

Abstract
The CaaX tetrapeptide motif typically directs three sequential posttranslational modifications, namely, isoprenylation, proteolysis, and carboxyl methylation. In all eukaryotic systems evaluated to date, two CaaX proteases (Rce1 and Ste24/Afc1) have been identified. Although the Trypanosoma brucei genome also encodes two putative CaaX proteases, the lack of detectable T. brucei Ste24 activity in trypanosome cell extracts has suggested that CaaX proteolytic activity within this organism is solely attributed to T. brucei Rce1 (J. R. Gillespie et al., Mol. Biochem. Parasitol. 153:115-124. 2007). In this study, we demonstrate that both T. brucei Rce1 and T. brucei Ste24 are enzymatically active when heterologously expressed in yeast. Using a-factor and GTPase reporters, we demonstrate that T. brucei Rce1 and T. brucei Ste24 possess partially overlapping specificities much like, but not identical to, their fungal and human counterparts. Of interest, a CaaX motif found on a trypanosomal Hsp40 protein was not cleaved by either T. brucei CaaX protease when examined in the context of the yeast a-factor reporter but was cleaved by both in the context of the Hsp40 protein itself when evaluated using an in vitro radiolabeling assay. We further demonstrate that T. brucei Rce1 is sensitive to small molecules previously identified as inhibitors of the yeast and human CaaX proteases and that a subset of these compounds disrupt T. brucei Rce1-dependent localization of our GTPase reporter in yeast. Together, our results suggest the conserved presence of two CaaX proteases in trypanosomatids, identify an Hsp40 protein as a substrate of both T. brucei CaaX proteases, support the potential use of small molecule CaaX protease inhibitors as tools for cell biological studies on the trafficking of CaaX proteins, and provide evidence that protein context influences T. brucei CaaX protease specificity.
Certain isoprenylated proteins are synthesized as precursors having a highly degenerate C-terminal tetrapeptide CaaX motif (C, cysteine; a, aliphatic amino acid; X, one of several amino acids). This motif typically directs three posttranslational modifications that include covalent attachment of an isoprenoid lipid to the cysteine residue, followed by endoproteolytic removal of the terminal three residues (i.e., aaX), and lastly, carboxyl methyl esterification of the farnesylated cysteine (49, 50). Relevant examples of proteins subject to the above modifications, also referred to as CaaX proteins, include the Ras and Ras-related GTPases, Gγ subunits, prelamin A, members of the Hsp40 family of chaperones, and fungal mating pheromones.
Isoprenylation of CaaX proteins is performed by either the farnesyltransferase (FTase) or the geranylgeranyl transferase I (GGTase I). The particular isoprenoid attached, C15 farnesyl or C20 geranylgeranyl, respectively, depends in part on the sequence of the CaaX motif (8, 26, 31). Proteolysis of isoprenylated intermediates is carried out by the otherwise unrelated Rce1p (Ras converting enzyme 1) and Ste24p (sterile mutant 24) enzymes, collectively referred to as CaaX proteases, which are integral membrane proteins residing within the endoplasmic reticulum (3, 40, 45). Studies to elucidate the specificities of the CaaX proteases have often involved reporters designed from biological substrates (e.g., Ras GTPases) (2, 3, 16, 21, 22, 24, 34). Although these studies suggest that isoprenylated CaaX tetrapeptides alone are sufficient for recognition as a substrate, insufficient evidence exists to assert whether this sequence contains all of the necessary information for substrate specificity. Reporters are typically cleaved by either Rce1p or Ste24p. The Saccharomyces cerevisiae a-factor mating pheromone is a rather unusual biological reporter since it is cleaved by both yeast CaaX proteases. Orthologs of the CaaX proteases from humans, worms, and plants can also cleave a-factor when heterologously expressed in yeast, thereby making a-factor a convenient reporter for comparative analyses of CaaX protease activities (3, 5, 6, 36). Where evaluated using the a-factor reporter, Rce1p and Ste24p display partially overlapping target specificity, and this is an expected property of CaaX proteases in all eukaryotic systems (5, 6, 36, 47). Unlike the isoprenylation and proteolysis steps, carboxyl methyl esterification exclusively relies on a single enzyme, the isoprenylcysteine carboxyl methyltransferase (ICMT) (23, 50). A farnesylated cysteine appears to be the sole recognition determinant of the endoplasmic reticulum-localized ICMT (10, 23, 38).
Disruption of the posttranslational modifications associated with CaaX proteins is often perceived as an anticancer strategy because of the prominent role of CaaX proteins in cellular transformation (i.e., the Ras GTPases) (49). To date, the most advanced drug discovery efforts have focused on farnesyltransferase inhibitors (FTIs) (9, 53). Inhibitors of the CaaX proteases and ICMT are also being developed (1, 11, 28, 37, 39, 48). Disrupting CaaX protein modifications has therapeutic application to other diseases as well. The relief of prelamin A toxicity by FTIs is a well-documented example (51). Accumulation of the farnesylated but unproteolysed precursor of lamin A results in a progeroid phenotype in individuals lacking ZmpSte24 proteolytic activity. The treatment of parasitic disease is another area under investigation (13). A number of FTIs have been developed that inhibit protozoan FTases, and in vivo testing is a continued effort (15, 32). Although research is less advanced with respect to CaaX protease and ICMT inhibitors, RNA interference experiments on the bloodstream form of Trypanosoma brucei indicate that CaaX processing enzymes are required for viability and proliferation of the parasite (20).
In the present study, we evaluated the enzymatic properties of the trypanosomal CaaX proteases. We establish through the use of a variety of in vivo and in vitro assays that T. brucei Rce1 and T. brucei Ste24 are active when heterologously expressed in S. cerevisiae and have partially overlapping substrate specificities. The assays rely on various reporters, specifically the yeast a-factor mating pheromone, a K-Ras4B-based fluorogenic peptide, a green fluorescent protein (GFP)-GTPase fusion, and a T. brucei Hsp40 protein. All but the GTPase reporter could be effectively cleaved by both T. brucei CaaX proteases. We also demonstrate that the trypanosomal CaaX proteases can be targeted for inhibition by small molecules both in vitro and when heterologously expressed in yeast, suggesting that the trypanosomal CaaX proteases may be attractive drug targets for pharmacological inhibition.
MATERIALS AND METHODS
Yeast strains and media.
