

Abstract
As ever more protease sequences are uncovered through genome sequencing projects, efficient parallel methods to discover the potential substrates of these proteases becomes crucial. Herein we describe the first use of fluorous-based microarrays to probe peptide sequences and begin to define the scope and limitations of fluorous microarray technologies for the screening of proteases. Comparison of a series of serine proteases showed that their ability to cleave peptide substrates in solution was maintained upon immobilization of these substrates onto fluorous-coated glass slides. The fluorous surface did not serve to significantly inactivate the enzymes. However, addition of hydrophilic components to the peptide sequences could induce lower rates of substrate cleavage with enzymes such as chymotrypsin with affinities to hydrophobic moieties. This work represents the first step to creating robust protease screening platforms using noncovalent microarray interface that can easily incorporate a range of compounds on the same slide.
Keywords: fluorous-linked peptides, fluorous microarrays, proteases
1. Introduction
Proteases are found in all life forms and are involved in a multitude of physiological processes from blood-clotting to apoptosis and inflammation [1]. These enzymes catalyze the specific hydrolytic breakdown of proteins into peptides or amino acids. This directed degradation is a tool for instance in the post-translational modification of proteins, viral protein separation, and food digestion. Given the crucial role of these hydrolytic enzymes in a range of biological operations, the discovery of new proteases and the design of protease inhibitors, such as the HIV-protease inhibitors, are important for unraveling biological pathways and validating new therapeutic targets [2]. As ever more protease sequences are uncovered through genome sequencing projects, efficient parallel methods to discover the potential substrates of these proteases becomes essential [3].
One promising method to screen a range of peptide sequences for their ability to serve as substrates for a given protease is peptide microarrays [4]. Microarrays have the advantage of using little material in comparison to microtiter well-based traditional assays and allow the parallel screening of multiple substrates using minimal enzyme quantities. Peptide microarray formation has been achieved through various methods including on surface synthesis of the peptide probes [5] or through covalent binding of peptides with modified surfaces [6]. The Ellman group [7a] and the Yao group [7b] independently reported microarray–based protease assays using 7-amino-4-carbamoylmethyl coumarin (ACC)-linked peptides. As shown in Fig. 1, protease cleavage of a substrate at the 7 position of the ACC-linked peptide results in the development of an enhanced fluorescent signal at that particular slide location.
Fig. 1.

Fluorogenic 7-amino-4-carbamoylmethyl coumarin (ACC).
Ellman and coworkers initially immobilized their substrates through an oxime bond directly onto aldehyde-functionalized slides (Fig. 2); however, they observed inefficient hydrolysis of the labeled peptides. To solve this problem, they immobilized the substrates on BSA-coated slides [8], and obtained enhanced rates of the hydrolysis that match rates observed in the corresponding solution-phase reactions. Yao and coworkers employed aminoacyl-ACC-glycine (Fig. 2) immobilized onto an aminopropyl-modified slide through an amide bond. They observed the desired hydrolysis of the substrate with lysine (R = (CH2)4NH2) by the protease trypsin; however, with the substrate derived from aspartic acid (R = CH2CO2H), the expected hydrolysis with Caspases was not observed.
Fig. 2.

Reported ACC substrates used in microarray assays.
Their experiments clearly suggest that the slide surface and attachment strategy play important roles to develop effective microarray-based protease assays. However, the process requires optimization of the slide coupling conditions and slide washing prior to the enzymatic assays to make slide preparation tedious. More recently, the Ellman’s group reported a noncovalent array strategy that uses glycerol nanodroplets of ACC-modified substrates probed with aerosolized protease solutions followed by monitoring of the fluorescence intensity [4c]. The approach circumvents the issues related to slide coupling conditions, but requires specialized aerosolization equipment and is limited to enzymes that remain stable and active under aerosolization conditions and glycerol.
The recent introduction of fluorous-based microarrays provides a surface that limits nonspecific protein interactions but still allows the facile noncovalent attachment of C8F17-modified substrates for screening [9]. Initial work described the arraying of carbohydrates using robotics standard in DNA microarray facilities and probing of the microarray with carbohydrate-binding proteins [9–12]. Remarkably, despite the noncovalent attachment strategy, immobilized carbohydrates withstood washing with buffer solutions for screening even in the presence of detergents. Subsequent work has shown the method to also be viable for the screening of small molecules for their ability to inhibit histone deacetylases [12]. In addition, the fluorous slides can be rinsed and reused multiple times, unlike traditional microarray slides [13]. Herein we describe the first use of fluorous-based microarrays to probe peptide sequences and begin to define the scope and limitations of fluorous microarray technologies for the screening of proteases.
