


Abstract
This article presents a comprehensive review of large and highly diverse superfamily of nucleotidyltransferase fold proteins by providing a global picture about their evolutionary history, sequence-structure diversity and fulfilled functional roles. Using top-of-the-line homology detection method combined with transitive searches and fold recognition, we revised the realm of these superfamily in numerous databases of catalogued protein families and structures, and identified 10 new families of nucleotidyltransferase fold. These families include hundreds of previously uncharacterized and various poorly annotated proteins such as Fukutin/LICD, NFAT, FAM46, Mab-21 and NRAP. Some of these proteins seem to play novel important roles, not observed before for this superfamily, such as regulation of gene expression or choline incorporation into cell membrane. Importantly, within newly detected families we identified 25 novel superfamily members in human genome. Among these newly assigned members are proteins known to be involved in congenital muscular dystrophy, neurological diseases and retinal pigmentosa what sheds some new light on the molecular background of these genetic disorders. Twelve of new human nucleotidyltransferase fold proteins belong to Mab-21 family known to be involved in organogenesis and development. The determination of specific biological functions of these newly detected proteins remains a challenging task.
INTRODUCTION
Nucleotidyltransferase (NTase) fold proteins constitute large and highly diverse superfamily of proteins (1). Almost all known members of this superfamily transfer nucleoside monophosphate (NMP) from nucleoside triphosphate (NTP) to an acceptor hydroxyl group belonging to protein, nucleic acid or small molecule. They are characterized by the presence of common α/β-fold structure composed of three-stranded, mixed β-sheet flanked by 4 α-helices with αβαβαβα topology. This common core corresponding to minimal NTase fold structure is usually decorated by various additional structural elements depending on the family (Figure 1). Sequence analyses of distinct members of this large superfamily revealed common sequence patterns that include conserved active site residues: hG[GS], [DE]h[DE]h and h[DE]h (h indicates a hydrophobic amino acid). Three conserved aspartate/glutamate are involved in coordination of divalent ions and activation of acceptor hydroxyl group of the substrate. Two of them (from [DE]h[DE]h motif) are located on the second core β-strand, while third carboxylate (from h[DE]h motif) is placed on the structurally adjacent third β-strand. The hG[GS] pattern is placed at the beginning of short, second core α-helix and have a crucial role in harboring of substrates within the active site (1).
Figure 1.

NTase fold superfamily structures. An example of NTase fold structure is presented together with the observed variation of secondary structure elements and additional domains in representative superfamily proteins of known structure. Positions of conserved active site motifs involved in catalysis ([DE]h[DE]h, h[DE]h) and substrate binding (hG[GS]) are marked above corresponding secondary structure elements. Side-chains of critical motifs residues (aspartates and serine) are shown as balls and sticks. Suffixes ‘_CT’ and ‘_NC’ refer to catalytic and non-catalytic NTase domains.
Known members of NTase fold superfamily contribute to many important biological functions, such as: (i) RNA polyadenylation, (ii) RNA editing, (iii) DNA repair, (iv) chromatin remodeling, (v) somatic recombination in the B cells, (vi) regulation of protein activity, (vii) antibiotic resistance and (viii) intracellular signal transduction (1,2). Specifically, they can bind and modify RNA (functional groups i-ii), DNA (iii–v), proteins (vi–vii), aminoglycosides (viii) and NTPs (viii). Proteins belonging to these functional groups are described shortly below.
PolyA polymerases (PAPs) play a central role in posttranscriptional modifications of pre-mRNA by adding few or few dozens of adenosine monophosphates to its 3′-end (i). This modification protects maturated mRNA against degradation from its 3′-end. CCA-adding enzymes (CCases) and Terminal Uridil Transferases (TUTases) participate in RNA editing (ii). CCases catalyze non-template dependent transfer of two CMPs and one AMP onto the 3′ terminus of immature tRNAs. Adding triad of nucleotides by CCases is necessary for subsequenct aminoacylation and further interactions of tRNAs with the ribosome during protein synthesis (3). TUTases take part in insertion of uridine monophosphate in various RNAs and may have different specific functions. TUTase 1 (RET1) is a processive transferase involved in uridylylation of guide RNA (gRNA), while TUTase 2 (RET2) modifies mRNA by gRNA-dependent insertion of uridine (4,5). Various NTase fold proteins are involved in DNA processing by participating in DNA repair, chromatin remodeling or somatic recombination (iii–v). Specifically, polymerases Pol β, Pol λ, Pol μ and terminal deoxynucleotidyltransferase (TdT) take part in numerous DNA repair processes (6). Pol λ and Pol μ fill shorts gaps in a template-dependent manner, while Pol μ has ability to catalyze template-independent synthesis (7). Additionally, Pol λ, Pol μ and TdT play an enormous role in creation changeability of antibodies (immunoglobulins), used by immune system to identify and neutralize antigens. Pol λ takes part in V(D)J recombination at immunoglobulin heavy-chain loci, Pol μ promotes accurate immunoglobulin κ light-chain recombination (7), while TdT is a template-independent DNA polymerase that synthesizes N regions (stretches of randomly added nucleotides) at junctions of immunoglobulin genes during somatic recombination. Other DNA polymerases belonging to NTase fold superfamily are TRF4/5 family proteins that possess a function related to DNA Topoisomerase I. The TRF enzymes are chromatin-associated NTases and are essential for condensation and segregation of chromatin in all eukaryotes. These proteins are required for repair of gaps that may be introduced as a result of topological manipulations during DNA condensation (1,2,8). Some members of NTase fold superfamily such as glutamine synthetase adenyltransferase can modulate activity of other proteins by either adding or removing of covalently bound NMP (vi) (9). Addition or removal of NMP to/from glutamine synthetase by glutamine synthetase adenyltransferase is crucial for assimilation of nitrogen in both prokaryotes and eukaryotes. Kanamycin nucleotidyltransferase (KNTase) and streptomycin nucleotidyltransferase are involved in metabolism of xenobiotics and are responsible for resistance of bacteria to antibiotics (vii). These plasmid encoded enzymes transfer a NMP to the hydroxyl group of aminoglycosides what weakens interactions of antibiotics with 30S subunit of ribosome (10). NTase fold proteins involved in intracellular signal transduction can be exemplified by bi-functional enzymes from Rel/Spo family (viii). Proteins belonging to this family enable bacteria to survive prolonged periods of nutrient limitation by taking part in metabolism of (p)ppGpp, important intracellular signaling alarmone. (p)ppGpp is associated with many biological processes such as quorum sensing, antibiotic synthesis, cell differentiation, bacteriocin production or virulence and stress-induced defense system. NTase fold superfamily also includes adenylate cyclases (ACs), which take part in synthesis of intracellular second messenger (cAMP) from ATP (1). ACs, through their ability to generate cAMP, regulate many cellular processes such as activation of protein kinases, modulation of calcium transport and regulation of gene activity. Another NTase fold proteins taking part in signal transduction are 2′–5′-oligoadenylate synthetases (OASes). These interferon-induced antiviral proteins participate in the regulation of apoptosis, cell growth and differentiation in vertebrates. OASes are activated through binding to dsRNA and then convert ATPs to 2′–5′-linked oligoadenylates (2–5A). 2–5A binds to a latent RNase L and triggers the formation of active dimeric enzyme, which degrades viral and cellular RNAs (2,11).
