


Abstract
To characterize the residues that participate in the catalysis of DNA cleavage and rejoining by the site-specific recombinase Tn3 resolvase, we mutated conserved polar or charged residues in the catalytic domain of an activated resolvase variant. We analysed the effects of mutations at 14 residues on proficiency in binding to the recombination site (‘site I’), formation of a synaptic complex between two site Is, DNA cleavage and recombination. Mutations of Y6, R8, S10, D36, R68 and R71 resulted in greatly reduced cleavage and recombination activity, suggesting crucial roles of these six residues in catalysis, whereas mutations of the other residues had less dramatic effects. No mutations strongly inhibited binding of resolvase to site I, but several caused conspicuous changes in the yield or stability of the synapse of two site Is observed by non-denaturing gel electrophoresis. The involvement of some residues in both synapsis and catalysis suggests that they contribute to a regulatory mechanism, in which engagement of catalytic residues with the substrate is coupled to correct assembly of the synapse.
INTRODUCTION
Site-specific recombinases promote programmed DNA rearrangements in a wide variety of biological contexts. The tyrosine recombinases and the serine recombinases are two structurally and mechanistically distinct enzyme families, each with hundreds of known members; their names refer to the conserved amino acid residue whose sidechain attacks specific DNA phosphodiesters to bring about transient strand cleavage. Studies on the structures and mechanisms of tyrosine recombinases and a related group of enzymes, the Type IB topoisomerases, have led to a quite detailed picture of the active site where the chemical steps of DNA strand cleavage and rejoining take place (1–3), and the roles of several conserved residues in catalysis have been elucidated. In contrast, much less is known about the active site of serine recombinases, the subject of this work.
Serine recombinases share a catalytic domain which is conserved in size (∼140 amino acids) and in sequence (Figure 1). In one well-studied group of serine recombinases which includes transposon resolvases (such as that of Tn3) and DNA invertases, the catalytic domain is at the N-terminus of the protein and is followed by a small helix-turn-helix DNA-binding domain. Other groups which include transposases and bacteriophage integrases have different arrangements and types of domains (4,5). The closely related Tn3 and γδ resolvases function to resolve (that is, split into two circles) cointegrate intermediates in replicative transposition, by promoting a site-specific recombination reaction between the two copies of the transposon in the cointegrate. The recombination site, res, contains three binding sites for resolvase dimers (Figure 2A). DNA strand breaking and rejoining are catalysed by the resolvase bound at site I, whereas the subunits bound at the accessory sites II and III have regulatory functions (1). Resolvase (185 amino acids) has two functional domains (Figure 2B). The N-terminal domain (residues 1–140) contains the residues that form the active site for catalysis of strand exchange, and is involved in subunit interactions. The C-terminal domain (residues 141–185) makes a sequence-specific interaction with motifs in the res DNA. During recombination, two resolvase dimers bound to site I come together to form a tetramer. All four strands of the two site Is are then cleaved by attack of the hydroxyl group of a conserved resolvase serine residue (S10) on specific phosphodiesters. The four resolvase subunits directly involved in the reaction all thus become covalently attached to the DNA via a phosphoserine linkage. Strand exchange is thought to involve a rotation that brings the cleaved DNA ends into a recombinant configuration, followed by rejoining of the strands by reversal of the cleavage reaction [(1), Figure 2C].
Figure 1.

Alignment of the amino acid sequences of selected serine recombinase catalytic domains. The proteins in the alignment have all been studied in vitro and shown to catalyse conservative site-specific recombination reactions. Accession numbers are given to the right of each protein's; name. Proteins whose names are in bold are in the resolvase-invertase group of serine recombinases (see text for details). Crystallographically determined secondary structure elements of γδ resolvase (24) are boxed and labelled above the alignment. The putative catalytic residues mutated in this study and their equivalents in the other proteins are highlighted in yellow, along with the conserved residue E124, which was not analysed here because it has already been shown to be non-essential for catalysis (30–32). NM resolvase contains mutations at the residues highlighted in red on the wild-type Tn3 resolvase sequence (see ‘Materials and Methods’ section for details).
Figure 2.

Site-specific recombination by resolvase. (A) The Tn3 (or γδ) recombination site res. The boxes represent binding sites for resolvase dimers. The lengths of the DNA segments are indicated (bp). The point within site I at which resolvase breaks and rejoins the DNA is marked by a staggered line. Wild-type resolvase requires the complete res site, but activated mutants such as NM resolvase can catalyse recombination at site I (outlined in blue), without sites II and III. (B) Left; crystal structure of a wild-type γδ resolvase dimer bound to site I [1GDT (24)]. Right; crystal structure of a synaptic tetramer of a γδ resolvase activated mutant with two cleaved site Is [1ZR4 (22); smaller scale]. The approximate positions of the active site residues are indicated by the red circles. The images were created with PyMOL. (C) Proposed strand exchange mechanism for serine recombinases. The reaction occurs in a synapse comprising a recombinase tetramer and two crossover sites (site I of res in the case of resolvase). Recombinase subunits are shown as red and blue ovals. The small circles mark the positions of the phosphodiester groups attacked by the recombinase serine residue (S10 for Tn3/γδ resolvase). Cleavage of all four DNA strands (resulting in covalent attachment of the recombinase to the DNA 5′-ends by a phosphoseryl linkage) is followed by exchange of the half-sites, then re-ligation [adapted from ref. (55)].
The mechanism outlined in Figure 2C involves eight chemical reaction steps (cleavage of four DNA strands followed by their re-ligation), as well as a conformational change to rearrange the cleaved DNA ends. How resolvase executes these transactions is as yet incompletely understood. The identification of S10 as the nucleophile that cleaves the phosphodiesters came from the finding that the 5′-phosphates of the cleaved DNA were covalently joined to a serine residue in γδ resolvase (6) and that S10, rather than the other well-conserved serine S39, is essential for catalytic activity [(7–9), Table 1]. More recently, S10 was observed to be linked to the 5′-ends of the cleaved site I DNA in crystal structures of reaction intermediates (22,23). Curiously, S10 is not buried in an ‘active site pocket’, but is part of a surface loop that adopts different conformations in different crystallographic γδ resolvase monomers (22–27). A number of other highly conserved residues with polar or charged sidechains are apparent in alignments of the amino acid sequences of serine recombinases (Figure 1), and most of these residues are quite close to S10, suggesting that they might contribute to the active site (Table 1). The importance of some of these residues in catalysis has been confirmed by mutation studies, and functions for some have been suggested [(1,22,28), Table 1]. However, the structure of the active site at the point of catalysis and the functions of a number of the putative active site residues remain obscure. Even in the recent structures of reaction intermediates (22,23), some of these residues are not in close contact with the DNA, and how they might collectively build a functional active site is not obvious.
Table 1.