2. Results and Discussion
The general structure of the fluorous-tagged peptide substrates for microarray-based protease assays on fluorous-coated slides is shown in Fig. 3. Although a single C8F17 group has been used successfully so far in all the fluorous microarray studies as the tag for immobilization of substrates [9–13], we employed a double C6F13 tag in this project to aid in the solution-phase synthesis of the peptide substrates using fluorous solid-phase extraction (F-SPE, see below). We decided to use a fluorous tag that has a significantly higher retention on fluorous silica gel than that of a single C8F17 tag since the target peptides could potentially be polar enough to cause undesired breakthrough of the fluorous-tagged compounds during the F-SPE procedure. The ACC and the C6F13 groups were designed to connect through a hydrophilic triethylenglycol spacer. We expected that the spacer would keep the peptide-ACC moiety away from the slide surface so that enzymes can interact with the peptide similar to a comparable solution-phase environment.
Fig. 3.

General structure of fluorous substrates.
Among the six known classes of proteases, the serine proteases are of particular interest given their ubiquity, and we selected five commercially available serine proteases: thrombin, plasmin, chymotrypsin, trypsin and Granzyme B. Thrombin [14], trypsin [15], and plasmin [16] prefer basic amino acid residues such as arginine and lysine at P1 (the first amino acid residue attached to the ACC). In contrast, granzyme B prefers acidic residues such as aspartic acid at P1 [17]. Chymotrypsin prefers hydrophobic amino acids such as phenylalanine at P1 [18]. The structures of the substrates—both fluorous and non-fluorous—are shown in Table 1. All the non-fluorous substrates were obtained from commercial sources and were used as the positive control in the solution-phase assays and also to check the activity of the corresponding enzymes.
Table 1.
Structures of the substrates.
| Compound Code |
Structure | Compound Code |
Structure | Substrate for |
|---|---|---|---|---|
| RC1 | Bz-FVR-ACC-[F-tag] | RM1 | Bz-FVR-AMC | Thrombin |
| RC2 | Bz-VPR-ACC-[F-tag] | RM2 | Bz-VPR-AMC | Thrombin |
| DC1 | Ac-IEPD-ACC-[F-tag] | DM1 | Ac-IEPD-AMC | Granzyme B |
| RC3 | Cbz-FR-ACC-[F-tag] | RM3 | Cbz-FR-AMC | Trypsin |
| KC3 | Suc-AFK-ACC-[F-tag] | KM3 | Suc-AFK-AMC | Plasmin |
| FC1 | Suc-AAPF-ACC-[F-tag] | FM1 | Suc-AAPF-AMC | Chymotrypsin |
Table One Footnote Graphic 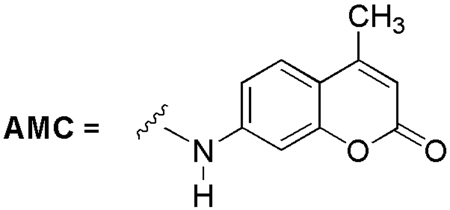
Suc = 3-Carboxy-propionyl, Bz = Benzoyl, Cbz = Benzyloxycarbonyl, F = phenylalanine, V = Valine, R = Arginine, P = Proline, I = Isoleucine, E = Glutamic acid, D = Aspartic acid, A = Alanine, K = Lysine
The synthesis of the fluorous tagged ACC is summarized in Scheme 1. Secondary alcohol 1 was prepared by the addition of a 2-(perfluorohexyl)ethyl Grignard reagent to ethyl formate in 93% yield. The alcohol was alkylated under basic conditions using a phase transfer catalyst to give tert-butyl ester 2 in 83% yield. The ester was hydrolyzed, and the carboxylic acid was activated as N-hydroxysuccinimidyl ester 4. The resulting compound was coupled to 4,7,10-trioxa-1,13-tridecanediamine to give fluorous tagged primary amine 5 in 66% yield from the acid 3. The amine was coupled with Fmoc-ACC [19] using diisopropylcarbodiimide (DIC) and hydroxybenzotriazole (HOBt) to give 6 in 92% yield. The Fmoc group was then removed by piperidine to give the desired fluorous tagged ACC 7 in 64% yield.