The yeast strains used in the present study are listed in Table 1. To assess in vivo T. brucei CaaX protease function, strains EG123 (ATCC 204278; MATa trp1 leu2 ura3 his4 can1), IH1793 (ATCC 204279; MATα lys1), and yWS164 (MATa trp1 leu2 ura3 his4 can1 mfa1-Δ1 mfa2-Δ1 rce1::TRP1 ste24::Kanr) were transformed with the appropriate plasmids (36). For isolation of membranes enriched for T. brucei CaaX proteases, the strain used was SM3614 (MATa trp1 leu2 ura3 his4 can1 rce1::TRP1 ste24::LEU2) transformed with the appropriate plasmid (45). This strain was also used for purification of heterologously expressed GST-Tbj1. To assess T. brucei Hsp40 function in vivo, strains BY4741 (MATa his3 leu2 met15 ura3) and a BY4741 derivative (MATa his3 leu2 met15 ura3 ydj1Δ) were obtained from the yeast haploid knockout collection (Research Genetics, Inc.) (19).
TABLE 1.
Yeast strains used in this study
| Strain | Genotypea | Reference |
|---|---|---|
| BY4741 | MATahis3 leu2 met15 ura3 | 19 |
| EG123 | MATatrp1 leu2 ura3 his4 can1 | 43 |
| IH1793 | MATα lys1 | 30 |
| SM3614 | MATatrp1 leu2 ura3 his4 can1 rce1::TRP1 ste24::LEU2 | 45 |
| yWS164 | MATatrp1 leu2 ura3 his4 can1 mfa1-Δ1 mfa2-Δ1 rce1::TRP1 ste24::Kanr | 6 |
| yWS304 | MATahis3 leu2 met15 ura3 ydj1Δ | 19 |
All strains are isogenic except BY4741 and yWS304, which are a separate isogenic pair.
Plasmid-transformed strains were created according to published methods (14). Unless otherwise noted, strains were routinely cultured at 30°C using yeast extract-peptone-dextrose (YEPD) or synthetic complete dropout (SC−) medium as previously described (25). Where temperature sensitivity was evaluated, yeast were simultaneously grown at 25 and 35.5°C for 3 and 2 days, respectively, on YEPD solid medium. Experiments involving galactose-induced GFP-Ras2p and GST-Tbj1 expression used synthetic media lacking glucose and uracil but containing 2% galactose, 1% glycerol, and 1% ethanol (SGal−ura).
Plasmids.
The plasmids used in the present study are listed in Table 2. Yeast expression plasmids encoding T. brucei Rce1 (pWS766) and T. brucei Ste24 (pWS767) were created by PCR-directed recombination-mediated plasmid construction (33). In brief, the open reading frames (ORFs) of T. brucei Rce1 and T. brucei Ste24 (accession nos. XP_843748 and XP_827211, respectively) were used to replace the ORFs of yeast RCE1 and STE24 encoded in pWS479 and pWS154, respectively (36, 45). The trypanosomal ORFs were amplified by PCR from T. brucei brucei genomic DNA (TREU 927). The appropriate PCR product was cotransformed into yeast with pWS479 or pWS154 that had been linearized with SphI and BglII/NotI, respectively, within the CaaX protease ORF. To facilitate recombination between PCR products and linearized plasmids, the PCR products were engineered to contain 39-bp extensions homologous to sequences immediately 5′ and 3′ of the yeast ORFs encoded in pWS479 and pWS154. The transformed yeast were plated on selective medium (SC−ura) to allow for growth of cells that had formed a circularized plasmid; a linearized plasmid is inefficiently propagated and does not support colony growth on selective media. Independent plasmids were isolated from yeast colonies, reamplified in E. coli, and subjected to restriction digest analysis and sequencing to verify the presence and sequence of the trypanosomal ORF.
TABLE 2.
Plasmids used in this study
| Plasmid | Genotype | Source or reference |
|---|---|---|
| pRS315 | CEN LEU2 | 42 |
| pRS316 | CEN URA3 | 42 |
| pSM1107 | CEN URA3 HA-STE24 | 18 |
| pSM1282 | 2μ URA3 PPGK10HIS-HA-STE24 | 46 |
| pSM1314 | CEN URA3 RCE1-HA | 40 |
| pSM1317 | 2μ URA3 STE14 | 38 |
| pSM1585 | 2μ URA3 HsHA-STE24 | 36 |
| pSM1605 | 2μ URA3 MFA1 | 41 |
| pWS28 | 2μ URA PPGK | 52 |
| pWS335 | 2μ URA3 PPGKHIS-HA-HsRce1Δ22 | 36 |
| pWS338 | 2μ URA3 leu2-d PCUP1GST-YDJ1 | 29 |
| pWS438 | 2μ LEU2 MFA1 | 6 |
| pWS439 | 2μ LEU2 MFA1(CAMQ) | This study |
| pWS440 | 2μ LEU2 MFA1(CASQ) | 6 |
| pWS441 | 2μ LEU2 MFA1(CTLM) | 6 |
| pWS442 | 2μ LEU2 MFA1(CTVM) | This study |
| pWS479 | 2μ URA3 PPGKRCE1-HAc | 36 |
| pWS523 | CEN URA3 PGALGST-Ydj1 | 19 |
| pWS750 | CEN LEU2 PGALGFP-RAS2 | 28 |
| pWS766 | 2μ URA3 PPGKTbRce1-HA | This study |
| pWS767 | 2μ URA3 PPGKHis-HA-TbSte24 | This study |
| pWS773 | 2μ LEU2 MFA1(CTML) | This study |
| pWS774 | 2μ LEU2 MFA1(CVIM) | This study |
| pWS775 | 2μ LEU2 MFA1(CTQQ) | This study |
| pWS776 | 2μ LEU2 MFA1(CVHQ) | This study |
| pWS782 | 2μ URA3 leu2-d PCUP1GST-Tbj1 | This study |
| pWS783 | 2μ URA3 leu2-d PCUP1GST-Tbj1(C401S) | This study |
| pWS798 | 2μ URA3 PPGK TbRce1(E151A)-HA | This study |
| pWS838 | 2μ URA3 PPGK TbRce1(H184A)-HA | This study |
| pWS800 | 2μ URA3 PPGK TbRce1(H254A)-HA | This study |
| pWS801 | 2μ URA3 PPGKHis-HA-TbSte24(H286A) | This study |
| pWS805 | 2μ URA3 PPGKHis-HA-TbSte24(E287A) | This study |
| pWS802 | 2μ URA3 PPGKHis-HA-TbSte24(H290A) | This study |
| pWS880 | 2μ LEU2 MFA1(CTAQ) | This study |
| pWS888 | 2μ URA3 PPGKTbj2 | This study |
| pWS889 | 2μ URA3 PPGKTbj3 | This study |
| pWS890 | 2μ URA3 PPGKTbj4 | This study |
| pWS900 | CEN URA3 PGALGST-Tbj1 | This study |
The process described above was used to create site-directed mutants of T. brucei Rce1 and T. brucei Ste24. To create the T. brucei Rce1 mutants, pWS766 was linearized with either AatII or BglII, as appropriate. Creation of T. brucei Ste24 mutants relied on SacI digestion of pWS767. To facilitate identification of plasmid candidates for sequencing, the oligonucleotides used for PCR were engineered to contain the intended mutation and a silent restriction site near the mutational site.