Published data on putative resolvase catalytic residues
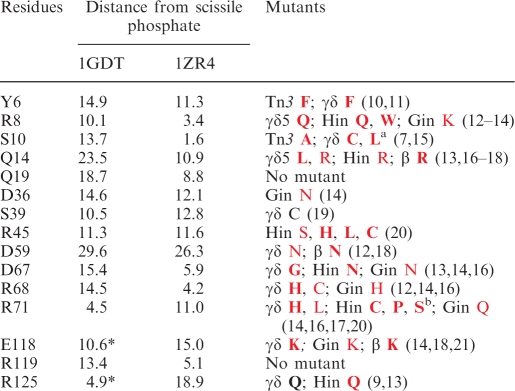 |
The first column lists the residues analysed here. The second and third columns give the distance (Å) of the sidechain functional groups (nearest non-hydrogen atom) from the phosphorus atom of the scissile phosphate, in the two structures shown in Figure 2B; the γδ resolvase dimer-site I structure 1GDT and the cleaved intermediate structure 1ZR4. In 1GDT, the P atom is that of chain C, residue A20, and the residues are in subunit B except for those marked with an asterisk, where the residue in subunit A is closer. In 1ZR4, the P atom is that of the phosphate attached to S10 of subunit A, and all the residues are in subunit A. The final column summarizes previously reported mutations of the residues. Those studied in vitro are in bold, and those studied only in vivo are in plain text. Mutations that abolished recombination activity are in red, and those that did not are in black. Relevant references are given in square brackets.
aMutants of the residue corresponding to S10 have been created for several serine recombinases; all are reported as being inactive in vivo and/or in vitro.
bA number of other mutations of this residue in Hin abolished activity in vivo (20).
The mechanism of catalysis by Tn3 resolvase and other serine recombinases is expected to share common features with the mechanisms of other enzyme-catalysed phosphoryl transfer reactions, including substrate orientation, general acid–base catalysis, and transition state stabilization. A general acid is typically proposed to donate a proton to the leaving group, and a general base to abstract a proton from the attacking hydroxyl group (28,29). The acid and base may be amino acid sidechain functional groups, although water is sometimes proposed to fulfil either function. The general acids or bases involved in catalysis by serine recombinases are still unconfirmed, if indeed they exist (28).
Studies on the mechanism of serine recombinases are made more difficult by their elaborate substrate requirements; for Tn3/γδ resolvase the substrate must have two 114 bp res sites complete with ‘accessory’ resolvase-binding sites II and III (Figure 2A), a directly repeated arrangement of the sites, and negative supercoiling. It can be difficult to analyse the mechanism of catalysis without interference from effects on other features of such a complex system. However, ‘activated’ resolvase multiple mutants that do not require the accessory binding sites, directly repeated sites, or supercoiling have been isolated (30–33). Some of these mutants catalyse rapid recombination between two double-stranded oligonucleotide site Is in vitro (34). Activated resolvase variants, and analogous mutants of the DNA invertases Hin and Gin, have been used to study structural and mechanistic aspects of recombination by the serine recombinases (1). One major advance was the crystal structure of a synaptic intermediate comprising a tetramer of activated mutant γδ resolvase subunits and two cleaved site Is, which revealed a flat hydrophobic interface between pairs of subunits that is proposed to support strand exchange by a rotation mechanism [(22), Figure 2B].
Here, we adopted a mutation-based strategy to analyse the roles of the putative catalytic residues of Tn3 resolvase. Analogous approaches have been used very successfully to characterize the active sites of other DNA-cleaving enzymes including tyrosine recombinases (1,2) and Type I topoisomerases (3,35,36). Following identification of a set of candidate catalytic residues on the basis of their conservation and sidechain properties, we mutated them in the context of an activated Tn3 resolvase variant, then analysed the effects of the mutations on binding to site I, formation of a synaptic complex, and catalysis of cleavage and recombination.
MATERIALS AND METHODS
Purification of resolvases and plasmid DNA; in vivo assays
Methods for the construction of plasmids for expression of resolvase mutants, resolvase overexpression and purification, and plasmid substrate preparation were as described in ref. (34). The activated variant ‘NM resolvase’ used here contains six mutations from wild-type Tn3 resolvase: R2A E56K G101S D102Y M103I Q105L (32). Further mutations were introduced by cloning appropriate synthetic double-stranded oligonucleotides into pFO2, an NM resolvase low-level expression plasmid. pFO2 was created by replacement of the wild-type resolvase reading frame in pMS140 with that of NM resolvase by exchange of a NdeI–Asp718 fragment. pMS140 is similar to pMA6811 (32) except that some undesired restriction sites have been removed. Full sequences of the plasmids are available on request from WMS. Colony colour assays for recombination activity in Escherichia coli were as in ref. (32).
In vitro assays
Plasmid recombination assays were as described in ref. (34). The recombination reaction buffer (before addition of resolvase) contained 50 mM Tris–HCl (pH 8.2), 10 mM MgCl2, 0.1 mM EDTA and 20 µg/ml plasmid DNA. Addition of 0.1 volume of resolvase, diluted in a buffer containing 20 mM Tris–HCl (pH 7.5), 1 mM DTT, 0.1 mM EDTA, 1 M NaCl and 50% v/v glycerol, resulted in a final NaCl concentration of 100 mM. The final concentration of resolvase was 400 nM, and reactions were at 37°C for 1 h, unless stated otherwise. Assays of resolvase-mediated DNA cleavage were similar except that the ‘EG buffer’ before resolvase addition contained 50 mM Tris–HCl (pH 8.2), 0.1 mM EDTA, 40% v/v ethylene glycol and 20 µg/ml plasmid DNA. Reactions were terminated by heating at 70°C for 5 min, or by adding loading buffer (for cleavage assays). The products were then analysed by 1.2% agarose gel electrophoresis (after digestion with restriction enzymes if required). Binding/synapsis reactions were carried out as described (34). The buffer prior to resolvase addition contained 20 mM Tris–HCl (pH 7.5), 10 µg/ml poly(dI/dC), 4% w/v Ficoll and 50 nM site I DNA. The final resolvase concentration was 400 nM unless otherwise stated. Samples were separated in 6.5% polyacrylamide gels (30:0.8, acrylamide: bisacrylamide). The buffer in the gel and tanks was TBE (100 mM Tris base, 100 mM boric acid, 1 mM EDTA; pH 8.3).
Single-strand cleavage assays
The ‘n-site I’ substrate used in single-strand cleavage assays was assembled by annealing a 5′-32P-labelled 70 bp site I top-strand oligonucleotide (sequence as in Figure 5A) with equimolar amounts of two bottom strand oligonucleotides, such that a nick is present at the scissile position on the bottom strand, and the 5′-hydroxyl at the nick is phosphorylated. For the cleavage assays, resolvase (8 µM; 2.2 µl), diluted in a buffer containing 20 mM Tris–HCl (pH 7.5), 1 mM DTT, 0.1 mM EDTA, 1 M NaCl and 50% v/v glycerol, was added to 20 µl aliquots containing 25 nM n-site I DNA, 50 mM Tris–HCl (pH 8.2), 10 µg/ml poly(dI/dC) and 8% w/v Ficoll, giving a final resolvase concentration of 800 nM. Reactions were incubated at 37°C, and were terminated by the addition of 0.25 volume of Stop Buffer [20% w/v Ficoll, 100 mM Tris base, 100 mM boric acid, 1 mM EDTA (pH 8.3), 0.5% w/v sodium dodecyl sulphate (SDS), 1 mg/ml protease K and 0.5 mg/ml bromophenol blue]. The samples were analysed on 6.5% polyacrylamide gels (30:0.8, acrylamide: bisacrylamide); the buffer in the gel and tanks was TBE–SDS (0.1% SDS, 100 mM Tris base, 100 mM boric acid, 1 mM EDTA; pH 8.3). Gels were pre-run for 20 min at 150 V, 22°C. Samples were then loaded and the gels were run for a further 2 h under the same conditions. Gels were dried, and bands were visualized by phosphor-imaging. The cleavage rates (k) presented in Table 2 were calculated from the data in Figure 4C using the equation:
assuming simple first-order kinetics (see Figure 4B).