It should be noted that various members of NTase fold superfamily frequently display little sequence similarity, despite retaining a common core fold and few active site residues. The lack of significant sequence similarity between the families and presence of various insertions make homology inference a challenging task and hinders new family identification with traditional sequence-based approaches. Although this superfamily was thoroughly studied using standard in silico tools in recent years (1,2), many of the families have remained undiscovered due to their extreme sequence and structure divergence. To extend the realm of NTase fold proteins, we performed comprehensive searches in publicly available databases of protein families and structures using the state-of-the-art distant homology detection approaches. As a consequence we identified 10 new families of NTase fold and collected the most complete set of evolutionary related proteins that adopt this common fold. Presented paper thus offers an up-to-date review of this highly diverse superfamily of proteins, providing a global insight into their evolutionary history, sequence and structure diversity, and fulfilled functional roles.
MATERIALS AND METHODS
Detection of novel NTase fold proteins
Initially, known NTase fold families and structures were identified from literature and various protein databases such as PFAM (12), KOG (13), COG (14) and SCOP (15). This set encompassing collected PFAM, KOG and COG families and PDB (16) structures was then used for comprehensive searches for novel so far unknown members of NTase fold superfamily using a distant homology detection method Meta-BASIC (17). Meta-BASIC is a consensus meta profile alignment method capable of finding very distant similarity between proteins through a comparison of sequence profiles enriched by predicted secondary structures (meta profiles).
Identification of novel families of NTase fold was carried out using Gene Relational DataBase (GRDB) system, which included precalculated Meta-BASIC connections between 10 340 PFAM, 4852 KOG and 4872 COG families and 20 540 proteins of known structure (PDB90, representatives from PDB filtered at 90% of sequence identity). Each family and each structure in the system was represented by its sequence (for PDB90) or consensus sequence (for PFAM, COG and KOG), sequence profile generated with PSI-BLAST (18) using NCBI non-redundant (NR) protein sequence database derivative (NR70), and secondary structure profile predicted with PSI-PRED (19).
The search strategy was based on the transitivity concept, where each newly identified PFAM, KOG and COG family or PDB structure was used for further Meta-BASIC searches until no new additional hits were found. Moreover, the high divergence of some NTase fold families that is likely reflected as lower than threshold (<40) Meta-BASIC scores was also considered. According to rigorous structural criteria used in LiveBench benchmarks (http://meta.bioinfo.pl/results.pl?comp_name=livebench-2008.2), predictions with scores above 40 have less than 5% probability of being incorrect. Specifically, in addition to high scoring (>40) Meta-BASIC hits, all below threshold hits with scores >20 were also analyzed to detect correct predictions placed among unreliable or incorrect ones.
Selection of potentially correct predictions was based on two essential defining criteria for NTase fold proteins, that were verified manually. First, family profile should include correctly aligned conserved acidic residues from catalytic motifs (with the exception of families that might loose their catalytic function) in addition to the presence of hG[GS] motif (that in some cases can be also less conserved). Second, family profile should include all secondary structure elements that correspond to the evolutionary core of NTase fold (αβαβαβα) and conserved critical hydrophobic positions responsible for forming a hydrophobic core of the structure. To confirm difficult predictions, consensus sequences and selected representative members of families that met the above mentioned criteria were submitted to the Protein Structure Prediction Meta Server (http://meta.bioinfo.pl) that assembles various top-of-the-line fold recognition methods. Models generated by these methods were analyzed with 3D-Jury, a consensus approach that uses structural comparisons to select potentially the best predictions (20). 3D-Jury system was run with default settings that included five servers used for consensus building: FFAS03 (21), mGenTHREADER (22), INBGU (23), FUGUE-2 (24) and Meta-BASIC (17).
Grouping of families and structures
Collected NTase fold proteins were assembled into groups of closely related families and structures. Families and structures within single group share relatively high sequence similarity detectable with both PSI-BLAST and RPS-BLAST with confident E-value lower than 0.001. Proteins belonging to different groups are more diverse and can not be linked by these standard homology detections tools.
Generation of superfamily alignment
Initially, to collect sequences that belong to analyzed NTase fold families, PSI-BLAST searches were performed against NR database using consensus sequences of the families. For each family, multiple sequence alignment was generated using PCMA program (25) and where necessary followed by manual adjustments. Simultaneously, all identified NTase fold structures were superimposed to derive structure-based alignment of their sequences for the conserved core regions of the fold that encompass three β-strands and four α-helices (αβαβαβα topology). Final alignment for whole NTase fold superfamily in the conserved regions was assembled from sequence-to-structure mappings between NTase fold families and the closest structures. In majority of cases, the closest structures were identified as the most common templates appearing in high scoring 3D-Jury predictions. The family-structure mappings were built manually based on Meta-BASIC and 3D-Jury alignments, PSI-PRED secondary structure predictions and conservation of critical active site residues and hydrophobic patterns using consensus alignment and 3D assessment approach (26) for representative family proteins.
