNA stem-loop. J. Mol. Biol. 2007;373:652–663. doi: 10.1016/j.jmb.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kontos H, Napthine S, Brierley I. Ribosomal pausing at a frameshifter RNA pseudoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol. Cell Biol. 2001;21:8657–8670. doi: 10.1128/MCB.21.24.8657-8670.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yusupova GZ, Yusupov MM, Cate JH, Noller HF. The path of messenger RNA through the ribosome. Cell. 2001;106:233–241. doi: 10.1016/s0092-8674(01)00435-4. [DOI] [PubMed] [Google Scholar]
- 14.Jenner L, Rees B, Yusupov M, Yusupova G. Messenger RNA conformations in the ribosomal E site revealed by X-ray crystallography. EMBO Reports. 2007;8:846–850. doi: 10.1038/sj.embor.7401044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takyar S, Hickerson RP, Noller HF. mRNA helicase activity of the ribosome. Cell. 2005;120:49–58. doi: 10.1016/j.cell.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 16.Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, Noller HF, Bustamante C, Tinoco I. Following translation by single ribosomes one codon at a time. Nature. 2008;452:598–603. doi: 10.1038/nature06716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plant EP, Dinman JD. Torsional restraint: a new twist on frameshifting pseudoknots. Nucleic Acids Res. 2005;33:1825–1833. doi: 10.1093/nar/gki329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Namy O, Moran SJ, Stuart DI, Gilbert RJ, Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature. 2006;441:244–247. doi: 10.1038/nature04735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen TM, Reihani SN, Oddershede LB, Sorensen MA. Correlation between mechanical strength of messenger RNA pseudoknots and ribosomal frameshifting. Proc. Natl Acad. Sci. USA. 2007;104:5830583–5830585. doi: 10.1073/pnas.0608668104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G, Wen JD, Tinoco I., Jr Single-molecule mechanical unfolding and folding of a pseudoknot in human telomerase RNA. RNA. 2007;13:2175–2188. doi: 10.1261/rna.676707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green L, Kim CH, Bustamante C, Tinoco I., Jr Characterization of the mechanical unfolding of RNA pseudoknots. J. Mol. Biol. 2008;375:511–528. doi: 10.1016/j.jmb.2007.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cornish PV, Stammler SN, Giedroc DP. The global structures of a wild-type and poorly functional plant luteoviral mRNA pseudoknot are essentially identical. RNA. 2006;12:1959–1969. doi: 10.1261/rna.199006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aupeix-Scheidler K, Chabas S, Bidou L, Rousset JP, Leng M, Toulme JJ. Inhibition of in-vitro and ex-vivo translation by a transplatin-modified oligo (2′-O-methylribonucleotide) directed against the HIV-1 gag–pol frameshift signal. Nucleic Acids Res. 2000;28:438–445. doi: 10.1093/nar/28.2.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas P, Jiang X, Pacchia AL, Dougherty JP, Peltz SW. The human immunodeficiency virus type 1 ribosomal frameshifting site is an invariant sequence determinant and an important target for antiviral therapy. J. Virol. 2004;78:2082–2087. doi: 10.1128/JVI.78.4.2082-2087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staple DW, Venditti V, Niccolai N, Elson-Schwab L, Tor Y, Butcher SE. Guanidinoneomycin B recognition of an HIV-1 RNA helix. Chembiochem. 2008;9:93–102. doi: 10.1002/cbic.200700251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dulude D, Theberge-Julien G, Brakier-Gingras L, Heveker N. Selection of peptides interfering with a ribosomal frameshift in the human immunodeficiency virus type 1. RNA. 2008;14:981–991. doi: 10.1261/rna.887008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brierley I, Dos Ramos FJ. Programmed ribosomal frameshifting in HIV-1 and the SARS-CoV. Virus Res. 2006;119:29–42. doi: 10.1016/j.virusres.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le SY, Shapiro BA, Chen JH, Nussinov R, Maizel JV. RNA pseudoknots downstream of the frameshift sites of retroviruses. Genet. Anal. Tech. Appl. 1991;8:191–205. doi: 10.1016/1050-3862(91)90013-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du Z, Giedroc DP, Hoffman DW. Structure of the autoregulatory pseudoknot within the gene 32 messenger RNA of bacteriophages T2 and T6: a model for a possible family of structurally related RNA pseudoknots. Biochemistry. 1996;35:4187–4198. doi: 10.1021/bi9527350. [DOI] [PubMed] [Google Scholar]