Figure 5.

Binding and synapsis by NM resolvase and its mutants. (A) The 70 bp double-stranded site I oligonucleotide used in these experiments. The site I sequence is boxed. The top strand was 5′-end-labelled with 32P as shown (asterisk). (B) Binding/synapsis assay. The concentration of the resolvase was 400 nM in all samples. Bands corresponding to unbound site I DNA, complexes of site I with 1 or 2 resolvase subunits, and site I synapse are indicated by U, 1, 2 and S, respectively (34). Lanes are collated from multiple gels. (C) Concentration dependence of binding/synapsis by NM resolvase and selected mutants. Assays were as in B, except that the resolvase concentrations were varied (100, 200, 400, 800 nM).
Table 2.
In vivo recombination and in vitro cleavage rates of NM resolvase mutants
| NM mutant | In vivo resolution | In vitro cleavage rate |
|---|---|---|
| NM | ++ | 1.00 |
| Y6A | − | 0.0002 |
| Y6F | − | 0.0002 |
| R8A | − | <0.0001 |
| R8K | − | 0.0002 |
| S10A | − | <0.0001 |
| Q14A | − | 0.0087 |
| Q14E | − | 0.0039 |
| Q14R | − | 0.016 |
| Q14K | − | 0.016 |
| Q14N | − | 0.018 |
| Q14L | − | 0.014 |
| Q19A | − | 0.0067 |
| Q19E | − | 0.0014 |
| Q19R | − | <0.000l |
| Q19K | − | <0.0001 |
| Q19N | − | 0.0093 |
| Q19L | − | 0.0002 |
| D36A | − | <0.0001 |
| D36N | − | 0.0004 |
| S39A | ++ | 0.025 |
| R45A | − | 0.0034 |
| R45K | − | 0.022 |
| D67A | − | 0.017 |
| D67N | − | 0.018 |
| R68A | − | 0.0014 |
| R68K | − | 0.0016 |
| R71A | − | 0.0007 |
| R71IC | − | 0.0009 |
| E118A | − | 0.015 |
| E118Q | − | 0.019 |
| R119A | ++ | 0.033 |
| R119K | ++ | 0.038 |
| R125A | + | 0.033 |
| R125K | + | 0.028 |
(++) indicates high resolution activity in the E. coli colony colour assay (i.e. pale colonies), (+) indicates partial resolution (pink colonies), and (−) indicates no observable activity (red colonies) [see ref. (32)]. The single-strand cleavage rates of the mutants were estimated from the data shown in Figures 4C and 6B as described in ‘Materials and Methods’ section, and are expressed relative to the rate of NM resolvase estimated from the 800 nM data shown in Figure 4B. Note that the initial rates of those mutants that gave high levels of cleavage in Figures 4C and 6B may be underestimated, due to deviations from first-order kinetics.
Figure 4.

Single-strand cleavage assay. (A) Cleavage of n-site I by NM resolvase. The 70 bp n-site I has a nick at the scissile position on the bottom strand; its sequence is otherwise identical to the site I substrate shown in Figure 5A. The 5′-end at the nick is phosphorylated (incorporated during chemical synthesis), and the 5′-end of the top strand was end-labelled with 32P (asterisk in diagram). Cleavage of n-site I by resolvase generates a 35 bp labelled half-site product, and an unlabelled resolvase–DNA covalent complex, as shown. (B) Left panel: time course of cleavage of n-site I by NM resolvase (concentration 400 nM). Aliquots were stopped at the times shown, and separated on a 6.5% polyacrylamide gel. Right panel: data from the gel shown on the left, and a corresponding time course at 800 nM NM resolvase, were quantitated and plotted. (C) Cleavage of n-site I by NM resolvase and mutants (800 nM). Reactions were for 30 min at 37°C. The percentage of cleavage product is indicated below each lane.
Structure analysis
The following crystal structures which include serine recombinase catalytic domains are available: wild-type γδ resolvase [two structures, 2RSL and 1GDR, containing four structurally distinct subunits; (25–27)]; a wild-type γδ resolvase dimer bound to site I DNA [1GDT; two distinct subunits; (24)]; an activated mutant γδ resolvase N-terminal domain tetramer [2GM5; four distinct subunits; (23)]; γδ resolvase mutants in synaptic complexes with cleaved site I DNA [three structures, with six distinct subunit forms; 1ZR2, 1ZR4 and 2GM4; (22,23)]; a dimer of Sin recombinase bound to DNA [2R0Q; two distinct subunits (37)]; the catalytic domain of TP901 integrase [3BVP; two distinct subunits (38)]; the catalytic domain of a Clostridium recombinase [3G13; two distinct subunits (39)]; and the catalytic domain of a Streptococcus recombinase [3GUV; one subunit (40)]. Structures were visualized and analysed using PyMOL (41).
RESULTS
Mutation of candidate active site residues of Tn3 resolvase
Mutations of putative active site residues were made in an activated Tn3 resolvase variant (‘NM resolvase’) which catalyses efficient site I × site I recombination in vivo and in vitro (32,34). The selection of amino acid residues which might contribute to the resolvase active site was based on sequence alignment of serine recombinases [(4,5) and M.R. Boocock, personal communication; Figure 1], and findings from previous studies on mutants of Tn3 resolvase, γδ resolvase, and other serine recombinases [reviewed by Grindley (9) and Johnson (42); see Table 1]. The following criteria were used for selection: (i) The same residue (or a conservative substitution with a similar sidechain functional group) must be present at an equivalent point in the sequence in most serine recombinases, and in all the biochemically well-characterized members of the resolvase/invertase group shown in Figure 1. (ii) The residue must have a charged or polar sidechain which might participate in catalysis. These criteria led to selection of 15 residues, including all those implicated in catalysis by previous studies: Y6, R8, S10, Q14, Q19, D36, S39, R45, D59, D67, R68, R71, E118, R119 and R125 (Table 1). Conservative mutations of these residues were made in order to minimize disruption of local and global protein structure; thus, arginine residues were changed to lysine (R → K), aspartate to asparagine (D → N), glutamate to glutamine (E → Q), glutamine to glutamate (Q → E), tyrosine to phenylalanine (Y → F) and serine to alanine (S → A). Each selected residue (except D59) was also mutated to alanine, to probe the effect of loss of the polar sidechain. The mutant resolvases were purified, and their biochemical properties were characterized as described in the following sections. In preliminary studies, the purified D59N mutant protein proved to be very insoluble. As D59 is far from the other active site residues (Table 1) and an alternative function has been proposed for it [stabilization of a sharp turn in the polypeptide; (25)], we decided not to carry out any further work on it.
Catalytic properties of NM resolvase mutants
Recombination
A preliminary characterization of site I × site I recombination activity of the mutants was carried out using a colony colour-based assay in E. coli [(30), Table 2]. All mutations at eleven of the fourteen chosen residues resulted in red colonies on MacConkey agar plates, indicating defective resolution. Only S39A, R119A, R119K, R125A and R125K had observable resolution activity (pink or pale colonies).
