


Abstract
HIV-1 integrase integrates retroviral DNA through 3′-processing and strand transfer reactions in the presence of a divalent cation (Mg2+ or Mn2+). The α4 helix exposed at the catalytic core surface is essential to the specific recognition of viral DNA. To define group determinants of recognition, we used a model composed of a peptide analogue of the α4 helix, oligonucleotides mimicking processed and unprocessed U5 LTR end and 5 mM Mg2+. Circular dichroism, fluorescence and NMR experiments confirmed the implication of the α4 helix polar/charged face in specific and non-specific bindings to LTR ends. The specific binding requires unprocessed LTR ends—i.e. an unaltered 3′-processing site CA↓GT3′—and is reinforced by Mg2+ (Kd decreases from 2 to 0.8 nM). The latter likely interacts with the ApG and GpT3′ steps of the 3′-processing site. With deletion of GT3′, only persists non-specific binding (Kd of 100 μM). Proton chemical shift deviations showed that specific binding need conserved amino acids in the α4 helix and conserved nucleotide bases and backbone groups at LTR ends. We suggest a conserved recognition mechanism based on both direct and indirect readout and which is subject to evolutionary pressure.
INTRODUCTION
HIV-1 cDNA integration into the host cell chromosome is catalyzed by the virus enzyme integrase (IN) (1,2). The reaction involves two separate steps: 3′ processing of the newly synthesized cDNA in the cytoplasm, and strand transfer in the nucleus (3). Processed cDNA and IN are imported into the nucleus via a preintegration complex (PIC) including viral and host proteins (4).
The 3′-processing and the strand transfer reactions have been modeled in vitro using purified recombinant IN and a double stranded DNA fragment (21 base pairs) mimicking either the U5 or the U3 LTR end. IN alone, in the presence of divalent cations, performs both the 3′ processing and the DNA strand transfer reactions (2). The DNA fragment plays the role of both the donor (virus DNA) and acceptor (cell DNA).
The IN monomer is a 32-kDa protein comprising three structural domains (5,6). The N-terminal domain (Nt: residues 1–49) has several helices and adopts a compact structure fixed by zinc atom coordination (7). The central domain of the catalytic core (CC: residues 50–212) bearing the active site acidic residues, Asp64, Asp116 and Glu152 (the so-called DDE catalytic triad) (8–12), belongs to a sup family of DNA/RNA strand transferases/nucleases (13–15). The C-terminal domain (Ct: residues 213–288) incorporates a Src-like domain and is involved in DNA host recognition (16).
The high resolution 3D structure of the entire enzyme bound or unbound to DNA has not yet been resolved. The main handicap to obtaining crystals for X-ray studies or in performing an NMR analysis of IN is the weak solubility of the protein. However, a low resolution structure of IN at the DNA contact level has been derived by electronic microscopy (17). This reveals an asymmetric tetrameric IN assembly, contrasting the symmetric structure provided by theoretical or semi-empirical models (18–20).
IN uses a divalent cation (either Mn2+ or Mg2+) as co-factor (21), similar to several enzymes that perform nucleic acid phosphoryl-transfer reactions. Mg2+ may be the relevant factor for IN function in vivo as its intracellular concentration is much higher than that of Mn2+ (1 mM vs 10−4 mM). Moreover, Mn2+ augments both the non-specific nuclease activity of IN (22) and the acceptance of sequence variations at the LTR extremities (23), and several mutations affecting Mn2+ are ineffective, which is not the case with Mg2+ (23,24). This difference in the selection of Mn2+ and Mg2+ also affects the efficiency of IN inhibitors and has been taken into account in the design of raltegravir and elvitegravir drugs (25). How does the divalent cation in IN function? Many uncertainties still exist. The cation may introduce conformational changes to the catalytic site, thus conferring an active structure, but it could also serve as an intermediate permitting the binding of IN to the DNA substrate (26,27).
Previously, to study binding of IN to DNA we have used a model approach involving an analogue (K156) of the amphiphile α4 helix lying at the surface of the IN CC (Figure 1A), and an oligonucleotide corresponding to the U5 LTR end (28). Results have highlighted the roles of Lys156 and Lys159 of the α4 helix and the need for an unprocessed LTR DNA end to achieve specific interaction (28). In a following paper we have shown that the α4 helix is the DNA recognition helix of the HTH (helix turn helix) motif (29). Here we aim to provide greater details on the interaction of the α4 helix with LTR ends in the presence of Mg2+. Our approach involved circular dichroism (CD), fluorescence (quenching and anisotropy) and 1H-NMR spectroscopy. Results were consistent with IN recognizing viral DNA via both direct and indirect readout, in which the binding is optimal only when LTR ends are unprocessed and divalent cations are present.
Figure 1.

(A) Crystal structure of the dimeric catalytic core domain showing the α4 helix at the protein surface [after Dyda et al. (10)]. (B) Peptides used in this study: sequences of α4 and analogues K156Y and K156W with the substituted and added residues in bold. (C) Wheel representations of α4 (top) and K156 (bottom) with the substituted and added residues in bold. (D) Oligonucleotides used in this study: unprocessed LTR34 under the hairpin form (three thymine loop and 17 base pair stem) numbered from 5′ to 3′, the processed oligonucleotide LTR32, and their fluoresceinated versions LTR34fm and LTR32fm.
MATERIALS AND METHODS
The peptides and oligonucleotides used in this study are shown in Figure 1. Some of their characteristics are also presented.
Peptides
Two versions of the peptide K156 (K156-Y and K156-W) (Figure 1B) were synthesized as previously reported (28). K156 is a helix-stabilized version of the helical α4 peptide (residues 147 to residues 169 in CC) (28). Briefly, several residues were replaced in parts of the helix deemed not important for DNA recognition by residues promoting helix formation. The K156 peptide backbone conformation had greater resemblance to the α4 helix in the protein context than the α4 peptide taken in isolation. It was also less aggregation prone and more adapted to the study of specific interactions, that are highly conformation dependent. The Tyr (Y) or Trp (W) aromatic residues added to the C-terminus enabled peptide concentration estimation from absorbances in UV spectra, using molar absorption coefficients of 1280 and 5600 M−1cm−1 at 280 nm for the tyrosine-containing peptide and the tryptophan-containing peptide, respectively.
The wheel presentation of the α4 helix and K156 peptide provided an illustration of the hydrophobic and hydrophilic domains and showed the positions of the substitutions made in the helix (Figure 1C).