- 30.Dinman JD, Richter S, Plant EP, Taylor RC, Hammell AB, Rana TM. The frameshift signal of HIV-1 involves a potential intramolecular triplex RNA structure. Proc. Natl Acad. Sci. USA. 2002;99:5331–5336. doi: 10.1073/pnas.082102199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girnary R, King L, Robinson L, Elston R, Brierley I. Structure–function analysis of the ribosomal frameshifting signal of two human immunodeficiency virus type 1 isolates with increased resistance to viral protease inhibitors. J. Gen. Virol. 2007;88:226–235. doi: 10.1099/vir.0.82064-0. [DOI] [PubMed] [Google Scholar]
- 32.Leger M, Sidani S, Brakier-Gingras L. A reassessment of the response of the bacterial ribosome to the frameshift stimulatory signal of the human immunodeficiency virus type 1. RNA. 2004;10:1225–1235. doi: 10.1261/rna.7670704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiss RB, Dunn DM, Shuh M, Atkins JF, Gesteland RF. E. coli ribosomes re-phase on retroviral frameshift signals at rates ranging from 2–50% New Biol. 1989;1:159–169. [PubMed] [Google Scholar]
- 34.Brunelle MN, Payant C, Lemay G, Brakier-Gingras L. Expression of the human immunodeficiency virus frameshift signal in a bacterial cell-free system: influence of an interaction between the ribosome and a stem-loop structure downstream from the slippery site. Nucleic Acids Res. 1999;27:4783–4791. doi: 10.1093/nar/27.24.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartz D, McPheeters DS, Traut R, Gold L. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988;164:419–425. doi: 10.1016/s0076-6879(88)64058-4. [DOI] [PubMed] [Google Scholar]
- 36.Yoshizawa S, Fourmy D, Puglisi JD. Recognition of the codon–anticodon helix by ribosomal RNA. Science. 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]
- 37.Powers T, Noller HF. A functional pseudoknot in 16S ribosomal RNA. EMBO J. 1991;10:2203–2214. doi: 10.1002/j.1460-2075.1991.tb07756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stern S, Moazed D, Noller HF. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol. 1988;164:481–489. doi: 10.1016/s0076-6879(88)64064-x. [DOI] [PubMed] [Google Scholar]
- 39.Moazed D, Noller HF. Interaction of tRNA with 23S rRNA in the ribosomal A, P, and E sites. Cell. 1989;57:585–597. doi: 10.1016/0092-8674(89)90128-1. [DOI] [PubMed] [Google Scholar]
- 40.Krisch HM, Allet B. Nucleotide sequences involved in bacteriophage T4 gene 32 translational self-regulation. Proc. Natl Acad. Sci. USA. 1982;79:4937–4941. doi: 10.1073/pnas.79.16.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson KS, Noller HF. Mapping the position of translational elongation factor EF-G in the ribosome by directed hydroxyl radical probing. Cell. 1998;92:131–139. doi: 10.1016/s0092-8674(00)80905-8. [DOI] [PubMed] [Google Scholar]
- 42.Liphardt J, Onoa B, Smith SB, Tinoco IJ, Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–737. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 43.Tinoco I., Jr Force as a useful variable in reactions: unfolding RNA. Ann. Rev. Biophys. Biomol. Struc. 2004;33:363–385. doi: 10.1146/annurev.biophys.33.110502.140418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seol Y, Skinner GM, Visscher K, Buhot A, Halperin A. Stretching of homopolymeric RNA reveals single-stranded helices and base-stacking. Phys. Rev. Lett. 2007;98:158103. doi: 10.1103/PhysRevLett.98.158103. [DOI] [PubMed] [Google Scholar]
- 45.Hartz D, McPheeters DS, Gold L. Selection of the initiator tRNA by Escherichia coli initiation factors. Genes Dev. 1989;3:1899–1912. doi: 10.1101/gad.3.12a.1899. [DOI] [PubMed] [Google Scholar]
- 46.Joseph S, Noller HF. EF-G-catalyzed translocation of anticodon stem-loop analogs of transfer RNA in the ribosome. EMBO J. 1998;17:3478–3483. doi: 10.1093/emboj/17.12.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fredrick K, Noller HF. Accurate translocation of mRNA by the ribosome requires a peptidyl group or its analog on the tRNA moving into the 30S P site. Mol. Cell. 2002;9:1125–1131. doi: 10.1016/s1097-2765(02)00523-3. [DOI] [PubMed] [Google Scholar]