Following purification of all the mutant proteins, we assayed their in vitro recombination activity on a site I × site I plasmid substrate (pAL225; Figure 3A). Activated resolvases promote recombination between the two site Is in a single pAL225 molecule to give inversion and resolution products. Smaller amounts of intermolecular recombination products may be observed. Resolvase may fail to complete recombination, leaving species with double-strand breaks at one or both of the site Is, and a resolvase subunit attached via its active site serine residue (S10) to each DNA 5′-end (in the experiments below, the attached resolvase is degraded by treatment with protease). When the DNA is analysed without restriction enzyme digestion, products of topoisomerization may be observed as ‘ladders’ of bands between the supercoiled and nicked plasmid bands. Topoisomerase activity requires transient cleavage and re-ligation of at least one DNA strand. The reactions shown in Figure 3B were carried out for 16 h at 37°C (except for the ‘parent’ NM resolvase; 1 h reaction) to allow even very slow recombination, cleavage or topoisomerase activity to be detectable. The results were consistent with the in vivo assay described above, but revealed low levels of activity by some of the defective mutants. Under these conditions, NM resolvase consumes more than half the pAL225 substrate in <1 min (34), so the predominance of substrate even after 16 h in many of the reactions shown in Figure 3B indicates very severe inhibition of recombination and cleavage activity. No recombination products were detectable in the restriction digests of reactions with the S10, Y6, R8, Q14, Q19, D36, R68 and R71 mutants, whereas all the other mutants gave observable recombination products, accompanied in some cases by products of DNA cleavage at site I. Electrophoresis of the DNA without restriction enzyme digestion revealed low cleavage activity (a full-length linear band) and topoisomerase activity by some recombination-defective mutants, notably Q14A and the R68 and R71 mutants.
Figure 3.

Recombination and DNA cleavage by NM resolvase active site mutants. (A) Diagram of the substrate plasmid pAL225, and its cleavage/recombination products. DNA segments between the recombination sites and restriction sites are denoted with letters A–D, and their sizes are given below the diagram. (B) Recombination of pAL225 by NM resolvase and mutants. Reactions were for 16 h at 37°C (1 h for the parent NM resolvase). Upper panel: samples were loaded on the gel after stopping the reaction, without further treatment. Lower panel: samples were first digested with PstI and HindIII. (C) Cleavage of pAL225 by NM resolvase and mutants in EG buffer, which contains 40% ethylene glycol. Reactions were for 16 h at 37°C. Samples were treated with protease K/SDS loading buffer prior to electrophoresis (no restriction enzyme treatment). The order of the samples is as in Figure 3B. Annotation: sc, supercoiled plasmid substrate; nc, nicked circular substrate; rc, resolution products (free circles); int, intermolecular recombination/cleavage product; 4.9 kb, full-length linear product of cleavage at one of the two site Is; 2.45 kb, products of cleavage at both site Is; nr, non-recombinant restriction fragment; inv, inversion product restriction fragment; res, resolution product restriction fragment; cp, cleavage product restriction fragment.
DNA cleavage
We also compared the activities of the resolvase mutants in a reaction buffer that contains 40% ethylene glycol (EG buffer); these conditions inhibit rejoining of the pAL225 DNA ends after double-strand cleavage by resolvase, leading to a 4.9 kbp linear product if only one site is cleaved, or two linear fragments, both 2.45 kbp, if both sites are cleaved (Figure 3A). In EG buffer NM resolvase cleaves more than half of the pAL225 DNA in <1 min at 37°C (our unpublished results). All the mutants that catalysed detectable recombination (see above) also gave large amounts of cleavage products after reaction for 16 h (Figure 3C); in general, the extent of cleavage was higher than the extent of recombination. The 2.45 kbp band (indicating high cleavage activity) was clearly visible in the products of the reactions with S39A and both mutants of R45, D67, E118, R119 and R125. For mutants with lower activity, only the 4.9 kbp full-length linear band was obvious. All the mutants of Y6, R8, S10, Q14, Q19, D36, R68 and R71 were either inactive or had only very weak cleavage activity, reflecting the results of the recombination assay (Figure 3B).
Single-strand cleavage assay
The effects of mutations on the rates of recombination and double-strand cleavage in plasmid substrates may be amplified because formation of the observed products requires multiple steps, each of which might be affected (at least two single-strand cleavages to linearize the substrate, or four cleavages followed by one or more re-ligation steps to make a recombinant product). Strand cleavage activity might also be masked by re-ligation of the cleaved DNA strands. We therefore devised an assay in which products are observed following cleavage of only one DNA strand, and re-ligation is suppressed. Oligonucleotides were annealed to form a 70 bp site I-containing DNA molecule (‘n-site I’) with a nick in the bottom strand at the position where cleavage/re-ligation would occur. Cleavage of the top strand of this substrate by resolvase creates a ‘left’ half-site which does not undergo re-ligation (Figure 4A), possibly because it has no covalently attached resolvase subunit. The other, ‘right’ half-site, with its covalently attached resolvase subunit, is able to make recombinant products (WMS and FJO, unpublished results). Here, we end-labelled the left half-site to observe cleavage of the top strand.
The n-site I substrate was cleaved rapidly by NM resolvase (t1/2 <1 min; Figure 4B). In contrast, Q19E and both mutants of Y6, R8, S10, D36, R68 and R71 cleaved <10% of the substrate after 30 min of reaction (Figure 4C), corresponding to an estimated rate reduction of ∼103-fold or more relative to NM resolvase (Table 2).
Binding and synapsis by NM resolvase mutants
A 70 bp site I double-stranded oligonucleotide (sequence shown in Figure 5A) was treated with NM resolvase or the mutants, and the samples were separated by polyacrylamide gel electrophoresis under conditions that favour observation of a ‘site I synapse’ comprising two copies of site I bound together by a resolvase tetramer (34) (Figure 5B). All the mutants were proficient in binding to site I (as can be seen by the reduced intensity of the unbound band U, and the presence of retarded bands corresponding to complexes with 1 or 2 resolvase subunits), but some gave lower amounts of the site I synapse band S than NM resolvase. Some of these synapsis-defective mutants (most clearly, the mutants of Y6 and D36) gave increased amounts of complexes of a single site I with one or two resolvase subunits. For other mutants (including Q19E, R119A and R119K), a smeary band running between the positions of the site I synapse and the complex of site I bound to 2 subunits suggested partial dissociation of the site I synapse during electrophoresis.
We analysed the effects of resolvase concentration on complex formation by NM resolvase and selected mutants with defective synapsis properties (Figure 5C). The site I synapse was the predominant bound species at all the NM resolvase concentrations tested; there was only a trace of the complex of site I with a resolvase monomer and no distinct band corresponding to the 2-subunit complex. In contrast, with the Y6A, Q19E and D36N mutants, the site I synapse band was very faint even at very high resolvase concentration, most of the DNA being in the complex of a single site I with 2 resolvase subunits (and a smeary higher band in the case of Q19E, as in Figure 5B). Instability of the synapse might therefore contribute to the catalytic deficiency of these mutants. The Y6F mutant was intermediate in its behaviour, making substantial amounts of synapse at high resolvase concentration.