Oligonucleotides
The two oligonucleotides were purchased from Eurogentec (Belgium) (Figure 1D). The choice of monomolecular hairpin-forming oligonucleotides rather than bimolecular duplex-forming oligonucleotides was motivated by the need for double helix stability under the low concentrations used in fluorescence and CD experiments (10−9 to 10−5 M). The oligonucleotide sequences reproduce an U5 LTR end that is unprocessed and processed (deletion of GT3′ on the upper strand). The central thymine of the three thymine-loop bears a fluorescein reporter allowing fluorescence measurements. The fluorescein is thought not to interfere with the binding of IN to LTR ends. Nonetheless, a version without fluorescein was prepared for CD and NMR studies.
Fluorescence measurements
The intrinsic fluorescence quantum yield and fluorescence anisotropy studies were carried out with a Jobin-Yvon Fluoromax II instrument (HORIBA Jobin Yvon, France) equipped with an Ozone-free 150 W xenon lamp. Samples (800 µl) were placed at 5°C in thermally jacketed 1 cm × 0.5 cm quartz cells. At least ten measurements for each titration point were recorded with an integration time of 1 s. Fluorophores were either tryptophan or fluorescein purposely fixed to the peptide or the oligonucleotide, respectively.
In fluorescence anisotropy, (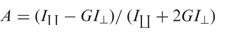 ), parallel (
), parallel (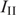 ) and perpendicular (
) and perpendicular (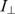 ) emission components were measured in L-format with 4 nm excitation and emission slit widths. With fluorescein as the fluorophore, the excitation was performed at 488 nm and the emission was recorded at 516 nm in the case of LTR34 and at 515 nm in the case of LTR32. Fluorescein-labeled oligonucleotides were diluted to the desired concentration (10 nM) in 800 µl of assay buffer (Na/Na2 phosphate, pH 6, I = 0.1 in the presence or absence of 5 mM MgCl2). Peptides were stepwise diluted. For each anisotropy measurement, the parallel
) emission components were measured in L-format with 4 nm excitation and emission slit widths. With fluorescein as the fluorophore, the excitation was performed at 488 nm and the emission was recorded at 516 nm in the case of LTR34 and at 515 nm in the case of LTR32. Fluorescein-labeled oligonucleotides were diluted to the desired concentration (10 nM) in 800 µl of assay buffer (Na/Na2 phosphate, pH 6, I = 0.1 in the presence or absence of 5 mM MgCl2). Peptides were stepwise diluted. For each anisotropy measurement, the parallel 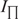 and the perpendicular
and the perpendicular  intensities of the background solution (i.e. buffer and protein contributions) were automatically subtracted from the sample value, calculating the G-value correction each time.
intensities of the background solution (i.e. buffer and protein contributions) were automatically subtracted from the sample value, calculating the G-value correction each time.
In quenching experiments, intrinsic fluorescence of K156-W was measured at a concentration of 400 nM in 800 µl of reaction buffer (with or without 5 mM MgCl2). Excitation at 290 nm provided an emission between 300 and 480 nm, using 2 and 5 nm excitation and emission slit widths, respectively. Maximal emission was measured at 355 nm. Titration isotherms illustrating the binding of Mg2+ to K156 were expressed as 1 − F/F0, where F0 is the fluorescence in the absence of Mg2+.
CD spectroscopy
CD spectra were recorded on a CD6 dichrograph (HORIBA Jobin Yvon, France). Measurements were calibrated with (+)-10-camphorsulfonic acid. Oligonucleotide and peptide concentrations varied from 6 to 12 µM in phosphate buffer pH 6, I = 0.1, with and without Mg2+. Samples were placed in jacketed cells with a 1 mm path length, minimizing thermal drift. To allow the solutions to reach their equilibrium state, these were incubated for 10 min at the chosen temperature. Spectra, recorded in 1 nm steps, were averaged over ten scans and corrected for the base line. They were presented as differential molar absorptivity per residue, Δε (M−1 cm−1), as a function of wavelength, between 260 and 185 nm for peptides and between 200 and 330 nm for DNA, peptide–DNA complexes or peptide alone. In the latter case, aliquots of peptide were added to LTR34 or LTR32 (12 µM) and the control spectrum of the LTR without ligand was subtracted from that of the complex. Effects of β-hydroxycarbonyl compounds were estimated by adding aliquots of the drug in the presence and absence of Mg2+. The α-helical content of peptides was obtained using the relation: Pα = −[Δε222 × 10] (Pα: percentage of α-helix; Δε222: CD per residue at 222 nm) (30).
NMR spectroscopy
Proton NMR spectra were recorded on Bruker Avance 500 and 700 MHz (equipped with a TCI cryo-probe) spectrometers and were processed with the SPARKY program. Samples (500µl) were prepared with 1 mM K156 and LTR34 diluted in 90% phosphate buffer (10 mM Na/Na2 pH 6.5)−10% 2H2O. TSP was used as an internal chemical shift reference. Standard two-dimensional NMR experiments were recorded at 10°C (31). Spectral widths were enlarged to 12 (for the peptide) and 20 p.p.m. (for the DNA and the complexes). The NOESY mixing times were fixed at 150, 200, 300 and 450 ms. The Clean-TOCSY (32) sequences were collected with MLEV-17 spin-locking fixed at 40, 60, 80, 100 and 120 ms. Solvent suppression was achieved by applying a WATERGATE pulse sequence. NOE relative intensities were measured from 300 ms mixing time NOESY spectra with solvent presaturation. Intensities (volume and height of connectivity) were determined by the Sparky program (Goddard and Kneller, University of California, San Francisco) and were confirmed by the TopSpin program (Bruker). The 3JαHNH coupling constants were measured on NH peaks in 1D NMR spectra. Chemical shifts were determined from both TOCSY and NOESY spectra.