Yeast expression plasmids encoding a-factor CaaX variants were also created by recombination-based plasmid construction. In most instances, PstI and MluI gapped pWS654 was used as the recipient vector for PCR products amplified from pWS438 (6). The PCR products contained regions of 5′ and 3′ homology for recombination, the desired mutation, and a silent restriction site that was used to facilitate identification of the appropriate recombinant clone for confirmatory DNA sequencing. The a-factor variants containing CAMQ and CTVM motifs were constructed in two steps. First, intermediate plasmids were created by recombination of MluI-gapped pSM1605 with PCR products amplified from pSM1605 using mutagenic oligonucleotides. The resultant plasmids were then modified by standard methods to replace the existing URA3 marker with LEU2.
Constructs encoding glutathione S-transferase (GST)-Tbj1 (pWS782) and GST-Tbj1(C401S) (pWS783) behind the constitutive CUP1 promoter were created by replacement of YDJ1 with Tbj1 using recombination within a GST-YDJ1 expression construct (pWS338) recovered from a GST fusion library (29). To facilitate recombination, pWS338 was linearized with NarI and Bsu36I. The construct encoding GST-Tbj1 behind the inducible GAL promoter (pWS900) was created similarly but used a GST-YDJ1 expression vector (pWS523) obtained from a different GST fusion library (19). pWS523 was linearized with NarI and PstI. The constructs encoding other T. brucei Hsp40 proteins (Tbj2-4) behind the constitutive phosphoglycerate kinase (PGK) promoter were also created by recombination using pWS28 linearized with XmaI as the recipient vector. PCR products for the above constructs were derived from TREU 927 genomic DNA. Candidate clones identified by restriction digest were sequenced to confirm the presence of the appropriate Hsp40 gene.
Mating assay.
The serial dilution yeast mating assay was performed as previously described (36). In brief, MATa yeast cells expressing the indicated CaaX protease were cultured for 36 h at 30°C in selective media. The cultures were normalized to an A600 of 1.0 ± 0.05 by the addition of sterile media. The MATα mating partner was cultured in YEPD and normalized in parallel using YEPD. In wells of a 96-well microtiter plate, individual MATa strains were mixed with MATα yeast using 10- and 90-μl volumes, respectively, of each normalized culture. The mating suspensions were serially diluted by repeatedly transferring 10 μl of each mixture into a new well containing 90 μl of MATα cells until five dilutions were prepared. All mixtures were subsequently spotted (5-μl volumes) onto an SD plate, and the plates were incubated for 72 to 96 h before the results were recorded by using a flatbed scanner. The cell suspensions were also spotted onto SC−lysine solid media, which is selective for MATa and diploid growth, to confirm that MATa cell dilutions were appropriately prepared.
GFP-Ras2p localization assay.
An inducible GFP-Ras2p reporter (pWS750) was used to determine the ability of yeast, human, and trypanosomal Rce1 (pWS479, pWS335, and pWS766, respectively) to promote proper localization of GFP-Ras2p in yeast and to evaluate the effect of chemical agents on Ras2p localization (28). Where yeast and trypanosomal Ste24 were evaluated, pSM1282 and pWS767 were used, respectively. In brief, mid-log yeast cells were harvested, washed twice with sterile H2O, and incubated in SGal−ura for 6 to 7 h at 30°C to induce expression of GFP-Ras2p. Where applicable, cells were incubated with compounds that were sonicated prior to use with a Fisher Scientific model 100 sonic dismembrator (10 min, maximum setting) at predetermined concentrations (0.6 to 55.6 μM) for 1 h at 30°C. In general, the compounds were used at doses where minimal cellular toxicity was observed (i.e., EC10; 90% cell survival). The doses were based on previously reported dose-response toxicity profiles (S. P. Manandhar, E. Hildebrandt, W. H. Jacobsen, G. Santangelo, and W. K. Schmidt, unpublished data). Evaluation of compound 3 required the addition of a nontoxic amount of sodium dodecyl sulfate (0.003%) during the incubation period to maximize effectiveness. The induced cells were mounted on a microscope slide, and the expression pattern of the GFP fusion was visualized by using a Zeiss Axioskop 2 Mot Plus microscope equipped with fluorescence optics and a ×100 Plan Apochromat objective lens (numerical aperture 1.4). Images were captured by using an ORCA-AG digital camera (Hamamatsu, Japan) and IPLab Spectrum software. At least five cell fields were taken from which representative images were selected. Using the above protocol, ca. 80% of the cells examined demonstrated GFP fluorescence.
To quantify the degree of GFP-Ras2p plasma membrane localization conferred by CaaX proteases, a confocal laser scanning microscopy protocol was implemented. In brief, yeast were cultured and induced to express GFP-Ras2p as described above, and images were collected by using a Zeiss LSM510 META microscope with a ×100 alpha Plan Fluar 1.45 NA oil objective lens at a resolution of 0.04 μm per pixel. GFP excitation was performed with a 488-nm laser, and the light was emitted was captured through a 505- to 530-nm BP filter. The relative association of GFP-Ras2p with the plasma membrane was determined by using Zeiss LSM imaging software from a minimum of five pictures and is reported relative to the total fluorescence associated with the image. Delocalized fluorescence was defined as fluorescence at ≥0.75 μm from the peak boundaries. The error of the associated bar graphs was calculated as the standard error of the mean for each value.
In vitro CaaX proteolysis fluorescence assay.
Cleavage of a synthetic quenched fluorogenic farnesylated peptide was used to monitor the in vitro activity of the yeast, human, and trypanosomal CaaX proteases (21, 37). In brief, a yeast strain devoid of endogenous CaaX protease activity (yWS164) was transformed with a heterologous expression vector encoding a particular CaaX protease (as defined in the figure or table legends). The resulting transformants were lysed and subjected to a differential centrifugation protocol optimized for the recovery of CaaX protease-enriched membranes as described below. The membranes (0.5 mg/ml) were incubated with a fluorogenic substrate (20 μM), and the relative fluorescence was monitored over a 90-min time course by using a BioTek Synergy HT microtiter plate fluorometer equipped with a 320/420-nm excitation/emission filter set. The substrates used were all internally quenched fluorogenic peptides based on K-Ras4B (21).