Domain architecture and genomic context analyses
To detect other protein domains in identified NTase fold families, collected family sequences were analyzed with Meta-BASIC after removal of the conserved NTase domain. Non-trivial assignments were further verified with 3D-Jury method. NTase fold proteins were also searched for transmembrane segments [TMHMM2 (27)], signal peptides [SignalP (28)], low compositional complexity [SEG (29)] and coiled coil regions [Coils2 (30)] using SMART server (31). Possible functional associations were investigated using genomic context analysis (neighborhood, co-occurrence, gene fusion and co-expression) with STRING database (32).
Identification of human superfamily members and associated genetic diseases
To identify human members of NTase fold superfamily, 43 570 human proteins (human proteome from ENSEMBL database) were included in the GRDB system. Similar search strategy was applied to that described above, starting with all known and newly identified families of NTase fold. Genetic disorders associated with genes encoding NTase fold proteins were identified using OMIM database (33). For selected human proteins high-quality 3D models were built with Modeller (34) followed by mapping of mutations observed in analyzed genetic disorders.
RESULTS AND DISCUSSION
In this study we applied similar approach to that used in our recent work where we identified a number of novel but highly diverged PD-(D/E)XK nuclease families (35). The initial set of known NTase fold proteins was used as the starting point for transitive Meta-BASIC searches for new superfamily members among all PFAM, KOG and COG families and PDB structures. To consider the observed high sequence divergence in NTase fold superfamily that is likely reflected in lower than confidence cut-off Meta-BASIC scores, we analyzed also below threshold hits. This resulted in identification of many novel families of NTase fold that escape detection with standard homology search methods. These potentially novel superfamily members were subjected to further extensive analyses including fold recognition with 3D Jury to confirm initial predictions. Final selection of correct but highly non-trivial assignments was based on a consistency of a predicted secondary structure pattern with that of NTase fold, general conservation of critical hydrophobic residues and—for potentially active enzymes—presence of active site motifs critical for maintaining of NTase function (Figure 2).
Figure 2.

Multiple sequence alignment for NTase fold superfamily. Only conserved regions of the fold core are shown for representative proteins of known structure and single representative sequences for groups lacking experimentally solved structure. Sequences are labeled according to PDB code or NCBI gene identification (gi) number and the number of group they belong to (XVII–XXVI, newly detected NTase fold proteins). Multiple NTase fold domains occurring in some proteins are denoted by consecutive roman numbers. The numbers of excluded residues are specified in square brackets. Conservation of residues is denoted with the following scheme: uncharged, highlighted in yellow; small, letters in red; critical active site aspartates/glutamates, highlighted in black. Locations of secondary structure elements (E, β-strand; H, α-helix) are marked above sequences.
On the basis of their sequence similarity (evolutionary distance), all collected NTase fold families and structures were assembled into 26 distinct groups (Table 1). Families within single group can be easily linked both with PSI-BLAST and RPS-BLAST (closely related), in contrast to those belonging to different groups that can not be connected with these standard homology detection methods (distantly related). Figure 3 illustrates detected similarity network for NTase fold families and structures using Meta-BASIC method. It should be noted that the Meta-BASIC connections between diverse members of this superfamily were established transitively. This shows clearly the importance of the concept of transitivity applied in our approach, where newly identified families were used for detection of other novel families of NTase fold in further searches. Moreover, many established connections between families of unknown structure are not detectable even with advanced fold recognition methods that require a known reference protein structure. In addition, by considering Meta-BASIC hits also with scores below a confident threshold of 40, we were able to identify the most divergent sequence groups. Although we investigated all Meta-BASIC predictions with scores above 20, subsequent analysis revealed that a more stringent cutoff of 28 would be sufficient to identify transitively all members of NTase fold superfamily (Figure 3).
Table 1.
NTase fold groups, their representatives in human and connections to genetic diseases
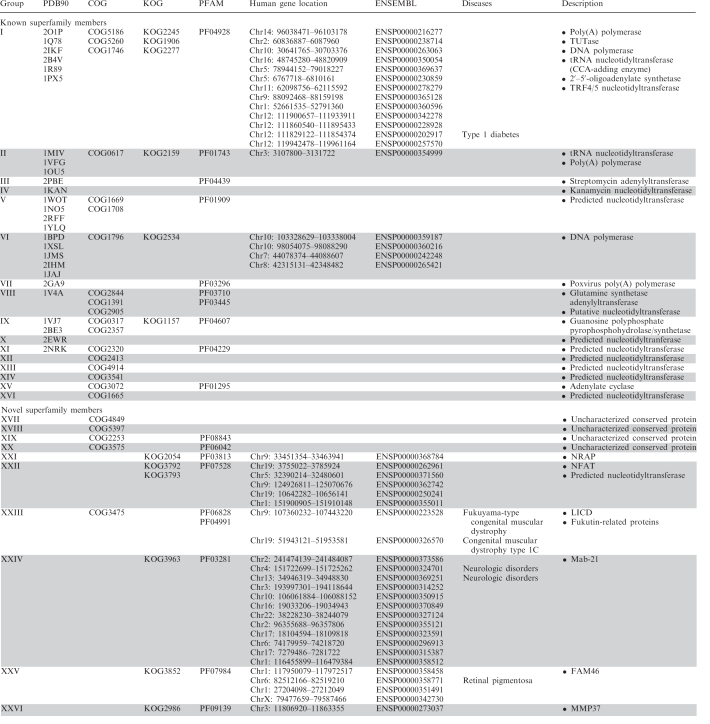 |
Proteins belonging to single group share relatively high sequence similarity detectable by both PSI-BLAST and RPS-BLAST. The columns provide: the group number (column ‘Group’); representative proteins (at 90% seq id) with solved structure (column ‘PDB90’); protein families catalogued in COG, KOG and PFAM databases (columns ‘COG’, ‘KOG’ and ‘PFAM’, respectively); human members in ENSEMBL database and their gene loci (columns ‘ENSEMBL’ and ‘Human gene location’, respectively), human genetic diseases associated with mutations found in corresponding genes (column ‘Diseases’) and functional description of the families (column ‘Description’).
Figure 3.

Connectivity network for NTase fold superfamily. Groups of closely related proteins that can be linked by both PSI-BLAST and RPS-BLAST are surrounded by black (known members of NTase fold superfamily) and red (newly detected) circles. Detected Meta-BASIC connections between PFAM, KOG, COG families, PDB structures and human proteins are shown as lines colored according to the Meta-BASIC score: pink, confident scores above 40; grey, below threshold scores between 28 and 40. For each group short description and taxonomy distribution is provided.