Scheme 1.

Synthesis of fluorous tagged ACC.
The peptide portions of the fluorous tagged substrates were constructed from 7 according to Fmoc-peptide synthesis methods [20] in solution-phase. At each step, the appropriate Fmoc-protected amino acid was activated by O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophophase (HATU) as its 7-azabenzotriazol-1-yl ester (Fmoc-AAn-OAt) in another flask, and then it was coupled to the amino group of the intermediate (Scheme 2). In a typical solid-phase Fmoc-peptide synthesis [20], after each coupling step, the remaining activated amino acid and reagents are removed prior to the removal of the Fmoc protecting group; however, in our solution-phase peptide synthesis, after the completion of the coupling reaction, the activated amino acid was first quenched with 1-octylamine, and then the Fmoc group was removed by piperidine. The reaction mixture was loaded on a FluoroFlash F-SPE cartridge to separate the desired intermediate from all other non-fluorous materials including the reagent by-product, the base, piperidine, and the quenched amino acid. The attachment of Fmoc-protected amino acid to 7, however, turned out to be inefficient even with HATU [21]. Therefore, after the first coupling reactions, the excess reagent and the activated amino acid were removed by F-SPE, and the second coupling was conducted. After the completion of the peptide synthesis part, the N-terminus of each peptide was capped with the appropriate protecting group such as a benzoyl (Bz) group in RC1 for example. In every step, the consumption of the starting material was conveniently monitored by LC-MS. The overall yields from 7 were 45 to 93%, and the purities of the products determined by HPLC were 80 to 93%.
Scheme 2.

Synthesis of peptides.
With the desired substrates in hand, we first conducted solution-phase reactions with thrombin using the same aqueous buffer that Ellman and coworkers used [7a]. A mixture of fluorous substrate RC2 and non-fluorous RM2 in DMF (3 mM each) was dissolved in the buffer solution (50 mM Tris, pH 8.0, 100 mM NaCl, 5 mM CaCl2, and 0.01% Tween-20), and thrombin was added. After 3 h at 23 °C, the reaction was analyzed by LC-MS. RM2 was completely hydrolyzed to give 7-amino-4-methylcoumarin; however, most of the RC2 remained. Increasing the concentration of the detergent (Tween-20) from 0.01% to 0.5 % seemed to increase the hydrolysis of RC2 by the same LC-MS analysis. Because the solubility of RC2 and the cleaved product 7 could be low in the aqueous buffer, the analysis of the reaction mixture by LC-MS may not represent the actual hydrolysis; however, these experiments confirmed that RC2 can be cleaved by thrombin. The LC-MS analysis also suggests that the fluorous substrate and the cleaved product can be in solution in the presence of the detergent, though it may also be possible that RC2 might have been in a micelle form [22] when its DMSO solution was diluted in the aqueous buffer. To be safe, in our microarray experiments, we decided not to include the detergent in the buffer, and repeated the solution-phase assays with all the combinations of the substrates and the proteases. The final buffer conditions are 50 mM Tris at pH 8.0, 100 mM NaCl, and 5 mM CaCl2.
The reactions were conducted in a microtiter plate at room temperature for 1h, and the fluorescence intensities were measured. The results with non-fluorous substrates and the fluorous tagged substrates are summarized in the left and the center columns in Graph 1, respectively. As shown in the graphs, the same peptide sequence, fluorous-tagged or not, qualitatively reacted with the protease set in the same manner. As mentioned earlier, thrombin [14], trypsin [15], and plasmin [16] prefer basic amino acid residues at P1. Consequently we expected to see the four sequences (RC1, RC2, RC3 and KC3) specific to these proteases all hydrolyzed, possibly at different rates, by these proteases. Indeed, thrombin, trypsin, and plasmin hydrolyzed the three sequences terminating with an arginine (RC1, RC2 and RC3); in addition, both trypsin and plasmin cut the lysine terminating sequence (KC3). In the case of chymotrypsin, which prefers hydrophobic amino acids at P1, FC1 and FM1 underwent the proteolysis reaction at a significantly higher rate than others. Granzyme B showed the hydrolysis of its specific sequence but somehow gave very low fluorescence intensities.
Graph 1.

Fluorescence intensities after each proteolysis reaction.