Inhibitory compounds were evaluated by using two methods. To determine values for percent inhibition of T. brucei Rce1 and T. brucei Ste24 using fixed concentrations of compounds, membranes prepared as described below were pretreated with compounds at 200 μM (FKBK [Z-Phe-Lys-2,4,6-trimethylbenzoyloxymethyl ketone], TPCK [tolylsulfonyl phenylalanyl chloromethyl ketone], EDTA, and EGTA) or 100 μM (all others) for 10 min at 30°C prior to substrate addition and activity analysis. Compounds were typically prepared as 10 mM stocks in water (EDTA and EGTA) or dimethyl sulfoxide (DMSO) (all others). Collected data were graphed (as the change in fluorescence versus time), and initial velocities were determined by using Microsoft Excel. These values were used to calculate percent activities relative to an untreated DMSO control. To determine 50% inhibitory concentration (IC50) values, the method described above was modified so that each condition contained 2.8 mg of bovine serum albumin/ml, and compound stocks were prepared at 100 mM in DMSO. Initial velocities were determined at various inhibitor concentrations ranging from 0.46 to 471 μM (11 points minimum). Compounds 1 to 9 were provided by the NCI Developmental Therapeutics Program and are as previously identified (28).
Isolation of yeast membranes.
Yeast membranes were essentially isolated as previously described using two slightly modified protocols to lyse cells (37). In general, membranes used for determination of the IC50 values and in vitro-coupled proteolysis methylation assays were isolated directly from cells by bead beating. In brief, mid-log cells were incubated in pretreatment buffer (100 mM Tris [pH 9.4], 10 mM NaN3 10 mM dithiothreitol; 10 A600/ml) for 10 min, resuspended in lysis buffer (50 mM Tris [pH 7.5], 0.2 M sorbitol, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 2 μg of aprotinin/ml, 3 μg each of leupeptin, chymostatin, and pepstatin/ml; 500 A600/ml), and lysed by using a Biospec mini-beadbeater and silica beads (5,000 rpm; three 20-s bursts; 3-min intervals on ice). For all other in vitro assays, membranes were isolated from spheroplasts. In brief, cells were pretreated as described above, and spheroplasts were generated by treatment with Zymolyase (4 μg/A600; Cape Cod, Inc.) for 30 min at 30°C in spheroplasting buffer (50 mM potassium phosphate [pH 7.5], 1.4 M sorbitol, 10 mM NaN3; 25 A600/ml), followed by 10 min on ice. Spheroplasts were harvested (3,000 × g, 10 min, 4°C), washed in spheroplasting buffer, resuspended in cold lysis buffer, and lysed by vortexing in the presence of silica beads (four 4-min vortex bursts at 4°C; 2-min intervals on ice). Independent of the method used for cell lysis, crude lysates were clarified twice (500 × g, 10 min, 4°C), and membranes were recovered (16,000 × g, 15 min, 4°C) from the clarified lysate. The membranes were resuspended in lysis buffer, reisolated by centrifugation, resuspended in lysis buffer to the original sample volume, adjusted to 1 mg of total protein/ml by dilution with lysis buffer, and frozen as aliquots at −80°C.
In vitro Tbj1 proteolysis assay.
TbJ1 (accession no. XP_951689) was heterologously expressed as a GST fusion protein (pWS900) in SM3614 (rce1Δ ste24Δ) and used for coupled proteolysis-methylation radiolabeling assays. In brief, cells from a 4-liter culture of yeast grown to an A600 of ∼1.0 in SGal−ura were harvested (3,000 × g for 10 min) and incubated with pretreatment buffer, and the cells were lysed directly as described above except that the lysis buffer was supplemented with 20 μg each of leupeptin, pepstatin, chymostatin, aprotinin, and trypsin inhibitor/ml. The resulting lysate was clarified (16,000 × g, 15 min) and incubated with a slurry of glutathione-Sepharose 4B resin (10 min, 25°C, 0.675 ml of resin/liter of starting culture). The unbound fraction was decanted after centrifugation of the mixture in a standard 15-ml conical tube.
Resin-bound GST-Tbj1 was used directly for in vitro assays. In brief, the resin was supplemented with ∼800 μl of lysis buffer, and the mixture was apportioned into nine 300-μl aliquots. Individual aliquots were supplemented with membranes lacking CaaX protease activity (yWS164/pRS316) or enriched for a specific trypanosomal CaaX protease (yWS164/pWS766 or yWS164/pWS767) and further supplemented with membranes enriched for the yeast ICMT (yWS164/pSM1317). The final sample was 550 μl and contained 0.54 mg of control or CaaX protease-enriched membranes/ml and 0.11 mg of ICMT-enriched membranes/ml. The membranes were isolated from cells lysed directly as described above. The samples also contained 13.9 μM S-adenosylmethionine (SAM; 19.6 mCi of 14C/mmol) derived by mixing appropriate amounts of SAM (Sigma) and [14C]SAM (56 mCi/mmol; GE Healthcare). After constant mixing for 12 h at 30°C, the samples were transferred to individual spin columns (GE Healthcare), centrifuged (1 min, 4,000 × g), and washed three times with lysis buffer supplemented with 1% N-octyl-β-d-glucopyranoside, and the recovered resin was quantitatively transferred to scintillation vials containing 10 ml of Bio-Safe II scintillation counting cocktail. Decays per minute were determined for each sample by using a Fisher/GP-200 Wallac 1409 scintillation counter.
RESULTS
Both of the trypanosomal CaaX proteases are functional enzymes.
The yeast CaaX proteases promote yeast mating, ostensibly through their ability to proteolyse the isoprenylated yeast a-factor precursor (3). CaaX proteases from distinct species can do the same when heterologously expressed in yeast (4-6, 36). Since a previous study had concluded that T. brucei Ste24 may be inactive, we first challenged T. brucei Rce1 and T. brucei Ste24 to cleave the CVIA CaaX motif present on yeast a-factor (20). This analysis revealed that the trypanosomal CaaX proteases could both promote yeast mating, as judged by quantitative serial dilution mating tests (Fig. 1A). This result suggests that the lack of activity previously reported for T. brucei Ste24 may be due to an assay specific or other issue, and not reflective of the intrinsic properties of this enzyme. By our analysis, the trypanosomal CaaX proteases performed as well if not better than their respective yeast counterparts in promoting yeast mating. In part, this observation can be explained by the fact that the yeast and T. brucei CaaX proteases were expressed using different plasmid systems. The yeast CaaX proteases were encoded on low-copy plasmids behind their respective promoters while the T. brucei CaaX proteases were encoded on high-copy plasmids behind the strong constitutive phosphoglycerate kinase promoter. Despite this issue, our result nonetheless indicates an activity for T. brucei Ste24 and suggests that both T. brucei Rce1 and T. brucei Ste24, like their counterparts in other organisms, have the common ability to recognize the a-factor precursor as a substrate (3-6, 36).
FIG. 1.