RESULTS AND DISCUSSION
Design of the peptide K156
The conformation of the α4 peptide in solution is mainly unordered (28) and is therefore very different of the α4 helix structure observed in the context of the protein (Figure 1A). Thus, the α4 peptide cannot be used to give an account of the role of the α4 helix in the recognition of DNA the enzyme. A more suitable model is peptide K156, a structural analogue of the α4 peptide displaying a higher helical content and thereby a pre-organized structure for interaction (Figure 1B). Most amino acid substitutions were made in ensuring that hydrophobic and hydrophilic surfaces were not significantly altered (Figure 1C). We also took into account the previously reported data of mutagenesis (33) to ensure that no amino acid important to 3′-processing was replaced. Substitutions were as follows: (i) Gly149–Ala, Gly163–Ala, Val151–Leu, Ile161–Leu, Ile162–Leu (alanine and leucine being strongly involved in helix formation, especially compared with side chain deprived glycine and β-branched valine/isoleucine, respectively); and (ii) Val150 Lys and Ser153 Glu, allowing the formation of two i–i + 3 pairs with stabilizing electrostatic interactions between positively and negatively charged side chains (Lys150 Glu153 and Glu153 Lys156). Examination of results of the variation in HIV-1 group M IN indicates that among all the substitutions performed in K156, only one, that of Ser153 (Ser153 Glu), confers resistance to raltegravir and elvitegravir (34).
Binding of Mg2+ to LTR34 and LTR32
The in vivo activity of Mg2+ is similar to that of a solution with 0.5–1.0 mM Mg2+ and 0.15 M monovalent salt (35). Both monovalent and divalent cations interact with the DNA surface and neutralize the phosphate negative charges, decreasing the repulsive forces between negative or positive charges. Mg2+ is a compact ion with a small atomic radius, facilitating its coordination with oxygen atoms in DNA (36,37). It is also a net hydrogen bond donor through its rigid octahedral primary solution shell (38); hydrogen bonds occur with bases in particular sequences (39,40). In fact, Mg2+ is involved in sequence-specific binding to the major and minor grooves of DNA, as well as non-specific binding to backbone phosphates (41). Often, the structures, the dynamics and the ligand interactions are modified by the cation, but each effect depends on the groove where the binding occurs.
We used CD and fluorescence spectroscopy to examine the effect of Mg2+ on both unprocessed LTR34 DNA and processed LTR32 DNA. We also studied 1H NMR data in the case of LTR34 DNA. In CD experiments addition of MgCl2 (to a final concentration of 5 mM) to a solution of LTR34 or LTR32 in phosphate buffer pH 6, at 5°C, did not produce noticeable change in the intensity or in the shape of spectra, which continued to present B DNA characteristics with a positive signal at ∼280 nm and a negative one at ∼250 nm (data not shown). The NMR analysis of LTR34 DNA in the absence of Mg2+ has been previously reported (42). Despite the large number and poor dispersion of proton resonances inherent to nucleic acids and the rather long size of our oligonucleotide (a stem of 17 base pairs), almost all the 1H-resonances were assigned. The addition of Mg2+ to LTR34 did not significantly modify the chemical shifts (Supplementary Table S1). LTR34 maintains the B-DNA form with or without Mg2+, which is consistent with the CD results.
Although Mg2+ does not visibly modify the DNA conformation, the titration isotherms obtained by fluorescence anisotropy show that the divalent cation binds to DNA. A deep transition in the curve occurs in the millimolar range for both LTR34 and LTR32 (data not shown). Curve analysis considering a simple two-state reversible equilibrium between Mg2+ ions and DNA, which is an oversimplified approach (43) using ‘GraphPad Prism’ (Non-linear ‘Least Squares’), provides Kd values of ∼60 mM for LTR34 and ≥80 mM for LTR32.
Binding of Mg2+ to K156
The interaction of Mg2+—which is assumed to act as a cofactor for the catalytic reaction and as a stabilizer of the IN–DNA complex—with the K156 peptide, was assessed by CD, NMR and fluorescence spectroscopy. The CD spectra of K156 in the presence and absence of Mg2+ are given in Supplementary Figure S1. In the absence of Mg2+, the CD spectra of K156 at 12 µM displayed two negative bands, at ∼225 and ∼208 nm, and a positive band, at ∼190 nm, typical of the α helix (28). The α helix content based on CD intensity at ∼225 nm (30) is of ∼25%. This partly depends on the composition and sequence of the peptide. There are several negatively charged Glu and positively charged Lys residues displaying i + 3 or i + 4 spacing within the K156 sequence. Their distribution allows either ion pair formation (Lys150–Glu153, Glu152–Lys156, Glu153–Lys156 and Glu157–Lys160) or ion pair repulsion (Glu153–Glu157, Lys156–Lys159 and Lys156–Lys160). The sum of the effects results either in a stabilization or destabilization of the helix, but salts modulate the intensity of effects.
We found that addition of MgCl2 to K156 slightly increased band intensities, and thus helix stability. Our previous NMR analysis, which mainly investigated the K156 backbone, has shown that K156 adopts a rather stable helix structure in buffer and aqueous media at pH 6 (28). Here, we extended the analysis to amino acid side chains groups, as side chains are directly implicated in the DNA–peptide interactions. We measured K156 chemical shifts in the presence and absence of Mg2+ (Supplementary Table S2). The addition of Mg2+ to K156 produced some weak chemical shift variation and a selective broadening of correlations, visible in the TOCSY spectra. The β and γ correlations of the catalytic acid residue Glu152 resulted in a weak increase in intensity. In contrast, the αH, NH and γH protons of Gln168, the βH protons of Lys159, the γH proton of Lys160, and the βH and γH protons of Leu161 displayed a weak decrease in intensity. One of the δ proton signals of Lys159 was no longer visible. The only noticeable chemical shift variation involved the γH proton of Glu153, which led to a shift of −0.1 p.p.m.
Ten 3JαHNH were accessible in the 1D spectra of K156 recorded in the presence and the absence of Mg2+. The residues analyzed were Ala149, Lys150, Glu152, Met154, Asn155, Asp157, Lys159, Lys160, Ala163 and Ala167, which are evenly distributed along the entire chain length and, thereby may be used as good indicators for the whole backbone structure. The average of 3JαHNH values in the absence of Mg2+ was 4.4 ± 0.3 Hz, a value consistent with that found for other helical structures (31). The addition of Mg2+ decreased the average value to 4.2 ± 0.3 Hz, suggesting a weak helix stabilization. The observed decrease in 3JαHNH values was accompanied by a variation in several typical NH(i)-NH(i + 1) and αH(i)-NH(i + 1) NOEs. For instance, NH(i)-NH(i + 1) correlations of the linked residues Ala149–Lys150, Glu153–Met154 and Asn155–Lys156, which show well-delineated cross-peaks, resulted in a noticeable increase of intensity (140, 70 and 40, respectively), consistent with helix stabilization.