According to our classification, 16 groups (Groups I–XVI, for a brief description of these groups see Supplementary Data) include well-known members of NTase fold superfamily while, importantly, remaining 10 groups of proteins (Groups XVII–XXVI) encompasses novel families of NTase fold that we identified among uncharacterized and poorly annotated proteins. Some of them probably do not exhibit NTase function as they lack important catalytic residues. Their specific function is not clear but we speculate that some of them may participate in signaling pathways or regulation of gene expression. To hint at additional functional information, both domain architecture (Supplementary Figure S1) and genomic context were derived for all NTase fold families. Newly identified NTase fold proteins are described in more details below.
Newly detected NTase fold superfamily members
Groups XVII–XX (uncharacterized proteins)
Four new groups of proteins of so far unknown structure and function that we assigned to NTase fold superfamily encompass: group XVII (uncharacterized protein conserved in bacteria COG4849), group XVIII (uncharacterized conserved protein COG5397), group XIX [uncharacterized conserved protein COG2253 and domain of unknown function DUF1814 (PF08843)], group XX [uncharacterized protein conserved in bacteria COG3575 and bacterial protein of unknown function DUF925 (PF06042)]. All these proteins have only single NTase domain and are probably active NTases, as they posses conserved catalytic acidic residues and hG[GS] motif. Unfortunately, no specific functions could be assigned for those enzymes through bioinformatics analyses and because of lack of any experimental data.
Group XXI (NRAP)
Nucleolar RNA-associated proteins (NRAP) (KOG2054 and PF03813) belong to another group of newly detected divergent members of NTase fold superfamily. High expression of NRAP is observed in tissues with most abundant ribosome biogenesis such as spleen, kidney, colon and testis. Usually, proteins from this group posses duplicated inactive NTase domain (lack of conserved catalytic amino acids), each followed by C-terminal PAP/OAS1 substrate binding domain, which is also found in OAS family proteins, TRF4/5 polymerases, trypanosomal urydylytransferases, poly(A) polymerases and archeal CCA-adding enzymes. The most probably both non-catalytic NTase and PAP/OAS1 substrate binding domains take part in nucleic acid binding. Accordingly, NRAP appears to be involved in ribosome biogenesis by interacting with pre-rRNA primary transcript (36). In addition, some proteins belonging to group XXI are classified as U3 snoRNP involved in maturation of pre-18S rRNA according to existing descriptions in NR sequence database generated by automatic computational analysis of large-scale protein–protein interactions (36). All these data suggests that NRAP proteins may participate in rRNA splicing.
Group XXII (NFAT)
Nuclear factor of activated T-cells (NFAT), subunits NF90 and NF45 (KOG3792 and KOG3793, respectively) together with DZF (PF07528) form a group of proteins newly assigned to NTase fold superfamily. These proteins are known to play an important role in regulation of antigen-response genes. Human NF45 (ILF2) and NF90 (ILF3) may contribute to RNA gene regulation at the levels of transcription, export and translocation (37,38). NF45 and NF90 also stabilize the association between the KU70, KU80 and the DNA-dependent protein kinase (DNA-PK). KU70, KU80 and DNA-PK each serve essential roles in development of lymphocytic immunity through their contributions to double-stranded DNA break repair and antibody and T-cell receptor diversity (38). Additionally, NF90 takes part in the formation of a circular conformation of the viral genomes, which is important for the coordination of viral translation and replication (39).
NTase domain in group XXII proteins is always followed by C-terminal PAP/OAS1 substrate binding domain, which is usually responsible for DNA or RNA binding. In addition to that, extra N-terminal (zinc finger domain) and C-terminal (double-stranded RNA binding motif—PF00035) domains may assist in binding nucleic acids. Importantly, only some proteins from this group [e.g. human proteins: spermatid perinuclear RNA-binding protein—STRBP and interleukin enhancer-binding factor 3 (ILF3)] share conserved [DE]h[DE]h motif in NTase domain and thus may have NTase activity. Similarly to DNA topoisomerase I—related function protein family, those NFAT proteins may function as a chromatin-associated NTases. Remaining proteins from group XXII that do not posses conserved amino acids responsible for catalysis could probably function as a transcription regulators. This is consistent with experimental data indicating that NFAT proteins may contribute to gene regulation (38).
Group XXIII (Fukutin/LICD)
Group XXIII includes LPS biosynthesis proteins (COG3475), Fukutin-related proteins (PF06828) and LICD protein family (PF04991). Proteins from this group are known to participate mostly in glycosylation and posttranslational protein modification. LicD2 from Streptococcus pneumoniae takes part in addition of phosphocholine to lipoteichoic acid (LTA) using CDP-choline as a substrate. Choline is an essential growth factor, which in the form of phosphocholine is incorporated in the cell wall by its attachment to teichioc (TA) and lipoteichoic acids. Choline participates in pneumococcal cell separation, transformation, autolysis and interactions with the host cell surface and choline-binding proteins. In Haemophilus influenzae licD is cotranscribed with licA, licB and licC, which encode choline kinase, choline transferase and phosphocholine citidyl transferase, respectively (Supplementary Figure S2). In S. pneumoniae, in contrast to H. influenzae, licD2 and licD1 (both homologous to licD) are cotranscribed with sugar transporter, while licA, licB and licC are regulated together in other operon (40,41). Finally, the yeast MNN4 (LICD family member) transfers mannosyl phosphate from GDP-mannose to N- and O-linked oligsosaccharides in proteins (42,43).
Predicted NTase domain encoded by licD from H. influenzae and licD1 and licD2 from S. pneumoniae is the most likely to be active what suggest its direct role in incorporation of phosphocholine (ChoP) to the cell wall LTA and TA (alone or in cooperation with N-terminal glycosyl transferase domain, see below). Specifically, some proteins from group XXIII possess additional domains including NAD(P) binding domain and newly identified but probably non-active glycosyl transferase family 2 domain (PF00535) that lacks conserved catalytic amino acids. Detection of glycosyl transferase domain was highly non-trivial and was possible by applying consensus of fold recognition (3D-Jury). Active glycosyl transferases form glycosyl bonds by catalyzing the transfer of sugar moieties from donor molecules such as UDP-glucose, UDP-N-acetyl-galactosamine, GDP-mannose or CDP-abequose to specific acceptor molecules (44). Probably, the non-active glycosyl transferase domain in group XXIII proteins can recognize specific molecules (choline—LicD or sugar—Fukutin) bound to NDP, transfer of which is carried by NTase domain. Additional NAD(P) binding domain has Rossmann fold structure but its specific function is not known.