Trypanosomal CaaX proteases promote yeast mating. (A) Yeast wild type (WT) or strains lacking Rce1p and Ste24p were evaluated for their ability to mate by using the serial dilution mating test. The wild-type strain (EG123) was cotransformed with the empty vectors marked with LEU2 (pRS315) and URA3 (pRS316). The CaaX protease-deficient strain (yWS164) was cotransformed with an a-factor encoding plasmid (pWS438) and a plasmid encoding the indicated CaaX protease from trypanosome or yeast. The CaaX protease encoding plasmids were pSM1107, pSM1314, pWS766, and pWS767. (B and C) Mutagenized forms of T. brucei Rce1 (B) and T. brucei Ste24 (C) were evaluated by the serial dilution mating test as described in panel A. Only the first row of data from the mating test is shown. The CaaX proteases were encoded in pWS766, pWS767, pWS798, pWS800, pWS801, pWS802, pWS805, and pWS838. (D) Trypanosomal, yeast, and human CaaX proteases were evaluated by the serial dilution mating test as in panel A to assess their specificities against a panel of CaaX motifs, some of which have previously been evaluated and others that naturally occur on trypanosomal proteins (see the text for details). Only the first row of data from the mating test is shown. The CaaX proteases were encoded in pSM1107, pSM1314, pSM1585, pWS335, pWS766, and pWS767. The a-factor variants were encoded in pWS439, pWS440, pWS441, pWS442, pWS773, pWS774, pWS775, pWS776, and pWS880.
Mutational analysis of the trypanosomal CaaX proteases.
To confirm that the mating observed in our genetic test was indeed dependent on the proteolytic activities of the T. brucei CaaX proteases and not some unrecognized activity, we next evaluated the effect of mutations predicted to inactivate the enzymes. Despite having a formally unresolved mechanism, a glutamate and two histidine residues are known to be essential for yeast Rce1p activity (12, 36). These residues are conserved in Rce1p orthologs, including T. brucei Rce1, but their functional importance in any ortholog outside of yeast had not been previously addressed (35). Mutation of T. brucei Rce1 at these conserved sites resulted in the loss of activity as measured by the yeast mating assay (Fig. 1B), further supporting a role for histidine and glutamate residues in the Rce1p mechanism. In the instance of yeast Ste24p, histidine and glutamate residues are also required (18). These amino acids are part of a putative HExxH zinc-coordination motif that is common among zinc-dependent metalloproteases. Expectedly, T. brucei Ste24 required this motif for activity (Fig. 1C). For both T. brucei CaaX proteases, the mutations had no observable effect on protein expression relative to the native proteins, as judged by immunoblots (D. Z. Mokry and W. K. Schmidt, unpublished data). Our observations as a whole thus support that both T. brucei Rce1 and T. brucei Ste24 have proteolytic activity.
Specificity of the trypanosomal CaaX proteases.
To gain additional insight into the enzymatic properties of the trypanosomal CaaX proteases, we applied our genetic approach to assess the target specificity of the enzymes by measuring their ability to recognize disparate CaaX motifs (Fig. 1D). Variants of the a-factor precursor were created that contained CaaX motifs previously documented to be Rce1-specific (e.g., CTLM and CTVM), Ste24-specific (e.g., CASQ), and nonspecific (e.g., CVIA and CAMQ) in the context of CaaX proteases from other species, namely, yeast and human (36, 47). Consistent with these prior observations, T. brucei Rce1 was observed to be specific for the CTLM and CTVM motifs, T. brucei Ste24 was specific for CASQ, and both were active against the native CVIA motif (see Fig. 1A). The CAMQ motif was recognized by both T. brucei CaaX proteases as predicted but was recognized preferentially by T. brucei Ste24.
We extended our analysis to include an evaluation of motifs associated with predicted trypanosomal CaaX proteins. Because trypanosomes do not appear to have a true Ras ortholog, the CTML and CVIM motifs found on the Ras-related proteins T. brucei RLP and T. brucei RHP, respectively, were evaluated as targets for T. brucei Rce1 activity (17). Although T. brucei Rce1 recognized both motifs, T. brucei Ste24 also recognized CVIM, indicating that the T. brucei CaaX proteases have overlapping specificity for this motif. Interestingly, CVIM is found on mammalian Ras and is considered to be an Rce1p-specific motif in mammalian systems. We also evaluated CaaX motifs found on putative trypanosomal Hsp40 proteins. This family of proteins was targeted because the CASQ motif, which is present on the farnesylated Hsp40 chaperone Ydj1p, is preferentially cleaved by various Ste24 orthologs in our genetic system. None of these motifs (CTQQ, CVHQ, and CTAQ) were cleaved by T. brucei Rce1, and only CVHQ was cleaved by T. brucei Ste24. This pattern was also observed in the context of yeast and human CaaX proteases, with the exception of the CTAQ motif, which was readily cleaved by Rce1 from these organisms. The reason for the lack of recognition of the CTQQ (found on two trypanosomal homologs of yeast Ydj1p) and CTAQ motifs by the T. brucei CaaX proteases is unknown but may relate to the protein context in which they were evaluated (see below and Discussion). When considering our results as a whole and in the context of the a-factor reporter, the trypanosomal CaaX proteases appear to have substantial but not complete conservation of substrate specificity with respect to that observed with CaaX proteases from other systems.
Trypanosomal Rce1 can cleave a yeast GTPase.
Both yeast and human Rce1p preferentially, if not exclusively, recognize GTPases of the Ras superfamily as substrates. Considering that the T. brucei CaaX proteases displayed target specificity profiles by our genetic test that were largely similar to the profiles displayed by the yeast and human CaaX proteases, we next evaluated whether T. brucei Rce1 had conserved the ability to recognize Ras GTPases as substrates. Attempts to heterologously express trypanosomal GTPases GFP-RLP and GFP-RHP in yeast did not yield a discernible membrane association for either GTPase, suggesting a potential defect with isoprenylation, so we addressed the ability of T. brucei Rce1 to interact with the yeast Ras2p GTPase. Ras2p normally decorates the cytosolic face of the yeast plasma membrane (Fig. 2A). This localization pattern is highly dependent on the status of Ras2p posttranslational processing (3, 28). In the absence of CaaX protease activity (rce1Δ ste24Δ), subcellular punctate structures are observed with a GFP-Ras2p reporter (Fig. 2B). As determined through qualitative and quantitative methods, plasmid-based expression of yeast Rce1p, but not Ste24p, can restore proper GFP-Ras2p localization in this genetic background (Fig. 2C and D, respectively, and Table 3). We took advantage of this observation to investigate whether either of the trypanosomal CaaX proteases could properly modify a GTPase. When the trypanosomal CaaX proteases were evaluated in the context of GFP-Ras2p, proper localization of the reporter was observed with T. brucei Rce1 but not T. brucei Ste24 (Fig. 2E and F, respectively, and Table 3).
FIG. 2.