We used fluorescence spectroscopy to determine the thermodynamic variables for Mg2+ and K156 binding. The signal of tryptophan (excitation at 290 nm, emission at 300–480 nm) purposely incorporated at the C-end of the peptide was used as the fluorophore. The change in fluorescence induced by Mg2+ was weak, indicating that the conformational change was also weak, and was likely limited to only some Mg2+-binding positions. Treatment of the titration curve yielded an apparent Kd value of 2.5 µM (data not shown).
CD analysis of the peptide binding to DNA
CD is a convenient method for analyzing both peptide and oligonucleotide conformations, and conformational changes accompanying complex formation. The CD spectra of LTR34 and LTR32 were typical of B DNA, and these spectra remained unchanged upon Mg2+ addition (data not shown). In contrast, there were slight changes in the K156 spectrum on Mg2+ addition, which is consistent with a more stable helix (Supplementary Figure S1). We investigated the binding of K156 to processed and unprocessed LTR ends in the presence of Mg2+. Previous experiments performed in the absence of Mg2+ have shown that the GT3′ dinucleotide deleted upon 3′-processing is essential for the specific binding between IN and virus DNA (28). Mixing LTR34 and K156 in the presence of Mg2+ resulted in a spectrum in the 190–260 nm region that clearly differed from the sum of individual K156 and LTR34 spectra. Since no changes were detected between 260 and 300 nm, an UV region rather specific to DNA, we deduced that the changes observed in the 190–260 nm UV region were due to conformational variations affecting the sole peptide. The difference spectra (i.e. spectra of DNA–K156 complexes at the 1:1 ratio minus the spectrum of unbound K156) showed that LTR34 stabilizes the K156 helix, but LTR32 does not, thereby confirming the contribution of the LTR GT3′ dinucleotide to complex formation (Figure 2A and B).
Figure 2.

CD of unbound and bound K156 in the presence of Mg2+. (A) Spectrum of K156 at 12 µM (Black line); difference spectrum [K156 + LTR34]—LTR34 at 12 µM each (Red line). (B) Spectrum of K156 at 12 µM (Black line); difference spectrum [K156 + LTR32]—LTR32 at 12 µM each (Red line). Phosphate buffer pH 6, I = 0.1, at 5°C, MgCl2 5 mM final concentration.
Analysis of the peptide binding to DNA by fluorescence
Direct implication of the α4 helix in interactions with LTR ends has been suggested in several in vitro and in vivo experiments (23,28,44–57). Here, the DNA–α4 peptide binding analysis was performed by monitoring the anisotropy signal of fluorescein linked to the hairpin oligonucleotides (Figure 1D). The binding isotherms were related to the total average amount of K156 binding (Figure 3A and B). The LTR34 isotherm had two phases: a small increase of anisotropy accompanied by an inflection in the nanomolar range, followed by a steeper increase with an inflection in the micromolar range. Curve analysis provided a Kd1 value of ∼0.8 nM (first binding) and a Kd2 value ≥100 µM (second binding). In contrast, the binding of K156 to LTR32 provided a monophasic isotherm with a Kd value ≥100 µM, which was similar to the LTR34 Kd2 value. Comparison of these values with our previous Kd values obtained in the absence of Mg2+ (i.e. Kd1 ∼2.1 nM and Kd2 ∼55 µM for LTR34 and Kd ∼65 µM for LTR32) (28) reveals that Mg2+ strengthens the high affinity binding between K156 and LTR34; however, it impairs low affinity binding in both LTR34 and LTR32.
Figure 3.

Fluorescence anisotropy titration of (A) LTR34 by peptide K156 in presence (Red line) and absence (Black line) of Mg2+; (B) LTR32 by peptide K156 in presence (Red line) and absence (Black line) of Mg2+. Phosphate buffer pH 6, I = 0.1, at 5°C, MgCl2 5 mM final concentration.
Thus, high affinity binding may be dependent on directional hydrogen bonds, which are known for their resistance to changes in salt concentration, and possible non-polar forces that are generally strengthened by salt addition (58). In contrast, low affinity binding that is impaired by salt addition is mediated by ionic interactions between negatively charged phosphate groups of the DNA backbone and positively charged side chain groups of the peptide.
Stabilization of high affinity binding in unprocessed LTR (LTR34) by Mg2+ suggests the binding of a divalent cation to the processing site CA↓GT3′. Mg2+ is known for its strong preference for the guanine base, although the adjacent base pair in the sequence usually cooperates in this interaction (38). Note that, in the crystal structure of the [d(CCAGTACTGG)]2 duplex, the divalent ion binds the 5′CAGT3′ sequence in the major groove, at either the ApG or GpT step (41,59). The solvated Mg2+ formed hydrogen bonds with guanine and adenine bases through its water molecules; the major groove was compressed at the ion binding site. This resulted in an opening of the minor groove, exactly where IN is supposed to perform its nucleophilic attack at LTR ends. Clearly, in a mechanism of two-ion catalysis, as the one generally assumed for IN, an Mg2+ ion positioned in the major groove may play the role of the second ion. Once detected and liganded by the carboxylate groups of catalytic residues this Mg2+ may assist the enzyme to cleave the appropriate phosphodiester bond. Note that in crystallographic studies the less compact Ca2+ also binds with high efficiency to the ApG and GpT steps, but its binding mode differs from that of Mg2+ (41). The same difference in binding may exist between Mg2+ and Mn2+, explaining the biological differences observed between these two ions in in vitro experiments.
Analysis of peptide binding to DNA by 1H NMR
Proton chemical shift deviations (CSDs) may aid the identification of interacting regions in structures of complexed proteins and nucleic acids (60). In general, the CSDs of only one partner are used to certify the interaction (61). Here we investigated CSDs of both K156 and LTR34 to map the interface of the peptide–DNA complex in the presence of Mg2+. Fluorescence experiments indicated a significant difference between the high affinity binding Kd (∼1 nM) and the low affinity binding Kd (≥100 µM); these conditions allowed an NMR analysis of binding in which the specific complex occurs more frequently than the non-specific complex. Yet, at the millimolar concentrations required by NMR, an equimolecular 1:1 complex was not attainable without material aggregation. To avoid a too much signal broadening and achieve a convenient mapping of the K156 and LTR interface, spectra were recorded at DNA–peptide ratios of 1:5 and 1:2.