Group XXIV (Mab-21)
This group contains small conserved eukaryotic family of ‘Male-abnormal 21’ (Mab-21) proteins (PF03281) and Mab-21-like cell fate specification proteins (KOG3963). They posses newly detected N-terminal NTase domain followed by C-terminal PAP/OAS1 substrate binding domain. In Caenorhabditis elegans Mab-21 protein is associated with cell fates and the formation of sensory organs in male nematodes, located in the tail and involved in copulation (45). D6 genes from Danio rerio and Xenopus laevis encode Mab-21 family proteins and are important during eye differentiation and neuronal development, respectively. In Mus musculus expression of the D6 genes is essential for development of the embryonic brain, eye, limbs and neural crest derivatives (46). Additionally, Mab-21 proteins are closely connected with signal transduction. In C. elegans Mab-21 is involved in transforming growth factor-β (TGF-β) signaling pathway (47), while human homologs interact with nuclear transducer SMAD1 in vertebrate bone morphogenetic protein 4 (BMP4) signaling pathway (46). It was shown that one of the human Mab-21 proteins (KIAA1754) may bind inositol 1,4,5-triphosphate (intracellular messanger) (48) and can act as a transcriptional repressor when targeted to a heterologous promoter (46). Our analyses indicate that only some Mab-21 family members have conserved catalytic residues in predicted NTase domain, while remaining homologs likely do not retain enzymatic function. On the basis of the presence of additional C-terminal domain (potentially responsible for nucleic acid binding) and available experimental data for the members of this family we hypothesize that Mab-21 proteins may act as a transcription regulators.
Group XXV (FAM46)
Group XXV consists of uncharacterized conserved FAM46 (C6orf37) proteins (KOG3852 and PF07984) with newly detected NTase domain. These proteins are expressed in the neural retina, adult skeletal muscle, thymus, liver, lung and brain (49). Majority of FAM46 proteins, similarly to other superfamily members from groups I, XXI, XXII and XXIV, possess additional C-terminal PAP/OAS1 substrate binding domain. FAM46 proteins are known to have different functions in many tissues at various stages of development. For instance, in human FAM46A is a SMAD signaling pathway related protein, where SMADs are intercellular effectors of transforming growth factor-β (TGF-β) (50). Consequently, similarly to Mab-21, FAM46 proteins seem to be involved in TGF-β signaling pathway. In addition, the presence of predicted potential phosphorylation sites in FAM46A (in regions corresponding to NTase and C-terminal PAP/OAS1 substrate binding domains) may suggest post-translational activation or regulation of its function (49). The most importantly, FAM46 proteins share all critical catalytic amino acids and thus might be active NTases which carry out some NTase reaction potentially crucial for cellular signaling.
Group XXVI (MMP37)
Another novel group of NTase fold proteins embraces uncharacterized conserved proteins (KOG2986) and mitochondrial matrix protein of 37 kDa (MMP37) family (PF09139). MMP37 family is involved in the translocation of proteins across the mitochondrial inner membrane via the TIM23-PAM complex. Gallas et al. hypothesized that MMP37 proteins enhance the early stages of the TIM23 matrix import pathway (51). Our analysis shows that although MMP37 proteins possess NTase fold, they have only one active site carboxylate (from [DE]h[DE]h motif) and thus probably are not able to carry out enzymatic reaction. These potentially non-active members of NTase fold superfamily may bind ATP, hydrolysis of which is necessary for the translocation of proteins through the membrane. This hypothesis is consistent with available experimental data and suggestions risen by Gallas et al.
Human NTase fold superfamily proteins
So far only 18 human proteins have been assigned to NTase fold superfamily. These NTases belong to three different groups: group I (PAPOLA, PAPOLG, PAPD1, PAPD5, PAPD4, POLS, TUT1, ZCCHC6, ZCCHC11, OAS1, OAS2, OAS3, OASL), group II (TRNT1) and group VI (POLB, POLM, POLL, DNTT). They participate in many important processes such as RNA maturation, transcription, translation and somatic recombination. Our comprehensive search in human proteome using Meta-BASIC and initiated with all classified above NTase fold families resulted in detection of 25 novel human superfamily members (Figure 4 and Supplementary Figure S3). These proteins of previously unknown structure and function have been placed in seven newly identified groups (Table 1): group XXI (one protein encoded by NOL6), group XXII (five proteins encoded by KIAA1085, ZFR, STRBP, ILF3 and ILF2), group XXIII (two proteins encoded by FKTN and FKTR), group XXIV (12 proteins encoded by C2ORF54, MAB21L2, MAB21L1, C3ORF59, KIAA1754, SMCR7L, SMCR7, C6ORF150, TMEM102, C1ORF161, ITPRIPL1 and ITPRIPL2), group XXV (four proteins encoded by FAM46A, FAM46B, FAM46C and FAM46D) and group XXVI (one protein encoded by C3ORF31). Assignment of these proteins to NTase fold superfamily combined with available experimental data allowed us to describe the molecular mechanisms of their action, e.g. in incorporation of choline into membrane or regulation of transcription and translation. It should be noted that genes encoding NTase fold proteins (43 human genes in total) are dispersed quite evenly in human genome and are located on majority of chromosomes with the exception of chromosomes 15, 18, 20, 21 and Y (Figure 4).
Figure 4.

NTase fold proteins in human. Locations of genes encoding NTase fold proteins in human genome are marked with triangles, while rectangles correspond to known genetic diseases associated with mutations found in some of these genes. ENSEMBL identifiers are provided for each protein in blue and red boxes that correspond to known and newly identified groups in our classification, respectively.