T. brucei Rce1 promotes proper Ras2p localization. Yeast strains that were wild type (EG123) (A) or deficient for endogenous yeast CaaX proteolytic activity (yWS164) (B to F) were each cotransformed with a plasmid encoding a GFP-Ras2p reporter (pWS750) and either an empty vector (A and B) or a plasmid encoding yeast Rce1p (C), yeast Ste24p (D), T. brucei Rce1 (E), or T. brucei Ste24 (F). For each condition, a small cluster of cells are shown as imaged after 6 h of induction of the reporter in SGal−ura media. The CaaX proteases were encoded in pSM1107, pSM1314, pWS766, and pWS767; pRS316 was used as the empty vector.
TABLE 3.
Quantification of GFP-Ras2p plasma membrane localization
| Backgrounda | CaaX protease | % PM localization ± SEMb |
|---|---|---|
| Wild type | Vector | 93.2 ± 1.5 |
| rce1Δ ste24Δ | Vector | 37.6 ± 4.2 |
| rce1Δ ste24Δ | S. cerevisiae Rce1 | 80.7 ± 1.2 |
| rce1Δ ste24Δ | T. brucei Rce1 | 83.0 ± 1.4 |
| rce1Δ ste24Δ | S. cerevisiae Ste24 | 42.3 ± 5.8 |
| rce1Δ ste24Δ | T. brucei Ste24 | 38.6 ± 2.0 |
Strains used were EG123 (wild type) or isogenic yWS164 (rce1Δ ste24Δ) that were cotransformed with plasmids encoding GFP-Ras2p (pWS750) and, as indicated, an empty vector (pRS316) or plasmid encoding a CaaX protease (pSM1107, pSM1314, pWS766, and pWS767).
Values for plasma membrane (PM) localization were calculated as s/√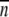 , where s is the sample standard deviation and n is number of observations.
, where s is the sample standard deviation and n is number of observations.
Trypanosomal Hsp40 protein Tbj1 can substitute for yeast Ydj1p and is cleaved by both Tb CaaX proteases.
Primarily through use of a-factor as a reporter molecule, the CaaX proteases have been demonstrated to possess partially overlapping substrate specificity (47). However, the only two established substrates for the Ste24p family of CaaX proteases are the precursors of the yeast a-factor mating pheromone and mammalian lamin A molecules. Since trypanosomes are not known to express orthologs of either of these molecules and, given the apparent specific ability of T. brucei Ste24 to cleave CaaX motifs associated with Hsp40 proteins from different organisms (i.e., CASQ of yeast Ydj1p and CVHQ of Tbj4), we postulated that isoprenylated Hsp40 proteins are substrates of Ste24p. In obvious conflict with this prediction is our observation that the T. brucei Hsp40-associated CaaX motifs CTQQ and CTAQ fail to promote a-factor production when coexpressed with either trypanosomal CaaX protease (see Fig. 1D). This leads to one of several possible hypotheses for these motifs: either they are not bona fide CaaX motifs (i.e., isoprenylated), they are farnesylated but not proteolysed, or they require a certain protein context in order to be recognized by Ste24 (i.e., CTQQ and CTAQ are unrecognizable by Ste24 in the context of yeast a-factor).
To differentiate between the above possibilities, we first determined whether any T. brucei Hsp40 protein having a CaaX motif could be isoprenylated by taking advantage of the observation that yeast are temperature sensitive when yeast Ydj1p is either absent or fails to be isoprenylated (7). A query of the T. brucei genome database yielded four putative Ydj1 orthologs, herein referred to as Tbj1 to Tbj4 (accession numbers XP_951689, XP_822483, XP_827509, and XP_845830, respectively). The degrees of sequence identity relative to yeast Ydj1p for Tbj1, Tbj2, Tbj3, and Tbj4 are 37, 18, 13, and 20%, respectively, as determined by CLUSTAL W2 analysis. Heterologous expression of Tbj1 and Tbj2 rescued growth of a ydj1Δ strain at elevated temperatures to an extent indistinguishable from the wild type, Tbj3 rescued growth less robustly and, for all practical purposes, Tbj4 failed to rescue growth (Fig. 3A). Tbj1 and Tbj2 both possess the CTQQ CaaX motif, whereas Tbj3 and Tbj4 possess the motifs CVHQ and CTAQ, respectively. The inability of Tbj4 to complement growth was surprising, since it contains more sequence homology to yeast Ydj1p than both Tbj2 and Tbj3. Sequence analysis of this clone and others isolated independently during construction of the Tbj4 expression plasmid consistently revealed an 18-nucleotide deletion near the amino terminus. While this may be the cause for the lack of complementation observed, it is also possible that the sequence for this protein is incorrectly annotated within the T. brucei genome database. The sequence we obtained for this clone has been submitted to the National Center for Biotechnology Information (accession no. FJ_611958).
FIG. 3.

Certain trypanosomal Hsp40 proteins rescue the temperature-sensitive phenotype of a Ydj1p-deficient yeast strain. (A) Inspection of the T. brucei genome identifies four trypanosomal Hsp40 proteins (Tbj1, Tbj2, Tbj3, and Tbj4) with homology to the yeast Hsp40 protein Ydj1p. The trypanosomal enzymes were heterologously expressed in ydj1Δ yeast (yWS304), and individual strains were assessed for growth at the indicated permissive and restrictive temperatures. In the case of Tbj1 alone, it was expressed as a GST fusion. The plasmids used were pWS782, pWS888, pWS889, and pWS890. (B) Plasmids encoding GST fusions of Ydj1p, Tbj1, and Tbj1(C401S) were transformed into ydj1Δ yeast (yWS304), and the transformed strains were assessed, along with ydj1Δ yeast and the isogenic wild-type strain (BY4741), for the ability to grow at the indicated temperatures. The plasmids used were pWS338, pWS782, and pWS783.
To specifically determine the impact of protein farnesylation on Tbj1 function, we mutated its CaaX motif cysteine to a serine and evaluated the effect. Tbj1(C401S) could not rescue the temperature sensitivity of the ydj1Δ yeast strain (Fig. 3B), which is the same effect observed for yeast Ydj1p when similarly mutated (7). We interpret this observation to indicate that the function of Tbj1 in trypanosomes has an absolute requirement for the cysteine residue within the CTQQ motif, thereby suggesting that Tbj1 is indeed isoprenylated.
To further test our hypothesis of protein context being important for specificity, we investigated whether T. brucei Rce1 and/or T. brucei Ste24 could cleave the CaaX motif of Tbj1 using a coupled proteolysis-carboxylmethylation assay. In this type of assay, the extent of CaaX proteolytic activity is indirectly monitored by the extent of ICMT-dependent carboxyl methylation, which is monitored by using a radioactive tracer. For the purposes of this experiment, the source of the Tbj1 substrate was a cell extract prepared from CaaX protease deficient yeast that expressed Tbj1 heterologously. Although the use of a purified form of the Tbj1 precursor would have been preferred, attempts to purify an adequate quantity of Tbj1 from yeast cell extracts were unsuccessful due to the instability of the precursor during purification. Use of the cell extract revealed that both T. brucei CaaX proteases can cleave the CTQQ motif of GST-TbJ1 (Table 4). This observation stands in contrast to that observed for the cleavage of the CTQQ motif in the context of the yeast a-factor reporter (see Fig. 1D), which is not recognized by either T. brucei CaaX protease. Hence, we propose that contextual information exists outside the CaaX motif that aids in directing CaaX processing.