Further analysis of mutants of Q14 and Q19
The functions of two glutamine residues, Q14 and Q19, are especially obscure (see ‘Discussion’ section). Data for the Q → A and Q → E mutants are presented above, but we decided to test the effects of a wider range of mutations of these two residues. Mutations to asparagine (Q → N) probed the effects of shortening the side chain by one methylene group while retaining the amide functional group; mutations to arginine or lysine (Q → R, Q → K) introduced a positive charge which might strengthen any interactions of the sidechain with the negatively charged DNA backbone; and mutations to leucine (Q → L) replaced the glutamine sidechain with a similar-sized but hydrophobic one.
None of the Q14 or Q19 mutants gave recombination products from pAL225, even after 16 h (Figure 6A, top panel). However, the assay in EG buffer (Figure 6A, lower panel) revealed cleavage activity by Q19N and all the Q14 mutants except Q14E. The active mutants also displayed some topoisomerase activity, and thus they can promote ligation as well as cleavage. The assay with n-site I (Figure 6B) revealed single-strand cleavage by all the Q14 mutants, along with Q19N, Q19A and Q19E.
Figure 6.

Properties of Q14 and Q19 mutants. Assays in parts A, B and C were as in Figures 3, 4 and 5 respectively. The results for Q14A, Q14E, Q19A and Q19E shown in Figures 3, 4 and 5 are included again here to allow comparison with the other mutants. (A) In vitro recombination (upper panel) and cleavage in EG buffer (lower panel) of plasmid substrate pAL225. (B) Single-strand cleavage assay on n-site I. (C) Binding/synapsis of site I.
The mutations of Q14 and Q19 did not seriously impair binding or synapsis (Figure 6C), except for Q14E and Q19E where glutamine is replaced by the negatively charged glutamate. Surprisingly, mutation of Q14 to the positively charged residues arginine and lysine (Q14R and Q14K) significantly increased the amount of site I synapse observed in this assay. Some Q19 mutants showed reduced binding of site I, but all formed a clear synapse band except Q19E (and a retarded band seen with Q19E can be interpreted as evidence for an unstable synapse; see above).
DISCUSSION
Mutations of several of the fourteen chosen candidate catalytic residues have very large effects on the rate of catalysis by NM resolvase. The effects of mutations of Y6, R8, S10, D36, R68 and R71 are especially strong, with complete abolition of recombination activity (Figure 3) and ∼103-fold reduction or more in the rate of single-strand cleavage (Figure 4 and Table 2), consistent with key roles of these sidechains in catalysis. Mutations of five other residues (Q14, Q19, R45, D67, E118) had large but less extreme effects. In previous studies on topoisomerases, a residue whose substitution by alanine resulted in a reaction rate reduction of ≥102-fold was deemed to be ‘essential’ for catalysis (35). By this criterion Q14, Q19 and R45 would be essential along with Y6, R8, S10, D36, R68 and R71, but we regard the latter group as being more important for catalysis because the effects of their mutations are about an order of magnitude larger. None of the mutations abolished binding of NM resolvase to site I, and none completely inhibited site I synapse formation, but there were some striking effects on the amounts of synapse observed (discussed further below).
A number of serine recombinase crystal structures are now available; some are of the proteins alone and others are of complexes with DNA (see ‘Materials and Methods’ section). Below, we discuss our results in the context of these structures, which should give us insight into the possible arrangement of residues at the active site for catalysis of DNA cleavage and rejoining, and how the active site could be assembled. It is worth noting here that the mutation-based strategy used in this study provides information on the roles of the amino acid sidechains, but not on any catalytic roles of main-chain atoms.
Residues S10, Y6, R8 and D36
The function of the S10 sidechain –OH as the nucleophile that attacks the scissile phosphodiester is well established (see ‘Introduction’ section). Therefore, as expected, the S10A mutant of NM resolvase was completely inactive in our assays for recombination, cleavage and topoisomerization. The S10A mutation also causes some reduction in the extent of binding and synapsis of site I (Figure 5 and data not shown), in accord with previous reports of effects of catalytic serine mutations on binding to the recombination site (7,20).
The functions of three other residues where mutations are very deleterious (Y6, R8 and D36) may be interconnected. All three residues are highly conserved in the serine recombinases, though the D36 equivalent is sometimes glutamate instead of aspartate, and the Y6 equivalent is a valine in one well-characterized enzyme [Bxb1 integrase (43); Figure 1]. In previous studies, the Y6F mutant of wild-type Tn3 resolvase was found to be almost inactive (10), but weak cleavage, ligation and topoisomerase activity by the γδ resolvase Y6F mutant was observed (11). Y6 is buried in the hydrophobic core of the catalytic domain, quite far from S10. In most of the structures the hydroxyl group of the Y6 sidechain is within hydrogen-bonding distance of the carboxyl group of D36 or its equivalent (as in Figure 7B). D36 is closer to the surface of the protein and also contacts mainchain atoms at the start of the B-helix. The Y6 hydroxyl is also close to the guanidinium moiety of the R8 sidechain, and in one structure (2GM5) appears to be interacting with it (Figure 7B). In turn, R8 is close to S10 and the DNA (in 1ZR2, 1ZR4 and 2GM4), its guanidinium group being within hydrogen-bonding distance of the non-bridging oxygen atoms of the phosphodiester attached to S10 (Figure 7A). It has been proposed that interactions by Y6 and D36 might help to maintain the tertiary structure of the active site region (24,11), but the very large effects of Y6 and D36 mutations on cleavage and recombination that we observe are also consistent with a more direct role in catalysis.
Figure 7.

Sidechain interactions in the resolvase active site. (A) View of the 1ZR4 crystal structure [subunit A; (22)] showing R8, Q19, D67, R68 and R71 clustered around S10 which is covalently attached to the 5′ phosphate of the cleaved site I DNA. The dashed lines indicate putative hydrogen-bonding interactions, and the dotted line indicates an apparent interaction between the sidechains of R8 and R68 (N–N distance 3.3 Å). (B) Interactions involving Y6, R8, D36 and R71 in one subunit (A) of the mutant γδ resolvase tetramer structure 2GM5 (23). The N–N distance between R8 and R71 (dotted line) is 3.1 Å. This structure contains no DNA. The protein has the catalytic residue mutation R68H.
A relationship between the roles of D36 and Y6 is supported by our results, which show that mutants of these residues behave very similarly in binding/synapsis assays; they give reduced amounts of the site I synapse band and increased amounts of complexes with a single site I (Figure 5), thus apparently reverting to more ‘wild-type’ behaviour compared to the synapsis-proficient NM resolvase (34). However, the Y6F mutant does give a strong synapse band at higher protein concentration (Figure 5C), so its catalytic defect is not simply due to lack of synapsis.