In all cases, the fluorescence intensities from the reactions with fluorous substrates were significantly lower than those obtained with non-fluorous substrates. We speculate that the cleaved ACC with the fluorous tag (7) have very little solubility in the solvent system without a detergent, and some of 7 might have adsorbed on the wall of the wells. It may also be possible that the fluorous tagged substrates have limited solubility in the solvent system, and that might have drastically slowed down the desired hydrolysis. Although inhibition of the catalytic activity by the fluorous tag (for example, competing binding to the catalytic site or allosteric inhibition) cannot be ruled out based on these results, the fluorous tag did not have any obvious deleterious effect on the selectivity at least in the solution-phase experiments, and we decided to move on to develop microarray assay procedure.
For the printing of the substrates, Ellman and coworkers used 200–500 µM of the peptide solution (Fig. 2), and the unreacted formyl group (the active site on the glass surface to covalently immobilize the substrates through oxime bond) had to be capped with methylhydroxyamine [7a]. In contrast, we simply spot solutions of the substrates in DMSO onto fluorous-coated glass slides, and, after drying, the slides were ready to use without any capping reaction. Also, because there is no chemical bond formation for immobilization of the substrates, no optimization of immobilization conditions was necessary. In addition, minimum amount of the substrates was needed for printing, and we could lower the concentration of the substrates to 15 µM in DMSO.
In our procedure, the six fluorous-tagged peptides in DMSO were individually printed (12 copies) on fluorous-coated glass slides using a DNA robotic arrayer. By immobilization of all substrates on one fluorous-coated slide, we expected that the positive substrate(s) for chymotrypsin could also serve as negative controls for the other proteases, and vise versa. Each protease (0.5 µM) in 50 mM TRIS buffer was then applied to the printed slide, and the array was incubated for 1 hour. After gentle aqueous washes (3 times), the slide was scanned at 350 nm (The common wavelength used to detect the aminocoumarin label). The fluorescence intensities were processed using ImaGene software, and are presented in the right column in Graph 1.
These microarray experiments showed that the thrombin, plasmin, and trypsin reacted with their specific sequences as would be expected despite the noncovalent immobilization of the substrates on a fluorous surface. Being consistent with the solution-phase results, thrombin degraded principally RC1, RC2 and RC3, as did trypsin and plasmin, in addition to the KC3 sequence. These results are also consistent with the previously reported microarray results with thrombin and plasmin [7]. In case of granzyme B, very low fluorescence increases were observed with all the substrates as we had anticipated from the solution-phase results. Chymotrypsin, on the other hand, hydrolyzed almost all the spotted peptides. Peptide substrates with arginine at P1 are known to be poorly hydrolyzed by chymotrypsin in general [18b, 18c]; therefore, it is surprising to see significant fluorescence increases from RC1, RC2, and RC3. We speculate that chymotrypsin’s interaction with the hydrophobic fluorous surface might have caused its conformational distortion, and it might have been able to adapt RC1, RC2, and RC3 into the catalytic site, though further experiments are needed to understand this interesting selectivity of chymotrypsin on the peptide substrates on fluorous surface.
3. Conclusion
This initial report on the utility of noncovalent fluorous-based microarrays for probing protease substrates shows the technique to be clearly viable for peptide immobilization and screening. The fluorescence intensities obtained by peptide cleavage in aqueous solutions and in microarray experiments are qualitatively consistent except chymotrypsin, though the fluorous surface did not serve to significantly inactivate the enzymes even in the case of chymotrypsin with its known affinity to hydrophobic moieties. In addition, the noncovalent attachment strategy allowed the detection of the hydrolysis with the spotting of peptides at almost 10–30 fold lower concentrations than the previously reported covalent peptide microarray strategy [7a]. However, an understanding of the choice of the spacers and tags for substrate immobilization on microarray surfaces is crucial for proper interpretation of data from surface-mediated enzyme reactions. For a protease that relies on hydrophobic interactions for substrate recognition, the fluorous tag and surface could serve as enough of a nonspecific recognition unit to induce cleavage of downstream amino acid sequences at low but observable rates. Interestingly, though, such information would still be valuable in the screening of proteases of unknown substrate specificity as long as data analysis protocols took into account compound information beyond just the amino acid sequences. A notable advantage to the noncovalent attachment strategy is that the exact compound can be tested in solution to deconvolute the effects of the spacer versus the slide surface on the activity. Clearly, incubation times of end-point type assays also matter for comparisons among substrates. This work represents the first step to creating robust protease screening platforms using noncovalent microarray platforms that can easily incorporate, without necessarily separate optimization of reaction conditions for each compound class, a range of compounds on the same slide.