TABLE 4.
Coupled proteolysis-carboxyl methylation in vitro assay reveals cleavage of Tbj1 by both T. brucei CaaX proteases
| Protease | cpma | SEMb | Pc |
|---|---|---|---|
| Vector | 345.5 | 18.8 | |
| T. brucei Rce1 | 499.2 | 31.2 | 0.002 |
| T. brucei Ste24 | 454.0 | 35.7 | 0.032 |
Values represent mean of six data points derived from two independent experiments (three replicates per experiment).
The standard error of the mean was calculated from the equation s/√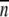 , where s is sample standard deviation and n is number of observations.
, where s is sample standard deviation and n is number of observations.
P values calculated by analysis of variance (single factor) relative to vector condition.
The trypanosomal CaaX proteases can be pharmacologically inhibited.
Protein isoprenylation is considered a target for antiparasitic drug discovery (13). Postisoprenylation enzymes appear to hold similar potential as drug discovery targets, as evidenced by the observation that RNAi-mediated gene silencing of T. brucei Rce1 or T. brucei ICMT impairs the growth of trypanosomes (20). Hence, we decided to determine the inhibitor profiles of the trypanosomal CaaX proteases using in vitro and in vivo approaches with small molecules known to inhibit CaaX proteases from other species (11, 12, 28).
For our in vitro approach, we first determined the utility of K-Ras4B-based, internally quenched, fluorogenic peptide substrates previously used to assess the function of the yeast and human CaaX proteases (21, 28, 37). Our previous studies indicated that quencher position within the CaaX motif can specify cleavage by Rce1 or Ste24, which prefer quencher placement at the a1 and X positions, respectively (37). We predicted a similar result for the trypanosomal CaaX proteases in part because of their differential ability to recognize a GTPase CaaX motif (see Fig. 2) and our observation that yeast and human orthologs have similar quencher position preferences (37; S. B. Porter and W. K. Schmidt, unpublished data). Surprisingly, this prediction was not supported by our findings. Unlike the profiles of yeast Rce1p and Ste24p, the trypanosomal CaaX proteases had largely overlapping profiles, with both proteases preferring the quencher at the X position (Fig. 4).
FIG. 4.

Trypanosomal CaaX proteases have similar specificities toward a synthetic K-Ras4B-based substrate. Yeast membranes enriched for the indicated trypanosomal (A) or yeast (B) CaaX protease were evaluated for their ability to cleave a farnesylated nonapeptide based on the K-Ras4B C terminus. The peptide contains an amino benzoic acid fluorophore that intensifies in fluorescence after cleavage of the CaaX motif (CVIM), which contains a dinitrophenol quencher that is coupled to a lysine (QL) placed at either the a1, a2, or X position. The activity for each CaaX protease is reported relative to the condition where maximal activity was observed. Closed bars represent Rce1 activity, and open bars represent Ste24 activity. The maximal activities were 15.16 and 11.02 relative fluorescence units/min for trypanosomal Rce1 and Ste24, respectively, and 93.27 and 25.10 relative fluorescence units/min for yeast Rce1 and Ste24, respectively. All membrane samples were prepared in the SM3614 background. The plasmids used were pSM1282, pWS479, pWS766, and pWS767.
Using an optimized fluorogenic reporter (CVIQL), we evaluated the relative in vitro sensitivities of the trypanosomal CaaX proteases to TPCK, a dipeptidyl (acyloxy)methyl ketone (FKBK), EDTA, EGTA, and a set of small molecule compounds previously demonstrated to inhibit yeast and human Rce1p (28, 37). This analysis revealed that T. brucei Rce1 was inhibited by all of the compounds, with the exception of EDTA and EGTA (Table 5). The observed inhibitor profile of T. brucei Rce1 was similar if not identical to that of yeast and human Rce1p (28). Unlike its yeast and human counterparts, T. brucei Ste24 was sensitive to TPCK. Otherwise, T. brucei Ste24 behaved as predicted, being relatively insensitive to the effects of EDTA, EGTA, weakly inhibited by compound 1, and strongly inhibited by the other compounds evaluated. To gain a more detailed understanding of the observed inhibitor effects on T. brucei Rce1, we determined IC50 values for the small molecule compound set (Table 6). The compounds largely had IC50 values of <10 μM, with the exception of compounds 2, 4, and 5, which had IC50 values of >80 μM.
TABLE 5.
Inhibitor profiles of trypanosomal CaaX proteases
| Compounda | Mean % activityb ± SEM for: |
|
|---|---|---|
| T. brucei Rce1 | T. brucei Ste24 | |
| TPCK | 21.6 ± 5.1 | 24.5 ± 5.0 |
| FKBK | 34.4 ± 6.0 | 30.0 ± 3.5 |
| EDTA | 92.0 ± 11.1 | 79.2 ± 10.2 |
| EGTA | 94.6 ± 5.5 | 75.7 ± 3.2 |
| 1 | 30.7 ± 4.8 | 59.4 ± 5.9 |
| 2 | 22.1 ± 4.7 | 24.1 ± 5.3 |
| 3 | 18.6 ± 2.7 | 14.9 ± 5.8 |
| 4 | 23.9 ± 9.5 | 31.3 ± 10.9 |
| 5 | 34.2 ± 9.4 | 35.1 ± 8.6 |
| 6 | 19.3 ± 4.0 | 28.0 ± 2.4 |
| 7 | 18.4 ± 3.8 | 28.6 ± 2.2 |
| 8 | 16.1 ± 3.3 | 14.6 ± 5.4 |
| 9 | 15.4 ± 3.7 | 20.6 ± 3.8 |
TPCK, FKBK, EDTA, and EGTA were used at 200 μM. Compounds 1 to 9 are as previously identified and were used at 100 μM (28).
Values are relative to an appropriate untreated control treated with H2O (EDTA and EGTA) or DMSO (TPCK, FKBK, compounds 1 to 9).
TABLE 6.