The base that abstracts a proton from the S10 hydroxyl group during DNA cleavage and from the deoxyribose 3′-hydroxyl in the re-ligation step has not been identified. We speculate that the guanidinium moiety of R8 might provide the strong base that would be necessary to deprotonate these rather non-acidic hydroxyl groups. R8 is positioned appropriately with respect to the scissile DNA phosphodiester, and might also interact with Y6 (see above). The role of Y6 might be to facilitate removal of a proton from R8 to form the base, and the D36 carboxylate might be the ultimate proton acceptor. The arginine sidechain has been deduced to act as a base in a number of enzyme active sites; in some cases, an interaction between an arginine guanidinium and a nearby carboxylate is proposed to lower the arginine pKa and thus facilitate deprotonation to form a base (44). In resolvase, the interaction of R68 and D67 conforms to this model, but the D67 sidechain is not essential (see below). However, R8 might have an analogous relationship with a carboxylate group, being linked to D36 via the Y6 hydroxyl. Interestingly, the guanidinium group of R8 also appears to associate with the guanidinium group of another arginine sidechain in 1ZR4 (R68; Figure 7A) and in 2GM5 (R71; Figure 7B).
The other conserved arginines
Unlike many enzymes that promote cleavage of phosphodiester bonds, resolvase-catalysed recombination does not require any divalent metal ions (45–47). Metal ions (usually Mg2+) are often assigned the role of withdrawing electrons from the phosphorus atom and stabilizing negative charge on the oxygen atoms of the phosphodiester in the transition state (29), but resolvase (and other serine recombinases) presumably uses positively charged amino acid sidechains such as that of arginine instead (1,9,28). The candidates for this role in Tn3 resolvase are R8, R45, R68, R71, R119 and R125.
R119 and R125 are quite distant from the scissile phosphate in the resolvase–DNA structures, and the R119A and R125A mutants are proficient for cleavage and recombination, so these residues apparently have non-catalytic roles. The mutants are however affected in synapsis (Figure 5). A regulatory function for R119 and R125 in the rotation step of strand exchange has been proposed (1,22).
R45 is closer to the scissile phosphate, though it does not contact it in any of the structures. R45 has been suggested to have a structural role, to ‘close a lid’ formed by the loop of residues 37–45 over Y6 and other active site residues by contacting the carboxylate of D75 (27). The NM resolvase mutants R45A and R45K retain cleavage and recombination activity (Figure 3), supporting the idea of a non-catalytic function.
Mutations of the other three arginine residues, R8, R68 and R71, have the most severe effects on catalysis. A possible role for R8 as a base is discussed above. The sidechains of both R8 and R68 are apparently in contact with the non-bridging oxygens of the phosphate covalently attached to S10 in the crystal structures of cleaved intermediates (1ZR2, 1ZR4 and 2GM4), and therefore are strong candidates for providing stabilizing positive charge during catalysis (1; Figure 7A). The R71 sidechain is also quite close to the scissile phosphate, but it has not been observed to contact it; in 1ZR4 it interacts with an adjacent phosphate. The mutants R71K and R71A are not inert but cleavage and recombination are severely reduced (Figures 3 and 4), so R71 may well also be involved in the mechanism of phosphate activation.
S39, D67 and E118
The mutants of these three residues retained substantial cleavage and recombination activities, suggesting non-catalytic roles [as was concluded previously for S39; (19)]. The contact between the D67 carboxylate [previously proposed as the general base; (48)] and the R68 guanidinium group (Figure 7A) might be involved in active site structural organization, as has been suggested by Yuan et al. (38) for the corresponding TP901 integrase residue D81. E118 is on the 1–2 dimer interface; the two E118 sidechains in the γδ resolvase dimer (and the corresponding residues in structures of other serine recombinases) apparently contact each other. The E118 sidechains have moved apart in the structures of cleaved intermediates, e.g. 1ZR4, and are quite far from the scissile phosphates in all the structures, but they might be much closer to the DNA immediately prior to cleavage, and thus might have an accessory role in catalysis.
Q14 and Q19
We were intrigued by the conservation of these two glutamine residues. The Q14 sidechain is on the surface of the protein and does not come close to the DNA nor any of the other potential active site residues in any of the structures, but it is clearly important; all the mutants were severely recombination-defective (Figure 6A and Table 1). On the other hand, all the Q14 mutants showed substantial activity in the single-strand cleavage assay (Figure 6B). Mutation of Q14 to a positively charged residue (Q14R, Q14K) increased the yield of site I synapse (Figure 6C, and data not shown), whereas a negatively charged sidechain (Q14E) decreased the amount of synapse and overall binding to site I, suggesting that Q14 might be close to the negative charge of the DNA backbone in complexes of resolvase with DNA. However, we still have no definite proposal for the function of this residue. The Q19A, Q19E and Q19N mutants retained some cleavage activity; Q19N was the most active, so H-bonding functionality at this position might be important. In the crystal structures, the carboxamide sidechain of Q19 is within H-bonding distance of the main-chain amino and carboxyl groups of R8, as well as the sidechain of another active site residue R68 (in 1GDT), so its role might be structural, to orient the R8 and R68 residues appropriately in the active site.
Involvement of active site residues in synapsis
A surprising finding of our study is that mutations of several of the putative active site residues of resolvase affect the amount of the site I synapse (and to a lesser extent, complexes with a single site I) (Figure 5 and 6). It might have been predicted that mutations of any of the six conserved arginine residues that remove the positive charge of the sidechain would reduce affinity for the negatively charged DNA, but this was not the case except for the E-helix residues R119 and R125, which are non-essential for catalysis. The strongest ‘synapsis-down’ effects were instead seen with the mutations of Y6 and D36. The Q14E and Q19E mutations also substantially reduced the amount of synapse observed on the gels, whereas other Q14 and Q19 mutations had smaller effects or even increased the amount of synapse (Q14K and Q14R). Reduced binding and synapsis by the S10A mutant was also noted (see above).
Synapsis of two site Is is proposed to occur by interaction of two site I-resolvase dimer complexes (22,34). The synaptic interface observed crystallographically involves residues that are far from the DNA, as does a hypothetical interface at initiation of synapsis (22,31,32), and none of the candidate catalytic residues mutated here are proposed to be involved. We hypothesize that assembly of the functional active site on the DNA is coupled to conformational changes in other parts of the resolvase tetramer which stabilize the site I synapse. Mutations of active site residues might thus disrupt this process and reduce synapse stability. In stable pre-cleavage synapses (such as those we see in our assays) the active site might be more fully assembled than it is in the current crystal structures, with the resolvase tetramer poised to initiate catalysis of strand cleavage. The pre-cleavage synapse, which has so far only been characterized at low resolution (15) is thus a very attractive target for future higher-resolution structural studies.
Substrate-induced organization of functional active sites might be common in enzymes that catalyse DNA phosphoryl transfer reactions; other proteins for which a similar paradigm has been proposed include EcoRI endonuclease (49), MutH (50), topoisomerase I (51) and the tyrosine recombinases (52). This proposed link between resolvase-mediated catalysis and site I synapsis may be only one of a series of ‘checkpoints’ in the mechanism of wild-type resolvase which serve to ensure that catalysis occurs only in the biologically appropriate circumstances. Cooperative assembly of Tn3 resolvase monomers to form dimers on the binding sites of res may ensure high specificity of binding (53); the requirement for a synaptic complex involving intertwining of the two res sites mediated by a complex network of protein–protein and protein–DNA interactions provides specificity for recombination between sites in the same DNA molecule and in direct repeat (54); assembly of Tn3 resolvase subunits into functional tetramers occurs only at synapsis of two site Is (34); following active site assembly, the catalytic events themselves (four strand cleavages, rotation, and four strand re-ligations) may be orchestrated to ensure an ‘all or nothing’ reaction; and features of the natural synaptic complex limit the strand exchange mechanism to a single round, giving the biologically relevant resolution products.