4. Experimental
4.1. Microtiter Plate-Based Protease Screening
4.1.1. 96-Well plate preparation
200 µL of fluorous-tagged peptide solution (12.5 µM) in 50 mM TRIS buffer (pH 8.0, 100 mM NaCl, 5 mM CaCl2) was dispensed in each well of an opaque 96-well plate.
4.1.2. Detection of protease reaction
50 µL of protease (thrombin, plasmin, chymotrypsin, trypsin, or granzyme B, 0.5 µM) in 50 mM TRIS buffer (pH 8.0, 100 mM NaCl, 5 mM CaCl2) was added to the fluorous-tagged peptide solution and incubated at ambient temperature for 1 h. The 96-well plate was scanned using a Varian Cary Eclipse Fluorescence Spectrophotometer with the microplate reader device set at 390/460 excitation/emission wavelength to detect the aminocoumarine label. The fluorescence intensities were determined using Cary Eclipse Software and the graphs were prepared with Prism4.
4.2. Fluorous-Based Peptide Microarray Preparation and Screening
4.2.1. Formation of fluorous-based peptide microarrays
Fluorous-tagged peptide compounds (15 µM) were dissolved in 100% DMSO. Each peptide was spotted (12 × 1) on the fluorinated glass slide (Fluorous Technologies, Inc.) using a robotic spotter with pin lifting technology (Cartesian PixSys 5500 Arrayer, Cartesian Technologies, Inc., Irvine, CA) at 60% relative humidity. The spotting pin stays in the peptide-containing solutions for 1 sec before spotting and stays on the slide for 25 µ sec per spot. Compounds are spotted 400 µm apart. The glass slide was dried in a humidified chamber for 0.5 h.
4.2.2. Detection of protease reaction
200 µL of protease (thrombin, plasmin, chymotrypsin, trypsin, or granzyme B, 0.5 µM) in 50 mM TRIS buffer (pH 8.0, 100 mM NaCl, 5 mM CaCl2) was applied to the printed glass slide. The array was incubated using a PC500 CoverWell incubation chamber (Grace Biolabs, Bend, OR) for 1 h. The slides were washed with deionized water 3 times and then dried in the absence of light. The glass slide, stored in a dark chamber, was scanned using Applied Precision’s ArrayWoRx Biochip Reader set at 350 nm, the common wavelength used to detect the aminocoumarine label. The spot intensities were determined using ImaGene software and the graphs were prepared with Prism4.
Supplementary Material
Acknowledgements
This material is based in part upon work supported by the National Institutes of Health, General Medical Sciences (1R41M075436-01 and 2R42GM075436-02).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.a) Campbell RD, Law SK, Reid KB, Sim RB. Annu. Rev. Immunol. 1988;6:61–195. doi: 10.1146/annurev.iy.06.040188.001113. [DOI] [PubMed] [Google Scholar]; b) Muller-Eberhard HJ. Annu. Rev. Biochem. 1988;57:321–347. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]; c) Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, Carroll MC, Verbeek JS, Botto M, Walport MJ, Molina H, Kalinke U, Acha-Orbea H, Aguzzi A. Nat. Med. 2001;7:488–492. doi: 10.1038/86567. [DOI] [PubMed] [Google Scholar]; d) Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Holgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]; e) Furie B, Furie BC. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]; f) Wang J, Doer S, Cereb J. Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 2.a) Lecaille F, Kaleta J, Brömme D. Chem. Rev. 2002;102:4459–4488. doi: 10.1021/cr0101656. [DOI] [PubMed] [Google Scholar]; b) Brömme D, Kaleta J. Curr. Pharm. Des. 2002;8:1639–1658. doi: 10.2174/1381612023394179. [DOI] [PubMed] [Google Scholar]; c) Sajid M, McKerrow JH. Mol. Biochem. Parasitol. 