Finally, we have also analyzed genetic mutations in these human proteins found in various diseases using data collected from OMIM database and manual literature searches. In addition to known connection between OASes and type 1 diabetes (52), we have shown that members of NTase fold superfamily may be also involved in congenital muscular dystrophy, neurologic diseases and retinal pigmentosa, what sheds some new light on the molecular background of these diseases (Figure 4 and Table 1).
Congenital muscular dystrophy
The congenital muscular dystrophies (CMDs) constitute a heterogeneous group of autosomal recessive disorders (53). According to Muntoi and Voit (54) there are three groups of proteins which take part in CMD disorder: extracellular proteins and/or receptors in extracellular matrix, O-mannosyl glycosyltransferases and endoplasmatic reticulum proteins. The second group includes O-mannosyl transferase, β1,2N-acetylglucosaminyltransferase, and Fukutin/Fukutin-related proteins which are newly predicted members of NTase fold superfamily.
Aravidin and Koonin observed that Fukutin (encoded by FCMD) and Fukutin related protein (encoded by FKRP) possess DxD motif and a distal aspartate residue suggesting that these proteins may coordinate a divalent cation similar to a number of NTases (43). Here we show that Fukutin-related protein is composed of N-terminal inactive glycosyltransferase domain followed by NTase domain, while Fukutin includes only NTase domain. It is possible that Fukutin and Fukutin-related protein possess enzymatic activity that contributes to the synthesis of the O-mannosyl glycans in α-dystroglycan. Our hypothesis is supported by experimental data which shows that CMD may be connected with reduction or abnormalities in glycosylated α-dystroglycan (55).
We analyzed all known mutations in FCMD and FKTR genes leading to Fukuyama-type congenital muscular dystrophy (FCMD) and congenital muscular dystrophy type 1C (MDC1C). MDC1C is caused by mutations in the gene encoding Fukutin-related protein (53), while retrotransposon insertion in the 3′ UTR of FCMD gene is known to lead to FCMD. Compounds heterozygotes have also been described with point mutations in one allele of the FCMD gene and the retrotransposon on the second allele. However, no patient had homozygous point mutations suggesting that the complete lack of functional Fukutin could result in embryonic lethality (53).
To analyze the role of single point mutations, we have built 3D models for NTase domain of Fukutin and Fukutin-related protein using the structure of putative NTase TM1012 from Thermotoga maritima (PDB: 2ewr) as the closest template identified by 3D-Jury. We have mapped analyzed mutations onto corresponding 3D models and found that they can be divided into two groups. First group includes mutations that cluster in the area of the active site (Q358P in FKTN and V405L in FKRP; green spheres on Figure 5) and probably impede catalysis or substrate binding. Remaining mutations (R307Q in FKTN and P316T, P448L, A455D in FKRP; red spheres on Figure 5) are also grouped on the surface but on the opposite side of the protein where they may impair protein–protein (e.g. with N-terminal substrate binding domain) or protein–membrane interactions. Specifically, these mutations may thus lead to improper orientation of NTase and N-terminal substrate binding domains and as a consequence may lead to misalignment of the bound substrate relative to the active site or alternatively weaken interaction of the protein with membrane.
Figure 5.

Genetic mutations in Fukutin and Fukutin related protein connected with congenital muscular dystrophy. Positions of single point mutations found in genes encoding Fukutin (in Fukuyama-type congenital muscular dystrophy, FCMD) and Fukutin related protein (in congenital muscular dystrophy type 1C, MDC1C) are shown as spheres on the respective 3D models of their structure. Mutations denoted with green spheres are clustered in the area of the active site and probably impede catalysis or substrate binding. Red spheres represent mutations grouped on the opposite side of the protein surface where they may impair protein–protein or protein–membrane interactions.
Neurological disorders
All human proteins from Mab-21 family consist of N-terminal NTase domain followed by C-terminal PAP/OAS1 substrate-binding domain and as hypothesized above they may function as transcription regulators. Although the detailed role of this protein family remains as of now undiscovered, there is an evidence on specific function of these proteins in the broad range of neurological disorders. The various phenotypes associated with this group of genes is related to the nature of the pathogenic condition. It was shown that CAG-trinucleotides repeats in the 5′-untranslated region of two Mab-21 proteins (C2orf54/ENSP00000373586 and MAB21L2/ENSP00000324701, both seem to be active NTases) may undergo the span what can lead to the accumulation of altered proteins in neurons. Previous reports suggested the involvement of this condition in neurologic disorders, such as affective disorders (56), or mental retardation (57). By the analogy to the other neurodegenerative disorders the specific function of these Ntase fold proteins is not critical for the altered phenotype, but is a result of the loco-specific pattern of expression of aberrant gene products.
Retinal pigmentosa
Retinal pigmentosa (RP) is a group of genetic eye conditions encompassing retinal disorders connected with rod retina degeneration (58). FAM46A, located on chromosome 6, was reported to be responsible for various types of dominant and recessive retinal dystrophies. FAM46A may be involved in signal transduction and experimental data shows that it may be a partner in protein–protein interaction map of the SMAD signaling pathway, specifically of SARA, a key regulator of SMAD activation. One of the SMAD-associated factors, BMP7, is known to increase a number of other segments process in chick cultured photoreceptors, while others such as interleukin 2 and STAT-1 are associated with photoreceptor survival and retinal degradation (50). This data combined with our fold and function prediction indicate that FAM46A is an important NTase in signal transduction, and lack of this NTase activity may lead to retinal dysfunction.
CONCLUSION
In this work we combined the state-of-the-art distant homology detection method with transitive similarity searches and fold recognition to identify currently the most complete realm of NTase fold proteins. To fulfill this task we analyzed also lower than threshold predictions to identify novel protein families distantly related to other members of NTase fold superfamily in terms of sequence and structure space. Using this approach we significantly increased the number of known NTase fold proteins and identified 10 new groups which include hundreds of proteins of previously unreported structures and molecular functions. These results clearly suggest that such approach can push limits further for distant homology detection in the midnight zone of homology and allows to study those superfamilies that were exposed in evolution to higher sequence divergence.