FUNDING
Biotechnology and Biosciences Research Council (No. BB/E022200) and a UK Commonwealth Academic Staff Scholarship Award (to F.J.O.). Funding for open access charge: Biotechnology and Biosciences Research Council (BB/E022200).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors are very grateful to Sean Colloms, Phoebe Rice, Sally Rowland and Martin Boocock for helpful comments and advice, and for critical reading of the manuscript draft. The authors also thank Bill Duddy, Elizabeth Kilbride and Arlene McPherson for preliminary studies on resolvase mutants.
REFERENCES
- 1.Grindley NDF, Whiteson KL, Rice PA. Mechanism of site-specific recombination. Ann. Rev. Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- 2.Jayaram M, Grainge I. Introduction to site specific recombination. In: Mullany P, editor. The Dynamic Bacterial Genome. New York, NY, USA: Cambridge University Press; 2005. pp. 33–81. [Google Scholar]
- 3.Schoeffler AJ, Berger JM. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Quart. Rev. Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 4.Smith MCM, Thorpe MM. Diversity in the serine recombinases. Mol. Microbiol. 2002;44:299–307. doi: 10.1046/j.1365-2958.2002.02891.x. [DOI] [PubMed] [Google Scholar]
- 5.Rowland SJ, Stark WM. Site-specific recombination by the serine recombinases. In: Mullany P, editor. The Dynamic Bacterial Genome. New York: Cambridge University Press; 2005. pp. 83–120. [Google Scholar]
- 6.Reed RR, Moser CD. Resolvase-mediated recombination intermediates contain a serine residue covalently linked to DNA. Cold Spring Harbor Symp. Quant. Biol. 1984;49:245–249. doi: 10.1101/sqb.1984.049.01.028. [DOI] [PubMed] [Google Scholar]
- 7.Hatfull GF, Grindley NDF. Analysis of γδ resolvase mutants in vitro: evidence for an interaction between serine-10 of resolvase and site-I of res. Proc. Natl Acad. Sci. USA. 1986;83:5429–5433. doi: 10.1073/pnas.83.15.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klippel A, Mertens G, Patschinsky T, Kahmann R. The DNA invertase of phage Mu: formation of a covalent complex with DNA via a phosphoserine at amino acid position 9. EMBO J. 1988;7:1229–1237. doi: 10.1002/j.1460-2075.1988.tb02935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grindley NDF. The movement of Tn3-like elements: transposition and cointegrate resolution. In: Craig NL, Craigie R, Gellert M, Lambowitz A, editors. Mobile DNA II. Washington, DC: American Society for Microbiology Press; 2002. pp. 272–302. [Google Scholar]
- 10.Arnold PH. University of Glasgow; 1997. Mutants of Tn3 resolvase. Ph.D. Thesis. [Google Scholar]
- 11.Leschziner AE, Boocock MR, Grindley NDF. The tyrosine-6 hydroxyl of γδ resolvase is not required for the DNA cleavage and rejoining reactions. Mol. Microbiol. 1995;15:865–870. doi: 10.1111/j.1365-2958.1995.tb02356.x. [DOI] [PubMed] [Google Scholar]
- 12.Hughes RE, Hatfull GF, Rice P, Steitz TA, Grindley NDF. Cooperativity mutants of γδ resolvase identify an essential interdimer interaction. Cell. 1990;63:1331–1338. doi: 10.1016/0092-8674(90)90428-h. [DOI] [PubMed] [Google Scholar]
- 13.Nanassy OZ, Hughes KT. In vivo identification of intermediate stages of the DNA inversion reaction catalyzed by the Salmonella Hin recombinase. Genetics. 1998;149:1649–1663. doi: 10.1093/genetics/149.4.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spaeny-Dekking L, Schlicher E, Franken K, van de Putte P, Goosen N. Gin mutants that can be suppressed by a Fis-independent mutation. J. Bacteriol. 1995;177:222–228. doi: 10.1128/jb.177.1.222-228.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nöllmann M, He J, Byron O, Stark WM. Solution structure of the Tn3 resolvase-crossover site synaptic complex. Mol. Cell. 2004;16:127–137. doi: 10.1016/j.molcel.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 16.Boocock MR, Zhu X, Grindley NDF. Catalytic residues of γδ resolvase act in cis. EMBO J. 1995;14:5129–5140. doi: 10.1002/j.1460-2075.1995.tb00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman BJ, Grindley NDF. Mutants of γδ resolvase: a genetic analysis of the recombination function. Cell. 1984;38:463–469. doi: 10.1016/0092-8674(84)90501-4. [DOI] [PubMed] [Google Scholar]
- 18.Canosa I, Ayora S, Rojo F, Alonso JC. Mutational analysis of a site-specifc recombinase: characterization of the catalytic and dimerization domains of the β recombinase of pSM19035. Mol. Gen. Genet. 1997;255:467–476. doi: 10.1007/s004380050519. [DOI] [PubMed] [Google Scholar]
- 19.Hatfull GF, Sanderson MR, Freemont PS, Raccuia PR, Grindley NDF, Steitz TA. Preparation of heavy atom derivatives using site-directed mutagenesis: Introduction of cysteine residues into γδ resolvase. J. Mol. Biol. 1989;208:661–667. doi: 10.1016/0022-2836(89)90156-3. [DOI] [PubMed] [Google Scholar]
- 20.Adams CW, Nanassy O, Johnson RC, Hughes KT. Role of arginine-43 and arginine-69 of the Hin recombinase catalytic domain in the binding of Hin to the hix DNA recombination sites. Mol. Microbiol. 1997;24:1235–1247. doi: 10.1046/j.1365-2958.1997.4141789.x. [DOI] [PubMed] [Google Scholar]
- 21.Hatfull GF, Noble SM, Grindley NDF. The γδ resolvase induces an unusual DNA structure at the recombinational crossover point. Cell. 1987;49:103–110. doi: 10.1016/0092-8674(87)90760-4. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Kamtekar S, Xiong Y, Sarkis GJ, Grindley NDF, Steitz TA. Structure of a synaptic γδ resolvase tetramer covalently linked to two cleaved DNAs. Science. 2005;309:1210–1215. doi: 10.1126/science.1112064. [DOI] [PubMed] [Google Scholar]
- 23.Kamtekar S, Ho RS, Cocco MJ, Li W, Wenwieser SVCT, Boocock MR, Grindley NDF, Steitz TA. Implications of structures of synaptic tetramers of γδ resolvase for the mechanism of recombination. Proc. Natl Acad. Sci. USA. 2006;103:10642–10647. doi: 10.1073/pnas.0604062103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang W, Steitz TA. Crystal structure of the site-specific recombinase γδ resolvase complexed with a 34 bp cleavage site. Cell. 1995;82:193–208. doi: 10.1016/0092-8674(95)90307-0. [DOI] [PubMed] [Google Scholar]
- 25.Sanderson MR, Freemont PS, Rice PA, Goldman A, Hatfull GF, Grindley NDF, Steitz TA. The crystal structure of the catalytic domain of the site-specific recombination enzyme γδ resolvase at 2.7 Å resolution. Cell. 1990;63:1323–1329. doi: 10.1016/0092-8674(90)90427-g. [DOI] [PubMed] [Google Scholar]