- 48.Ringquist S, MacDonald M, Gibson T, Gold L. Nature of the ribosomal mRNA track: analysis of ribosome-binding sites containing different sequences and secondary structures. Biochemistry. 1993;32:10254–10262. doi: 10.1021/bi00089a048. [DOI] [PubMed] [Google Scholar]
- 49.Philippe C, Eyermann F, Benard L, Portier C, Ehresmann B, Ehresmann C. Ribosomal protein S15 from Escherichia coli modulates its own translation by trapping the ribosome on the mRNA initiation loading site. Proc. Natl Acad. Sci. USA. 1993;90:4394–4398. doi: 10.1073/pnas.90.10.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ringquist S, Schneider D, Gibson T, Baron C, Bock A, Gold L. Recognition of the mRNA selenocysteine insertion sequence by the specialized translational elongation factor SELB. Genes Dev. 1994;8:376–385. doi: 10.1101/gad.8.3.376. [DOI] [PubMed] [Google Scholar]
- 51.Mazauric MH, Leroy JL, Visscher K, Yoshizawa S, Fourmy D. Footprinting analysis of BWYV pseudoknot-ribosome complexes. RNA. 2009;15:1775–1786. doi: 10.1261/rna.1385409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 53.Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J. Locking and unlocking of ribosomal motions. Cell. 2003;114:123–134. doi: 10.1016/s0092-8674(03)00476-8. [DOI] [PubMed] [Google Scholar]
- 54.Wilson KS, Nechifor R. Interactions of translational factor EF-G with the bacterial ribosome before and after mRNA translocation. J. Mol. Biol. 2004;337:15–30. doi: 10.1016/j.jmb.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 55.Spiegel PC, Ermolenko DN, Noller HF. Elongation factor G stabilizes the hybrid-state conformation of the 70S ribosome. RNA. 2007;13:1473–1482. doi: 10.1261/rna.601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huttenhofer A, Noller HF. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J. 1994;13:3892–3901. doi: 10.1002/j.1460-2075.1994.tb06700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hüttenhofer A, Westhof E, Böck A. Solution structure of mRNA hairpins promoting selenocysteine incorporation in Escherichia coli and their base-specific interaction with special elongation factor SELB. RNA. 1996;2:354–366. [PMC free article] [PubMed] [Google Scholar]
- 58.Wang MD, Yin H, Landick R, Gelles J, Block SM. Stretching DNA with optical tweezers. Biophys. J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seol Y, Skinner GM, Visscher K. Elastic properties of a single-stranded charged homopolymeric ribonucleotide. Phys. Rev. Lett. 2004;93:118102. doi: 10.1103/PhysRevLett.93.118102. [DOI] [PubMed] [Google Scholar]
- 60.Moffitt JR, Chemla YR, Izhaky D, Bustamante C. Differential detection of dual traps improves the spatial resolution of optical tweezers. Proc. Natl Acad. Sci. USA. 2006;103:9006–9011. doi: 10.1073/pnas.0603342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao S, Chen SJ. Predicting ribosomal frameshifting efficiency. Phys. Biol. 2008;5:16002. doi: 10.1088/1478-3975/5/1/016002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ringquist S, Shinedling S, Barrick D, Green L, Binkley J, Stormo GD, Gold L. Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol. Microbiol. 1992;6:1219–1229. doi: 10.1111/j.1365-2958.1992.tb01561.x. [DOI] [PubMed] [Google Scholar]
- 63.Vellanoweth RL, Rabinowitz JC. The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol. Microbiol. 1992;6:1105–1114. doi: 10.1111/j.1365-2958.1992.tb01548.x. [DOI] [PubMed] [Google Scholar]
- 64.Phelps SS, Gaudin C, Yoshizawa S, Benitez C, Fourmy D, Joseph S. Translocation of a tRNA with an extended anticodon through the ribosome. J. Mol. Biol. 2006;360:610–622. doi: 10.1016/j.jmb.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 65.Jacks T, Madhani HD, Masiarz FR, Varmus HE. Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell. 1988;55:447–458. doi: 10.1016/0092-8674(88)90031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baril M, Dulude D, Gendron K, Lemay G, Brakier-Gingras L. Efficiency of a programmed –1 ribosomal frameshift in the different subtypes of the human immunodeficiency virus type 1 group M. RNA. 2003;9:1246–1253. doi: 10.1261/rna.5113603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brierley I, Jenner AJ, Inglis SC. Mutational analysis of the “slippery-sequence” component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 1992;227:463–479. doi: 10.1016/0022-2836(92)90901-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.