The resulting comprehensive classification of NTase fold proteins provides a global picture of this superfamily and sheds some new light on sequence–structure–function relationships and evolution of these proteins that diverged from a common ancestor to adapt to various functional niches. Majority of NTase fold proteins are active NTase that participate in various important biological processes, including DNA repair, somatic recombination, intercellular signaling transduction, enzyme activity regulation and antibiotic resistance. Functional predictions for newly detected members of this superfamily suggest also novel, previously not observed activities such as regulation of gene expression or choline incorporation into cell membrane.
We also performed the systematic search of human proteome to detect 43 NTase fold proteins including 25 novel members of this superfamily. The new members belong to six of ten groups, with nearly a half of these (12) belonging to the group XXIV (Mab-21) in our classification. Some members of this family were reported to be actively involved in either transcription or secondary messenger signaling. Genetic data from OMIM database combined with sequence/structure analyses for newly detected human NTases and their inactive homologs, allowed to obtain a novel insight into molecular background of selected genetic disorders, including a congenital muscular dystrophy, retinal pigmentosa and neurological disorders. These data should inspire further experimental studies on predicted activity and potential substrates providing further insight into detailed biological roles of these highly important proteins in humans.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
EMBO Installation, National Institute of Health (1R01GM081680-01), Foundation for Polish Science and Ministry of Science and Higher Education (MNiSW) (N N301 159435) grants. L.R. acknowledges the support of European Community and MNiSW grants: OxyGreen (KBBE- 2007-212281) and PepLaser (HEALTH-F4-2009-223243). L.S.W. was supported by MNiSW grant N N518 331736. 85. Funding for open access charge: MNiSW (N N301 159435).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Aravind L, Koonin EV. DNA polymerase beta-like nucleotidyltransferase superfamily: identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999;27:1609–1618. doi: 10.1093/nar/27.7.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rogozin IB, Aravind L, Koonin EV. Differential action of natural selection on the N and C-terminal domains of 2′-5′ oligoadenylate synthetases and the potential nuclease function of the C-terminal domain. J. Mol. Biol. 2003;326:1449–1461. doi: 10.1016/s0022-2836(03)00055-x. [DOI] [PubMed] [Google Scholar]
- 3.Tomita K, Fukai S, Ishitani R, Ueda T, Takeuchi N, Vassylyev DG, Nureki O. Structural basis for template-independent RNA polymerization. Nature. 2004;430:700–704. doi: 10.1038/nature02712. [DOI] [PubMed] [Google Scholar]
- 4.Deng J, Ernst NL, Turley S, Stuart KD, Hol WG. Structural basis for UTP specificity of RNA editing TUTases from Trypanosoma brucei. EMBO J. 2005;24:4007–4017. doi: 10.1038/sj.emboj.7600861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stagno J, Aphasizheva I, Rosengarth A, Luecke H, Aphasizhev R. UTP-bound and Apo structures of a minimal RNA uridylyltransferase. J. Mol. Biol. 2007;366:882–899. doi: 10.1016/j.jmb.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. A closed conformation for the Pol lambda catalytic cycle. Nat. Struct. Mol. Biol. 2005;12:97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 7.Moon AF, Garcia-Diaz M, Bebenek K, Davis BJ, Zhong X, Ramsden DA, Kunkel TA, Pedersen LC. Structural insight into the substrate specificity of DNA Polymerase mu. Nat. Struct. Mol. Biol. 2007;14:45–53. doi: 10.1038/nsmb1180. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Castano IB, Adams C, Vu C, Fitzhugh D, Christman MF. Structure/function analysis of the Saccharomyces cerevisiae Trf4/Pol sigma DNA polymerase. Genetics. 2002;160:381–391. doi: 10.1093/genetics/160.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y, Zhang R, Joachimiak A, Carr PD, Huber T, Vasudevan SG, Ollis DL. Structure of the N-terminal domain of Escherichia coli glutamine synthetase adenylyltransferase. Structure. 2004;12:861–869. doi: 10.1016/j.str.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen LC, Benning MM, Holden HM. Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyltransferase. Biochemistry. 1995;34:13305–13311. doi: 10.1021/bi00041a005. [DOI] [PubMed] [Google Scholar]
- 11.Hartmann R, Justesen J, Sarkar SN, Sen GC, Yee VC. Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein 2′-5′-oligoadenylate synthetase. Mol. Cell. 2003;12:1173–1185. doi: 10.1016/s1097-2765(03)00433-7. [DOI] [PubMed] [Google Scholar]
- 12.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 16.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ginalski K, von Grotthuss M, Grishin NV, Rychlewski L. Detecting distant homology with Meta-BASIC. Nucleic Acids Res. 2004;32:W576–W581. doi: 10.1093/nar/gkh370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 20.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 21.Jaroszewski L, Slabinski L, Wooley J, Deacon AM, Lesley SA, Wilson IA, Godzik A. Genome pool strategy for structural coverage of protein families. Structure. 2008;16:1659–1667. doi: 10.1016/j.str.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGuffin LJ, Jones DT. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–881. doi: 10.1093/bioinformatics/btg097. [DOI] [PubMed] [Google Scholar]
- 23.Fischer D, Eisenberg D. Protein fold recognition using sequence-derived predictions. Protein Sci. 1996;5:947–955. doi: 10.1002/pro.5560050516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- 25.Pei J, Sadreyev R, Grishin NV. PCMA: fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics. 2003;19:427–428. doi: 10.1093/bioinformatics/btg008. [DOI] [PubMed] [Google Scholar]
- 26.Ginalski K, Rychlewski L. Protein structure prediction of CASP5 comparative modeling and fold recognition targets using consensus alignment approach and 3D assessment. Proteins. 2003;53(Suppl. 6):410–417. doi: 10.1002/prot.10548. [DOI] [PubMed] [Google Scholar]