αH-NH regions of the TOCSY spectra of K156 alone and K156 with DNA (DNA:peptide ratio of 1:5) are given in the Supplementary Figure S2. The specific K156 binding site on LTR34 was fully saturated at this ratio, although some non-specific binding was likely to occur. The CSDs for the base and ribose protons of LTR34 bound to K156 at a DNA: peptide ratio of 1:5, relative to unbound DNA (in the presence of Mg2+), are given in Supplementary Table S3. Assuming that only CSDs values ≥0.05 p.p.m. indicate interaction we can infer that eleven nucleotide bases interact with the K156 amino acid residues (Figure 4). These are asymmetrically distributed between the two strands, with eight of them on the upper strand (the one that undergoes processing) and only three on the lower strand. Four of the affected bases belong to the same base pairs, A35.T3 and A25.T13, whereas two other bases, C28 and A9, are in close spatial proximity in the double helix. The A35 nucleotide, constitutive of the highly conserved CpA step, essential to the viral DNA integration, is among these nucleotides. Once again, the role of GT3′ next to the conserved CpA step was highlighted: both the guanine and thymine base protons displayed significant CSDs. The large CSD of the guanine imino proton (0.23 p.p.m.) suggested a significant change in base arrangement; however, the corresponding signal in the NOESY spectrum showed no change, suggesting that stability of the G36.C2 base pair was not impaired by the interaction of LTR34 with K156.
Figure 4.

The LTR34 double helix affected by K156 interactions. The LTR structures presented are taken from Renisio et al. 2005. Purple and cyan circles indicate the bases whose chemical shifts are most affected (CSDs: purple ≥ 0.1 ppm; 0.05 ppm ≤ cyan < 0.1 ppm) by K156 (DNA:peptide ratio of 1:5).
The high number of desoxyribose sugars affected by the binding of K156 was also remarkable (Supplementary Table S3). Binding contacts extend inward, about 15 or 16 base pairs from the right end of the LTR (Figure 4). Remarkably, the binding pattern was similar to the 3′-processing pattern observed by Esposito and Craigie with 21 base pair LTR DNAs and the whole enzyme (23). There authors found that in addition to the six commonly outermost nucleotides AGCACT3′, the adenine (AAAA) tract in the distal LTR was also involved in viral DNA recognition by IN; our previous work (28) also suggested the implication of the adenine tract in binding between K156 and LTR. Furthermore, present findings resemble those on the stepwise increase in LTR length versus IN binding, mutational analysis, chemical modifications and photo cross-linking experiments, as well as those of DNA protection against DNase in the IN–viral LTR DNA complex (23,47,62–64). That the full enzyme recognizes the whole oligonucleotide (23) is conceivable, but how the relatively small α4 peptide recognizes both the terminal ACGT3′ and the internal A-tract remains unclear.
NMR data describing the binding of K156 were obtained at a DNA:peptide ratio of 1:2 (at 700 MHz). Under these conditions, ∼50% of K156 was bound to LTR34. Thus, the so measured CSDs should be weaker than CSDs obtained for an equimolecular complex. Both the side chain and backbone protons of several K156 residues underwent shifts in the complex. The backbone NH and αH protons may be influenced by local functional group contacts, and reflect to a good degree the changes to residues nearest the binding site (61,65). The chemical shifts for bound and unbound K156 are given in Supplementary Table S4. Overall, αH CSDs were larger than NH CSDs (an average of 0.09 ppm versus an average of 0.03 p.p.m.). The αHs and NHs of residues Asn155, Lys156, Lys159, Glu(Ser)153, Glu152, Leu(Ile)161 and Leu(Ile)162 were most affected during binding (Figure 5). Unfortunately, Gln148 could not be identified in our spectra. All of these residues, except Leu161(Ile) and Leu162(Ile), formed the polar/charged face of the α4 helix (Figure 6). The binding of residues Lys156, Lys159 and Gln148 to viral LTR ends has already been shown through cross-linking experiments (23,44–46,48,49,51), whereas the binding of the Asn155 residue has been predicted by molecular modeling (57). Among these, the two positively charged residues Lys156 and Lys159 are required for high affinity binding between K156 and the U5 LTR (28). Lys156, Lys159 and Asn155 have been further shown to interact with the L731–988 derivative (66) and 5CITEP (67), two IN inhibitors supposed to interact at the DNA–protein interface. On another hand the CSD exhibited by Leu161 could be attributed to changes caused by interactions occurring with residues in close proximity or by the dissociation of K156 multimers stabilized by hydrophobic residues (68).
Figure 5.

CSDs for the αHs (A) and NHs (B) of K156 bound to LTR34 at a DNA:peptide ratio of 1:2. Non-assigned residues are indicated by a dot.
Figure 6.

Backbone of the amphipathic K156 helix with residues of its polar/charged face (pink) that are most affected by LTR34, at a DNA: peptide ratio of 1:2. Yellow residues (Glu156, Leu161 and Leu162) are substituted residues showing CSD variations upon complex formation.
Possible DNA–peptide contacts
The LTR34 oligonucleotide is composed of 34 nucleotides, and 11 of the first 15 nucleotides interact with K156 via their bases (Figure 4). Most of these are important for 3′-processing (23). There is a good statistical correlation between the frequency of nucleobase conservation in DNAs and the interaction of these bases with protein amino acid side chains (69). At the same time, most of the α4 helix amino acids interacting with LTR DNA are themselves conserved residues, recognized for their role in the enzyme catalytic activity and virus infectivity (33,45). This suggests that DNA–protein recognition proceeds through the interaction of conserved complementary domains. Tight binding requires an optimal number of contacts between the amino acid chains and the bases, and also the backbone phosphates and sugars. These contacts include ionic interactions, van der Waals forces, and hydrogen bonds. Lys and Gln belong to a category of residues having side chains forming bidentate interactions with DNA bases, which in general provide higher specificity than those using single hydrogen bonds (70). The positively charged side chain of Lys is also frequently found in ionic interactions with DNA phosphate groups. For instance, the conserved Lys159 has been shown to interact with the adenine base and phosphate group of invariant CpA (47,48). Moreover, Lys159, together with Lys156, belongs to the Lys-rich sequence 156 Lys Glu Leu Lys Lys160, which has been implicated in the specific binding of IN to DNA (33). The base of adenine in CpA (and that of its complementary thymine) is also in contact with the catalytic Glu152 residue of the α4 helix (54). The Gln148 residue (not identified in our spectra) has been shown to interact with the adenine (23) and the cytosine (46) of the 5′AC overhang in processed LTR.