- 26.Rice PA, Steitz TA. Model for a DNA-mediated synaptic complex suggested by crystal packing of γδ resolvase subunits. EMBO J. 1994;13:1514–1524. doi: 10.2210/pdb1gdr/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rice PA, Steitz TA. Refinement of γδ resolvase reveals a strikingly flexible molecule. Structure. 1994;2:371–384. doi: 10.1016/s0969-2126(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 28.Mizuuchi K, Baker TA. Chemical mechanisms for mobilising DNA. In: Craig NL, Craigie R, Gellert M, Lambowitz A, editors. Mobile DNA II. Washington, DC: American Society for Microbiology Press; 2002. pp. 12–23. [Google Scholar]
- 29.Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: Two-Mg2+-ion catalysis and substrate specificity. Mol. Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Arnold PH, Blake DG, Grindley NDF, Boocock MR, Stark WM. Mutants of Tn3 resolvase which do not require accessory binding sites for recombination activity. EMBO J. 1999;18:1407–1414. doi: 10.1093/emboj/18.5.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkis GJ, Murley LL, Leschziner AE, Boocock MR, Stark WM, Grindley NDF. A model for the γδ resolvase synaptic complex. Mol. Cell. 2001;8:623–631. doi: 10.1016/s1097-2765(01)00334-3. [DOI] [PubMed] [Google Scholar]
- 32.Burke ME, Arnold PH, He J, Wenwieser SVCT, Rowland SJ, Boocock MR, Stark WM. Activating mutations of Tn3 resolvase marking interfaces important in recombination catalysis and its regulation. Mol. Microbiol. 2004;51:937–948. doi: 10.1046/j.1365-2958.2003.03831.x. [DOI] [PubMed] [Google Scholar]
- 33.Rowland SJ, Boocock MR, McPherson AL, Mouw KW, Rice PA, Stark WM. Regulatory mutations in Sin recombinase support a structure-based model of the synaptosome. Mol. Microbiol. 2009 doi: 10.1111/j.1365-2958.2009.06756.x. DOI: 10.1111/j.1365-2958.2009.06756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olorunniji FJ, He J, Wenwieser S.VCT, Boocock MR, Stark WM. Synapsis and catalysis by activated Tn3 resolvase mutants. Nucleic Acids Res. 2008;36:7181–7191. doi: 10.1093/nar/gkn885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shuman S. Vaccinia virus DNA topoisomerase: a model eukaryotic type IB enzyme. Biochim. Biophys. Acta. 1998;1400:321–337. doi: 10.1016/s0167-4781(98)00144-4. [DOI] [PubMed] [Google Scholar]
- 36.Krogh BO, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell. 2000;5:1035–1041. doi: 10.1016/s1097-2765(00)80268-3. [DOI] [PubMed] [Google Scholar]
- 37.Mouw KW, Rowland SJ, Gajjar MM, Boocock MR, Stark WM, Rice PA. Architecture of a serine recombinase-DNA regulatory complex. Mol. Cell. 2008;30:145–155. doi: 10.1016/j.molcel.2008.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan P, Gupta K, Van Duyne GD. Tetrameric structure of a serine integrase catalytic domain. Structure. 2008;16:1275–1286. doi: 10.1016/j.str.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 39.Bagaria A, Burley SK, Swaminathan S. Crystal structure of a putative conjugative transposon recombinase from Clostridium difficile. 2009 DOI:10.2210/pdb3g13/pdb. [Google Scholar]
- 40.Bonanno JB, Freeman J, Bain KT, Do J, Sampathkumar P, Wasserman S, Sauder JM, Burley SK, Almo SC. Crystal structure of a resolvase family site-specific recombinase from Streptococcus pneumoniae. 2009 DOI:10.2210/pdb3guv/pdb. [Google Scholar]
- 41.DeLano WL. The PyMOL Molecular Graphics System ( http://www.pymol.org/) 2002. (Last accessed date August 5, 2009)
- 42.Johnson RC. Bacterial site-specific DNA inversion systems. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, DC: American Society for Microbiology Press; 2002. pp. 230–271. [Google Scholar]
- 43.Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF. Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol. Microbiol. 2003;50:463–473. doi: 10.1046/j.1365-2958.2003.03723.x. [DOI] [PubMed] [Google Scholar]
- 44.Gullen-Schlippe YV, Hedstrom L. A twisted base? The role of arginine in enzyme-catalysed proton abstractions. Arch. Biochem. Biophys. 2005;433:266–278. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 45.Reed RR, Grindley NDF. Transposon-mediated site-specific recombination in vitro: DNA cleavage and protein-DNA linkage at the recombination site. Cell. 1981;25:721–728. doi: 10.1016/0092-8674(81)90179-3. [DOI] [PubMed] [Google Scholar]
- 46.Stark WM, Sherratt DJ, Boocock MR. Site-specific recombination by Tn3 resolvase: topological changes in the forward and reverse reactions. Cell. 1989;58:779–790. doi: 10.1016/0092-8674(89)90111-6. [DOI] [PubMed] [Google Scholar]
- 47.Castell SE, Jordan SL, Halford SE. Site-specific recombination and topoisomerization by Tn21 resolvase: role of metal ions. Nucleic Acids Res. 1986;14:7213–7226. doi: 10.1093/nar/14.18.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan B, Maciejewski MW, Marintchev A, Mullen GP. Solution structure of the catalytic domain of γδ resolvase. Implications for the mechanism of catalysis. J. Mol. Biol. 2001;310:1089–1107. doi: 10.1006/jmbi.2001.4821. [DOI] [PubMed] [Google Scholar]
- 49.Kurpiewski MR, Engler LE, Wozniak LA, Kobylanska A, Koziolkiewicz M, Stec WJ, Jen-Jacobson L. Mechanisms of coupling between DNA recognition specificity and catalysis in EcoRI endonuclease. Structure. 2004;12:1775–1788. doi: 10.1016/j.str.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 50.Lee JY, Chang J, Joseph N, Ghirlando R, Rao DN, Yang W. MutH complexed with hemi- and unmethylated DNAs: coupling base recognition and DNA cleavage. Mol. Cell. 2005;20:155–166. doi: 10.1016/j.molcel.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 51.Lesher DT, Pommier Y, Stewart L, Redinbo MR. 8-Oxoguanine rearranges the active site of human topoisomerase I. Proc. Natl Acad. Sci. USA. 2002;99:12102–12107. doi: 10.1073/pnas.192282699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwon HJ, Tirumalai R, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science. 1997;276:126–131. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blake DG, Boocock MR, Sherratt DJ, Stark WM. Cooperative binding of Tn3 resolvase monomers to a functionally asymmetric binding site. Curr. Biol. 1995;5:1036–1046. doi: 10.1016/s0960-9822(95)00208-9. [DOI] [PubMed] [Google Scholar]
- 54.Stark WM, Boocock MR. Topological selectivity in site-specific recombination. In: Sherratt DJ, editor. Mobile Genetic Elements (Frontiers in Molecular Biology series) Oxford, UK: Oxford University Press; 1995. pp. 101–129. [Google Scholar]
- 55.Stark WM, Boocock MR, Sherratt DJ. Catalysis by site-specific recombinases. Trends Genet. 1992;8:432–439. [PubMed] [Google Scholar]