2002;120:1–21. doi: 10.1016/s0166-6851(01)00438-8. [DOI] [PubMed] [Google Scholar]; d) Rosenthal PJ. Int. J. Parasitol. 2004;34:1489–1499. doi: 10.1016/j.ijpara.2004.10.003. [DOI] [PubMed] [Google Scholar]; e) Turk B, Turk D, Turk V. Biochim. Biophys. Acta. 2000;1477:98–111. doi: 10.1016/s0167-4838(99)00263-0. [DOI] [PubMed] [Google Scholar]; f) Nakagawa T, Roth W, Wong P, Nelson A, Farr A, Deussing J, Villadangos JA, Ploegh H, Peters C, Rudensky AY. Science. 1998;280:450–453. doi: 10.1126/science.280.5362.450. [DOI] [PubMed] [Google Scholar]; g) Castoldi E, Rosing J. Curr. Opin. Hematol. 2004;11:176–181. doi: 10.1097/01.moh.0000130315.41033.32. [DOI] [PubMed] [Google Scholar]; h) Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, et al. Proc. Natl. Acad. Sci. U. S. A. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Rawlings ND, Tolle DP, Barrett AJ. Nucleic Acids Res. 2004;32:D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Leung D, Abbenante G, Fairlie DP. J. Med. Chem. 2000;43:305–341. doi: 10.1021/jm990412m. [DOI] [PubMed] [Google Scholar]; k) Barret AJ, Rawlings ND, Woessner JF. Handbook of Proteolytic Enzymes. London: Academic Press; 1998. [Google Scholar]; l) Overall CM, Blobel CP. Nature Rev. 2007;8:245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]; m) Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Nat. Rev. Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 3.a) Leytus SP, Melhado LL, Mangel WF. Biochem. J. 1983;209:299–307. doi: 10.1042/bj2090299. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cai SX, Zhang HZ, Guastella J, Drewe J, Yang W, Weber E. Bioorg. Med. Chem. Lett. 2001;11:39–42. doi: 10.1016/s0960-894x(00)00590-4. [DOI] [PubMed] [Google Scholar]; c) Jeffery DA, Bogyo M. Curr. Opin. Biotechnol. 2003;14:87–95. doi: 10.1016/s0958-1669(02)00010-1. [DOI] [PubMed] [Google Scholar]; d) Jessani N, Cravatt BF. Curr. Opin. Chem. Biol. 2004;8:54–59. doi: 10.1016/j.cbpa.2003.11.004. [DOI] [PubMed] [Google Scholar]; e) Harris JL, Mason DE, Li J, Burdick MD, Backes BJ, Chen TC, Shipway A, van Heeke G, Gough L, Ghaemmagfurtherhami A, et al. Chem. Biol. 2004;11:1361–1372. doi: 10.1016/j.chembiol.2004.08.008. [DOI] [PubMed] [Google Scholar]; f) Speers AE, Cravatt, BF BF. Chembiochem. 2004;5:41–47. doi: 10.1002/cbic.200300721. [DOI] [PubMed] [Google Scholar]; g) Winnsinger N, Ficarro S, Schultz PG, Harris JL. Proc. Nat. Acad. Sci. 2002;99:11139–11144. doi: 10.1073/pnas.172286899. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Sun H, Panicker RC, Yao SQ. Biopolymers. 2006;88(2):141–149. doi: 10.1002/bip.20664. [DOI] [PubMed] [Google Scholar]
- 4.a) Hang HC, Ploegh H. Chem. & Biol. 2004;11:1328–1330. doi: 10.1016/j.chembiol.2004.09.005. [DOI] [PubMed] [Google Scholar]; b) Winssinger N, Damoiseaux R, Tully DC, Geierstanger BH, Kurdick KW, Harris JL. Chem. Biol. 2004;11:1351–1360. doi: 10.1016/j.chembiol.2004.07.015. [DOI] [PubMed] [Google Scholar]; c) Gosalia DN, Salisbury CM, Maly DJ, Ellman JA, Diamond SL. Proteomics. 2005;5:1292–1298. doi: 10.1002/pmic.200401011. [DOI] [PubMed] [Google Scholar]; d) Schutkowski M, Reineke U, Reimer U. ChemBioChem. 2005;6:513–521. doi: 10.1002/cbic.200400314. [DOI] [PubMed] [Google Scholar]