However, most importantly, Mg2+ is implicated in the specific recognition of U5 LTR by the α4 helix of IN. How Mg2+ impacts the recognition? Mg2+ displays a high preference for guanine in crystal structures of DNA duplexes that are in either the A or B form (41,71). In several oligonucleotide crystals, Mg2+ has been identified within the major and the minor groove of ApG and GpT steps (41), which in LTRs are constitutive to the 3′-processing site. The base binding of Mg2+ in the major groove may cause helix bending by base-roll compression toward the major groove (41), which may then favor DNA cleavage from the minor groove. In fact, the CSDs for both aromatic and sugar protons strongly suggest that both the base and the scissile phosphodiester group of adenine are involved in interactions. Thus, binding of the Glu 152 carboxylic group may be mediated by the preferential binding of Mg2+ to this group.
CONCLUSION
Our data support the idea of a tight complex between viral DNA and IN formed through both specific interactions, involving amino acid side chains with DNA bases, and non-specific interactions involving amino acid side chains and DNA backbone groups. This suggests a recognition of viral DNA at the U5 LTR end that combines a direct and an indirect readout mechanism. In the indirect readout mechanism, the DNA sequence governs the geometry of the binding site, and thus the quality of interaction among amino acid side chains and backbone sugar and phosphate groups is strongly related to the sequence of the DNA. The direct readout mechanism involves direct contacts between protein functional groups and the DNA bases in appropriate geometry. Overall, the precise recognition of LTRs by IN is the result of the formation of a large network of interactions between the two partners. In this respect, the mechanism presented here resorts to many of the same basic features commonly found in the formation of specific DNA–protein complexes. Yet, it significantly differs from the mechanism, mostly of indirect readout type, proposed on the bases of chemical modifications and nucleotide substitutions, as well as of a stepwise increase in DNA–ligand complexity (47,64,72). Such modifications in DNA may have effects on structural and energetic properties of local but also distant interactions, preventing a straight-forward analysis of the binding.
Obviously, the mechanism through which IN recognizes the LTR ends requires further understanding, and the analysis must be extended to other regions of the protein. Yet, our results already shed light on the implication of the α4 helix and the divalent Mg2+ ion in the specific binding between IN and viral LTR DNA. The specific binding involves complementary conserved amino acid residues and nucleotides, suggesting that the IN α4 helix contains most of the specificity features that are required for a precise recognition of viral DNA, especially at the 3′-processing site. Clearly, a model that would reproduce the relevant binding properties of the enzyme could improve our understanding of the mechanisms underlying complex formation and might also facilitate the design of drugs blocking viral integration.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
SIDACTION grant (to S.F.) and by the French-Lebanese program CEDRE (05 SF21/L14 to S.F. and R.M.). Funding for open access charge: CNRS.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Craigie R, Mizuuchi K, Bushman FD, Engelman A. A rapid in vitro assay for HIV DNA integration. Nucleic Acids Res. 1991;19:2729–2734. doi: 10.1093/nar/19.10.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asante-Appiah E, Skalka AM. Molecular mechanisms in retrovirus DNA integration. Antiviral Res. 1997;36:139–156. doi: 10.1016/s0166-3542(97)00046-6. [DOI] [PubMed] [Google Scholar]
- 3.Katz RA, Skalka AM. The retroviral enzymes. Annu. Rev. Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 4.Sherman MP, Greene WC. Slipping through the door: HIV entry into the nucleus. Microbes. Infect. 2002;4:67–73. doi: 10.1016/s1286-4579(01)01511-8. [DOI] [PubMed] [Google Scholar]
- 5.van Gent DC, Oude Groeneger AA, Plasterk RH. Identification of amino acids in HIV-2 integrase involved in site-specific hydrolysis and alcoholysis of viral DNA termini. Nucleic Acids Res. 1993;21:3373–3377. doi: 10.1093/nar/21.15.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelman A, Bushman FD, Craigie R. Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J. 1993;12:3269–3275. doi: 10.1002/j.1460-2075.1993.tb05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM. Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol. 1997;4:567–577. doi: 10.1038/nsb0797-567. [DOI] [PubMed] [Google Scholar]
- 8.Engelman A, Craigie R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 1992;66:6361–6369. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz RA, Skalka AM. The catalytic domain of avian sarcoma virus integrase: conformation of the active-site residues in the presence of divalent cations. Structure. 1996;4:89–96. doi: 10.1016/s0969-2126(96)00012-3. [DOI] [PubMed] [Google Scholar]
- 10.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 11.Kulkosky J, Jones KS, Katz RA, Mack JP, Skalka AM. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell Biol. 1992;12:2331–2338. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leavitt AD, Shiue L, Varmus HE. Site-directed mutagenesis of HIV-1 integrase demonstrates differential effects on integrase functions in vitro. J. Biol. Chem. 1993;268:2113–2119. [PubMed] [Google Scholar]
- 13.Rice P, Mizuuchi K. Structure of the bacteriophage Mu transposase core: a common structural motif for DNA transposition and retroviral integration. Cell. 1995;82:209–220. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- 14.Davies JF, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- 15.Katayanagi K, Okumura M, Morikawa K. Crystal structure of Escherichia coli RNase HI in complex with Mg2+ at 2.8 A resolution: proof for a single Mg(2+)-binding site. Proteins. 1993;17:337–346. doi: 10.1002/prot.340170402. [DOI] [PubMed] [Google Scholar]
- 16.Musacchio A, Noble M, Pauptit R, Wierenga R, Saraste M. Crystal structure of a Src-homology 3 (SH3) domain. Nature. 1992;359:851–855. doi: 10.1038/359851a0. [DOI] [PubMed] [Google Scholar]