- 27.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 28.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 29.Wootton JC. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput. Chem. 1994;18:269–285. doi: 10.1016/0097-8485(94)85023-2. [DOI] [PubMed] [Google Scholar]
- 30.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 31.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl Acad. Sci. USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. STRING 8 – a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007;80:588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 35.Knizewski L, Kinch LN, Grishin NV, Rychlewski L, Ginalski K. Realm of PD-(D/E)XK nuclease superfamily revisited: detection of novel families with modified transitive meta profile searches. BMC Struct. Biol. 2007;7:40. doi: 10.1186/1472-6807-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Utama B, Kennedy D, Ru K, Mattick JS. Isolation and characterization of a new nucleolar protein, Nrap, that is conserved from yeast to humans. Genes Cells. 2002;7:115–132. doi: 10.1046/j.1356-9597.2001.00507.x. [DOI] [PubMed] [Google Scholar]
- 37.Larcher JC, Gasmi L, Viranaicken W, Edde B, Bernard R, Ginzburg I, Denoulet P. Ilf3 and NF90 associate with the axonal targeting element of Tau mRNA. FASEB J. 2004;18:1761–1763. doi: 10.1096/fj.04-1763fje. [DOI] [PubMed] [Google Scholar]
- 38.Zhao G, Shi L, Qiu D, Hu H, Kao PN. NF45/ILF2 tissue expression, promoter analysis, and interleukin-2 transactivating function. Exp. Cell Res. 2005;305:312–323. doi: 10.1016/j.yexcr.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 39.Isken O, Grassmann CW, Sarisky RT, Kann M, Zhang S, Grosse F, Kao PN, Behrens SE. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J. 2003;22:5655–5665. doi: 10.1093/emboj/cdg562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang JR, Idanpaan-Heikkila I, Fischer W, Tuomanen EI. Pneumococcal licD2 gene is involved in phosphorylcholine metabolism. Mol. Microbiol. 1999;31:1477–1488. doi: 10.1046/j.1365-2958.1999.01291.x. [DOI] [PubMed] [Google Scholar]
- 41.Weiser JN, Shchepetov M, Chong ST. Decoration of lipopolysaccharide with phosphorylcholine: a phase-variable characteristic of Haemophilus influenzae. Infect. Immun. 1997;65:943–950. doi: 10.1128/iai.65.3.943-950.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jigami Y, Odani T. Mannosylphosphate transfer to yeast mannan. Biochim. Biophys. Acta. 1999;1426:335–345. doi: 10.1016/s0304-4165(98)00134-2. [DOI] [PubMed] [Google Scholar]
- 43.Aravind L, Koonin EV. The fukutin protein family–predicted enzymes modifying cell-surface molecules. Curr. Biol. 1999;9:R836–R837. doi: 10.1016/s0960-9822(00)80039-1. [DOI] [PubMed] [Google Scholar]
- 44.Campbell JA, Davies GJ, Bulone V, Henrissat B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997;326(Pt 3):929–939. doi: 10.1042/bj3260929u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho SH, So GM, Chow KL. Postembryonic expression of Caenorhabditis elegans mab-21 and its requirement in sensory ray differentiation. Dev. Dyn. 2001;221:422–430. doi: 10.1002/dvdy.1161. [DOI] [PubMed] [Google Scholar]
- 46.Nikolaidis N, Chalkia D, Watkins DN, Barrow RK, Snyder SH, van Rossum DB, Patterson RL. Ancient origin of the new developmental superfamily DANGER. PLoS ONE. 2007;2:e204. doi: 10.1371/journal.pone.0000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morita K, Chow KL, Ueno N. Regulation of body length and male tail ray pattern formation of Caenorhabditis elegans by a member of TGF-beta family. Development. 1999;126:1337–1347. doi: 10.1242/dev.126.6.1337. [DOI] [PubMed] [Google Scholar]
- 48.van Rossum DB, Patterson RL, Cheung KH, Barrow RK, Syrovatkina V, Gessell GS, Burkholder SG, Watkins DN, Foskett JK, Snyder SH. DANGER, a novel regulatory protein of inositol 1,4,5-trisphosphate-receptor activity. J. Biol. Chem. 2006;281:37111–37116. doi: 10.1074/jbc.M608760200. [DOI] [PubMed] [Google Scholar]
- 49.Lagali PS, Kakuk LE, Griesinger IB, Wong PW, Ayyagari R. Identification and characterization of C6orf37, a novel candidate human retinal disease gene on chromosome 6q14. Biochem. Biophys. Res. Commun. 2002;293:356–365. doi: 10.1016/S0006-291X(02)00228-0. [DOI] [PubMed] [Google Scholar]
- 50.Barragan I, Borrego S, Abd El-Aziz MM, El-Ashry MF, Abu-Safieh L, Bhattacharya SS, Antinolo G. Genetic analysis of FAM46A in Spanish families with autosomal recessive retinitis pigmentosa: characterisation of novel VNTRs. Ann. Hum. Genet. 2008;72:26–34. doi: 10.1111/j.1469-1809.2007.00393.x. [DOI] [PubMed] [Google Scholar]
- 51.Gallas MR, Dienhart MK, Stuart RA, Long RM. Characterization of Mmp37p, a Saccharomyces cerevisiae mitochondrial matrix protein with a role in mitochondrial protein import. Mol. Biol. Cell. 2006;17:4051–4062. doi: 10.1091/mbc.E06-04-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Field LL, Bonnevie-Nielsen V, Pociot F, Lu S, Nielsen TB, Beck-Nielsen H. OAS1 splice site polymorphism controlling antiviral enzyme activity influences susceptibility to type 1 diabetes. Diabetes. 2005;54:1588–1591. doi: 10.2337/diabetes.54.5.1588. [DOI] [PubMed] [Google Scholar]
- 53.Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB, Mercuri E, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul. Disord. 2004;14:635–649. doi: 10.1016/j.nmd.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Sciandra F, Gawlik KI, Brancaccio A, Durbeej M. Dystroglycan: a possible mediator for reducing congenital muscular dystrophy? Trends Biotechnol. 2007;25:262–268. doi: 10.1016/j.tibtech.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 56.Margolis RL, Stine OC, Ward CM, Franz ML, Rosenblatt A, Callahan C, Sherr M, Ross CA, Potter NT. Unstable expansion of the CAG trinucleotide repeat in MAB21L1: report of a second pedigree and effect on protein expression. J. Med. Genet. 1999;36:62–64. [PMC free article] [PubMed] [Google Scholar]
- 57.Potter NT. Meiotic instability associated with the CAGR1 trinucleotide repeat at 13q13. J. Med. Genet. 1997;34:411–413. doi: 10.1136/jmg.34.5.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inglehearn CF. Molecular genetics of human retinal dystrophies. Eye. 1998;12(Pt 3b):571–579. doi: 10.1038/eye.1998.147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.