- 5.a) Fodor SPA, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. Science. 1991;251:767–773. doi: 10.1126/science.1990438. [DOI] [PubMed] [Google Scholar]; b) Frank R. Tetrahedron. 1992;48:9217–9232. [Google Scholar]
- 6.a) Falsey JR, Renil R, Park S, Li S, Lam KS. Bioconj Chem. 2001;12:346–353. doi: 10.1021/bc000141q. [DOI] [PubMed] [Google Scholar]; b) Houseman BT, Huh JH, Kron SJ, Mrksich M. Nat. Biotechnol. 2002;20:270–274. doi: 10.1038/nbt0302-270. [DOI] [PubMed] [Google Scholar]; c) Houseman BT, Gawalt ES, Mrksich M. Langmuir. 2003;19:1522–1531. [Google Scholar]; d) Lesaicherre ML, Uttamchandani M, Chen GYJ, Yao SQ. Bioorg. Med. Chem. Lett. 2002;12:2079–2083. doi: 10.1016/s0960-894x(02)00379-7. [DOI] [PubMed] [Google Scholar]
- 7.a) Salisbury CM, Maly DJ, Ellman JA. J. Am. Chem. Soc. 2002;124:14868–14870. doi: 10.1021/ja027477q. [DOI] [PubMed] [Google Scholar]; b) Zhu Q, Uttamchandani M, Li D, Lesaicherre ML, Yao SQ. Org. Lett. 2003;5:1257–1260. doi: 10.1021/ol034233h. [DOI] [PubMed] [Google Scholar]
- 8.MacBeath B, Schreiber SL. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 9.Jaipuri FA, Collet BYM, Pohl NL. Angew. Chem. Int. Ed. 2008;47:1707–1710. doi: 10.1002/anie.200704262. [DOI] [PubMed] [Google Scholar]
- 10.Ko K-S, Jaipuri FA, Pohl NL. J. Am. Chem. Soc. 2005;127:13162–13163. doi: 10.1021/ja054811k. [DOI] [PubMed] [Google Scholar]
- 11.Mamidyala SK, Ko K-S, Jaipuri FA, Park G, Pohl NL. J. Fluorine Chem. 2006;127:571–579. [Google Scholar]
- 12.Vegas AJ, Bradner JE, Tang W, McPherson OM, Greenberg EF, Koehler AN, Schreiber SL. Angew. Chem. Int. Ed. 2007;46(42):7960–7964. doi: 10.1002/anie.200703198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholson RL, Ladlow ML, Spring DR. Chem. Commun. 2007:3906–3908. doi: 10.1039/b712906h. [DOI] [PubMed] [Google Scholar]
- 14.Chang J-Y. Eur. J. Biochem. 1985;151:217–224. doi: 10.1111/j.1432-1033.1985.tb09091.x. [DOI] [PubMed] [Google Scholar]
- 15.Baines NJ, Baird JB, Elmore DT. Biochem. J. 90;1964:470–476. doi: 10.1042/bj0900470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierzchala PA, Dorn CP, Zimmerman M. Biochem. J. 1979;183:555–559. doi: 10.1042/bj1830555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Harris JL, Peterson EP, Hudig D, Thornberry NA, Craik CS. J. Biol. Chem. 1998;273:27364–27373. doi: 10.1074/jbc.273.42.27364. [DOI] [PubMed] [Google Scholar]; b) Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 18.a) Zimmerman M, Yurewicz E, Patel G. Anal. Biochem. 1976;70:258–262. doi: 10.1016/s0003-2697(76)80066-8. [DOI] [PubMed] [Google Scholar]; b) Schellenberger V, Braune K, Hofmann H-J, Jakubke H-D. Eur. J. Biochem. 1991;199:623–636. doi: 10.1111/j.1432-1033.1991.tb16163.x. [DOI] [PubMed] [Google Scholar]; c) Ascenzi P, Menegatti E, Guarneri M, Bortolotti F, Antonini E. Biochemistry. 1982;21:2483–2490. doi: 10.1021/bi00539a030. [DOI] [PubMed] [Google Scholar]
- 19.Maly DJ, Leonetti F, Backes BJ, Dauber DS, Harris JL, Craik CS, Ellman JA. J. Org. Chem. 2002;67:910–915. doi: 10.1021/jo016140o. [DOI] [PubMed] [Google Scholar]
- 20.Chan WC, White PD. Basic Procedures. In: Chan WC, White PD, editors. Fmoc solid phase peptide synthesis. New York: Oxford University Press; 2000. pp. 41–76. [Google Scholar]
- 20.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7754–7759. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riess JG. Tetrahedron. 2002;58:4113–4131. and references therein. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.