- 17.Ren G, Gao K, Bushman FD, Yeager M. Single-particle image reconstruction of a tetramer of HIV integrase bound to DNA. J. Mol. Biol. 2007;366:286–294. doi: 10.1016/j.jmb.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Podtelezhnikov AA, Gao K, Bushman FD, McCammon JA. Modeling HIV-1 integrase complexes based on their hydrodynamic properties. Biopolymers. 2003;68:110–120. doi: 10.1002/bip.10217. [DOI] [PubMed] [Google Scholar]
- 19.Karki RG, Tang Y, Burke TR, Jr, Nicklaus MC. Model of full-length HIV-1 integrase complexed with viral DNA as template for anti-HIV drug design. J. Comput. Aided Mol. Des. 2004;18:739–760. doi: 10.1007/s10822-005-0365-5. [DOI] [PubMed] [Google Scholar]
- 20.Wielens J, Crosby IT, Chalmers DK. A three-dimensional model of the human immunodeficiency virus type 1 integration complex. J. Comput. Aided Mol. Des. 2005;19:301–317. doi: 10.1007/s10822-005-5256-2. [DOI] [PubMed] [Google Scholar]
- 21.Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelman A, Craigie R. Efficient magnesium-dependent human immunodeficiency virus type 1 integrase activity. J. Virol. 1995;69:5908–5911. doi: 10.1128/jvi.69.9.5908-5911.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esposito D, Craigie R. Sequence specificity of viral end DNA binding by HIV-1 integrase reveals critical regions for protein-DNA interaction. EMBO J. 1998;17:5832–5843. doi: 10.1093/emboj/17.19.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katzman M, Katz RA, Skalka AM, Leis J. The avian retroviral integration protein cleaves the terminal sequences of linear viral DNA at the in vivo sites of integration. J. Virol. 1989;63:5319–5327. doi: 10.1128/jvi.63.12.5319-5327.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 26.Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Asante-Appiah E, Seeholzer SH, Skalka AM. Structural determinants of metal-induced conformational changes in HIV-1 integrase. J. Biol. Chem. 1998;273:35078–35087. doi: 10.1074/jbc.273.52.35078. [DOI] [PubMed] [Google Scholar]
- 28.Zargarian L, Benleumi MS, Renisio JG, Merad H, Maroun RG, Wieber F, Mauffret O, Porumb H, Troalen F, Fermandjian S. Strategy to discriminate between high and low affinity bindings of human immunodeficiency virus, type 1 integrase to viral DNA. J. Biol. Chem. 2003;278:19966–19973. doi: 10.1074/jbc.M211711200. [DOI] [PubMed] [Google Scholar]
- 29.Merad H, Porumb H, Zargarian L, Rene B, Hobaika Z, Maroun RG, Mauffret O, Fermandjian S. An unusual helix turn helix motif in the catalytic core of HIV-1 integrase binds viral DNA and LEDGF. PLoS One. 2009;4:e4081. doi: 10.1371/journal.pone.0004081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong L, Johnson WC., Jr Environment affects amino acid preference for secondary structure. Proc. Natl Acad. Sci. USA. 1992;89:4462–4465. doi: 10.1073/pnas.89.10.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wüthrich K. NMR of Protein and Nucleic Acids. New York, NY: Wiley; 1986. [Google Scholar]
- 32.Griesinger C, Otting G, Wuthrich K, Ernst RR. Clean Tocsy for H-1 spin system-identification in macromolecules. J. Am. Chem. Soc. 1988;110:7870–7872. [Google Scholar]
- 33.Semenova EAMCaPY. HIV-1 integrase inhibitors: update and perspectives. Adv. Pharmacol. 2008;56:199–228. doi: 10.1016/S1054-3589(07)56007-2. [DOI] [PubMed] [Google Scholar]
- 34.Rhee SY, Liu TF, Kiuchi M, Zioni R, Gifford RJ, Holmes SP, Shafer RW. Natural variation of HIV-1 group M integrase: implications for a new class of antiretroviral inhibitors. Retrovirology. 2008;5:74. doi: 10.1186/1742-4690-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Draper DE. RNA folding: thermodynamic and molecular descriptions of the roles of ions. Biophys. J. 2008;95:5489–5495. doi: 10.1529/biophysj.108.131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sissi C, Palumbo M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009;37:702–711. doi: 10.1093/nar/gkp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowan JA. Structural and catalytic chemistry of magnesium-dependent enzymes. Biometals. 2002;15:225–235. doi: 10.1023/a:1016022730880. [DOI] [PubMed] [Google Scholar]
- 38.Subirana JA, Soler-Lopez M. Cations as hydrogen bond donors: a view of electrostatic interactions in DNA. Annu. Rev. Biophys. Biomol. Struct. 2003;32:27–45. doi: 10.1146/annurev.biophys.32.110601.141726. [DOI] [PubMed] [Google Scholar]
- 39.Misra VK, Draper DE. On the role of magnesium ions in RNA stability. Biopolymers. 1998;48:113–135. doi: 10.1002/(SICI)1097-0282(1998)48:2<113::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 40.Manning GS. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q Rev. Biophys. 1978;11:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 41.Chiu TK, Dickerson RE. 1 A crystal structures of B-DNA reveal sequence-specific binding and groove-specific bending of DNA by magnesium and calcium. J. Mol. Biol. 2000;301:915–945. doi: 10.1006/jmbi.2000.4012. [DOI] [PubMed] [Google Scholar]
- 42.Renisio JG, Cosquer S, Cherrak I, El Antri S, Mauffret O, Fermandjian S. Pre-organized structure of viral DNA at the binding-processing site of HIV-1 integrase. Nucleic Acids Res. 2005;33:1970–1981. doi: 10.1093/nar/gki346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Misra VK, Draper DE. The interpretation of Mg(2+) binding isotherms for nucleic acids using Poisson-Boltzmann theory. J. Mol. Biol. 1999;294:1135–1147. doi: 10.1006/jmbi.1999.3334. [DOI] [PubMed] [Google Scholar]
- 44.Gao K, Butler SL, Bushman F. Human immunodeficiency virus type 1 integrase: arrangement of protein domains in active cDNA complexes. EMBO J. 2001;20:3565–3576. doi: 10.1093/emboj/20.13.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu R, Limon A, Ghory HZ, Engelman A. Genetic analyses of DNA-binding mutants in the catalytic core domain of human immunodeficiency virus type 1 integrase. J. Virol. 2005;79:2493–2505. doi: 10.1128/JVI.79.4.2493-2505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson AA, Santos W, Pais GC, Marchand C, Amin R, Burke TR, Jr, Verdine G, Pommier Y. Integration requires a specific interaction of the donor DNA terminal 5′-cytosine with glutamine 148 of the HIV-1 integrase flexible loop. J. Biol. Chem. 2006;281:461–467. doi: 10.1074/jbc.M511348200. [DOI] [PubMed] [Google Scholar]
- 47.Agapkina J, Smolov M, Barbe S, Zubin E, Zatsepin T, Deprez E, Le BM, Mouscadet JF, Gottikh M. Probing of HIV-1 integrase/DNA interactions using novel analogs of viral DNA. J. Biol. Chem. 2006;281:11530–11540. doi: 10.1074/jbc.M512271200. [DOI] [PubMed] [Google Scholar]
- 48.Jenkins TM, Esposito D, Engelman A, Craigie R. Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 1997;16:6849–6859. doi: 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drake RR, Neamati N, Hong H, Pilon AA, Sunthankar P, Hume SD, Milne GW, Pommier Y. Identification of a nucleotide binding site in HIV-1 integrase. Proc. Natl Acad. Sci. USA. 1998;95:4170–4175. doi: 10.1073/pnas.95.8.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lutzke RA, Vink C, Plasterk RH. Characterization of the minimal DNA-binding domain of the HIV integrase protein. Nucleic Acids Res. 1994;22:4125–4131. doi: 10.1093/nar/22.20.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heuer TS, Brown PO. Photo-cross-linking studies suggest a model for the architecture of an active human immunodeficiency virus type 1 integrase-DNA complex. Biochemistry. 1998;37:6667–6678. doi: 10.1021/bi972949c. [DOI] [PubMed] [Google Scholar]
- 52.Dirac AM, Kjems J. Mapping DNA-binding sites of HIV-1 integrase by protein footprinting. Eur. J. Biochem. 2001;268:743–751. doi: 10.1046/j.1432-1327.2001.01932.x. [DOI] [PubMed] [Google Scholar]
- 53.Engelman A, Liu Y, Chen H, Farzan M, Dyda F. Structure-based mutagenesis of the catalytic domain of human immunodeficiency virus type 1 integrase. J. Virol. 1997;71:3507–3514. doi: 10.1128/jvi.71.5.3507-3514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerton JL, Ohgi S, Olsen M, DeRisi J, Brown PO. Effects of mutations in residues near the active site of human immunodeficiency virus type 1 integrase on specific enzyme-substrate interactions. J. Virol. 1998;72:5046–5055. doi: 10.1128/jvi.72.6.5046-5055.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harper AL, Skinner LM, Sudol M, Katzman M. Use of patient-derived human immunodeficiency virus type 1 integrases to identify a protein residue that affects target site selection. J. Virol. 2001;75:7756–7762. doi: 10.1128/JVI.75.16.7756-7762.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen A, Weber IT, Harrison RW, Leis J. Identification of amino acids in HIV-1 and avian sarcoma virus integrase subsites required for specific recognition of the long terminal repeat Ends. J. Biol. Chem. 2006;281:4173–4182. doi: 10.1074/jbc.M510628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perryman AL, McCammon JA. AutoDocking dinucleotides to the HIV-1 integrase core domain: exploring possible binding sites for viral and genomic DNA. J. Med. Chem. 2002;45:5624–5627. doi: 10.1021/jm025554m. [DOI] [PubMed] [Google Scholar]
- 58.Leberman R, Soper AK. Effect of high salt concentrations on water structure. Nature. 1995;378:364–366. doi: 10.1038/378364a0. [DOI] [PubMed] [Google Scholar]
- 59.Kielkopf CL, Ding S, Kuhn P, Rees DC. Conformational flexibility of B-DNA at 0.74 A resolution: d(CCAGTACTGG)(2) J. Mol. Biol. 2000;296:787–801. doi: 10.1006/jmbi.1999.3478. [DOI] [PubMed] [Google Scholar]
- 60.Varani G, Chen Y, Leeper TC. NMR studies of protein-nucleic acid interactions. Methods Mol. Biol. 2004;278:289–312. doi: 10.1385/1-59259-809-9:289. [DOI] [PubMed] [Google Scholar]
- 61.Fielding AJ, Usselman RJ, Watmough N, Simkovic M, Frerman FE, Eaton GR, Eaton SS. Electron spin relaxation enhancement measurements of interspin distances in human, porcine, and Rhodobacter electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO) J. Magn Reson. 2008;190:222–232. doi: 10.1016/j.jmr.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ellison V, Brown PO. A stable complex between integrase and viral DNA ends mediates human immunodeficiency virus integration in vitro. Proc. Natl Acad. Sci. USA. 1994;91:7316–7320. doi: 10.1073/pnas.91.15.7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vink C, Banks M, Bethell R, Plasterk RH. A high-throughput, non-radioactive microtiter plate assay for activity of the human immunodeficiency virus integrase protein. Nucleic Acids Res. 1994;22:2176–2177. doi: 10.1093/nar/22.11.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bugreev DV, Baranova S, Zakharova OD, Parissi V, Desjobert C, Sottofattori E, Balbi A, Litvak S, Tarrago-Litvak L, Nevinsky GA. Dynamic, thermodynamic, and kinetic basis for recognition and transformation of DNA by human immunodeficiency virus type 1 integrase. Biochemistry. 2003;42:9235–9247. doi: 10.1021/bi0300480. [DOI] [PubMed] [Google Scholar]
- 65.Hunter TM, McNae IW, Liang X, Bella J, Parsons S, Walkinshaw MD, Sadler PJ. Protein recognition of macrocycles: binding of anti-HIV metallocyclams to lysozyme. Proc. Natl Acad. Sci. USA. 2005;102:2288–2292. doi: 10.1073/pnas.0407595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sechi M, Carta F, Sannia L, Dallocchio R, Dessi A, Al-Safi RI, Neamati N. Design, synthesis, molecular modeling, and anti-HIV-1 integrase activity of a series of photoactivatable diketo acid-containing inhibitors as affinity probes. Antiviral Res. 2009;81:267–276. doi: 10.1016/j.antiviral.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 67.Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H, Davies DR. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl Acad. Sci. USA. 1999;96:13040–13043. doi: 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maroun RG, Zargarian L, Stocklin R, Troalen F, Jankowski CK, Fermandjian S. A structural study of model peptides derived from HIV-1 integrase central domain. Rapid Commun. Mass Spectrom. 2005;19:2539–2548. doi: 10.1002/rcm.2093. [DOI] [PubMed] [Google Scholar]
- 69.Mirny LA, Gelfand MS. Structural analysis of conserved base pairs in protein-DNA complexes. Nucleic Acids Res. 2002;30:1704–1711. doi: 10.1093/nar/30.7.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garvie CW, Wolberger C. Recognition of specific DNA sequences. Mol. Cell. 2001;8:937–946. doi: 10.1016/s1097-2765(01)00392-6. [DOI] [PubMed] [Google Scholar]
- 71.Egli M, Tereshko V, Mushudov GN, Sanishvili R, Liu X, Lewis FD. Face-to-face and edge-to-face pi-pi interactions in a synthetic DNA hairpin with a stilbenediether linker. J. Am. Chem. Soc. 2003;125:10842–10849. doi: 10.1021/ja0355527. [DOI] [PubMed] [Google Scholar]
- 72.Wang JY, Ling H, Yang W, Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001;20:7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.