IC50 values of Rce1p inhibitors observed with trypanosomal and human Rce1
| Species | Compound | Mean IC50 (μM) ± SEM |
|---|---|---|
| T. brucei | 1 | 3.24 ± 0.52 |
| 2 | 83.42 ± 7.18 | |
| 3 | 8.10 ± 3.21 | |
| 4 | 160.75 ± 14.21 | |
| 5 | 176.77 ± 34.16 | |
| 6 | 1.30 ± 0.07 | |
| 7 | 0.25 ± 0.22 | |
| 8 | 4.23 ± 1.46 | |
| 9 | 0.91 ± 0.13 | |
| H. sapiens | 7 | 2.38 ± 0.19 |
| 8 | 2.43 ± 0.77 |
We have observed that certain small molecule inhibitors described above can induce delocalization of GFP-Ras2p in vivo when applied to yeast cultures expressing yeast Rce1p and that this phenotype is similar to that observed in the absence of CaaX protease activity (28). Thus, we predicted that chemical treatment of yeast heterologously expressing trypanosomal Rce1 as the only CaaX protease would result in a similar phenotype. Indeed, six compounds induced delocalization (Fig. 5A). Compounds 4 and 9 strongly induced delocalization, with 91%, and 79% of the cell population responding, respectively. Compounds 3, 6, 7, and 8 also induced delocalization, but less dramatically, with 48, 47, 62 and 59% of the cell population responding, respectively. Examples of the delocalized patterns, regardless of being the majority or minority phenotype, are shown in the respective panels of the figure. Compounds 1, 2, and 5 had no effect on GFP-Ras2p localization at the concentrations evaluated, which may represent a lack of cell permeability by these compounds.
FIG. 5.

Chemical agents can disrupt trypanosomal Rce1 activity in vivo. (A) Effect of chemical agents on T. brucei Rce1 activity. yWS164 yeast expressing a GFP-Ras2p reporter and T. brucei Rce1 were transiently treated for 1 h with DMSO (subpanel D) or the indicated chemical agents prior to induction of GFP-Ras2p expression as described in Fig. 2. Compounds were used at doses that were largely nontoxic to liquid cultures of yeast (EC10). The EC10 doses for compounds 3, 4, 6, 7, 8, and 9 were 7.89, 6.78, 0.67, 11.1, 10.8, and 3.78 μM, respectively. All others were used at 55.6 μM. The plasmids used were pWS750 and pWS766. (B) Dose-dependent effects of compounds 7 and 8 on human and trypanosomal Rce1. yWS164 yeast expressing a GFP-Ras2p reporter (pWS750) and either the human (pWS335) or trypanosomal (pWS766) Rce1 ortholog were evaluated for sensitivity to compounds 7 and 8 by the protocol described in panel A using the doses of compounds indicated. The percentage of cells with a delocalized phenotype are reported from a minimum population of 60 cells from at least three independent cell fields.
We were intrigued by the observation that compounds 7 and 8 could induce GFP-Ras2p delocalization, even if only in a minority of the population, because of evidence suggesting that human Rce1 was less sensitive to these compounds (28). To further investigate the relative effects of these compounds on trypanosomal and human Rce1, we performed both in vivo and in vitro dose-response studies. Consistent with expectations, T. brucei Rce1 was more sensitive to compounds 7 and 8 than human Rce1 when evaluated using our GFP-Ras2p localization assay (Fig. 5B). We observed that a 5.3 μM concentration of compound 7 was sufficient to induce delocalization of GFP-Ras2p in the context of T. brucei Rce1, but this concentration had no apparent effect on human Rce1. In fact, an 11.2 μM concentration of compound was required to delocalize the reporter to the same extent in the context of human Rce1. Similarly, a lower concentration of compound 8 was needed to comparably delocalize GFP-Ras2p in the context of T. brucei Rce1 compared to human Rce1 (10.8 and 25 μM, respectively). Consistent with our in vivo results, in vitro IC50 determinations revealed that compound 7 was nearly 10-fold more selective for T. brucei Rce1 than its human counterpart (Table 6). The opposite, however, was true for compound 8, which was ∼2-fold less potent against T. brucei Rce1. The reason for the lack of correlation with this compound is unknown. Together, our observations indicate that GFP-Ras2p is an effective reporter for T. brucei Rce1 activity, that the subcellular distribution of GFP-Ras2p can be used as an indicator for determining the effectiveness of compounds for disrupting T. brucei Rce1 activity in vivo, and that the reporter can be used to demonstrate differential targeting specificity by Rce1 inhibitors (i.e., human versus trypanosomal Rce1).
DISCUSSION
The results of this study are consistent with the conclusion that two separate CaaX protease activities are present in T. brucei corresponding with genes encoding orthologs of Rce1p and Ste24p found in other eukaryotic systems. This observation can be contrasted with that of a previous investigation into the enzymatic properties of these proteins that revealed a proteolytic activity in association with T. brucei Rce1 but not T. brucei Ste24 (20). We propose that the lack of activity observed in the earlier study for T. brucei Ste24 may be a direct consequence of an inappropriate reporter for the enzyme. By our own evidence, we find that T. brucei Ste24 is active against several CaaX motifs when it is evaluated in the context of the yeast a-factor reporter, including the CVIM motif present on the substrate used in the prior trypanosomal study (Fig. 1). From these observations, we hypothesize that contextual information is present within the non-CaaX portion of reporters that helps specify T. brucei CaaX protease specificity. The idea of contextual information being required by the CaaX proteases has been previously proposed (47) and is further supported by our observation that CTQQ is suitable as a CaaX motif in the context of Tbj1, but not yeast a-factor (Fig. 1D and Table 4). Moreover, it has been demonstrated that yeast Ste24p cannot cleave the CIIS motif in its natural protein context (i.e., Ras2p), but can when appended to the a-factor precursor (3, 47). Collectively, these observations challenge the predictive utility of the a-factor reporter system, or any single reporter background for that matter, as a means by which to assess CaaX protease specificity.
Our most contextually correct data set for the T. brucei CaaX proteases establishes that both can mediate maturation of the T. brucei Hsp40-family protein Tbj1 (Table 4). However, it would be inappropriate to extrapolate that Tbj2, Tbj3, and Tbj4 are also processed in this manner because their CaaX motifs have not been assessed in their proper protein context. We also cannot infer that T. brucei Rce1 specifically mediates maturation of the T. brucei RLP GTPase, despite an obvious preference for cleavage of its CaaX motif (CTML) by T. brucei Rce1 in our a-factor assay. Likewise, T. brucei Rce1 might specifically cleave T. brucei RHP despite its motif (CVIM) being cleaved by both T. brucei CaaX proteases in our yeast assay. It appears that determination of specific CaaX protease processing preferences must be experimentally addressed on an individual basis within their native context using methods similar to those applied in this and other studies (4, 27, 34). This will certainly be a challenging task given that inspection of the T. brucei brucei genome using the TriTrypDB server reveals over 200 proteins with a canonical CaaX motif, which can be reduced to 61 by applying filters that eliminate pseudogenes, hypothetical proteins, and those with putative signal sequences that would not be expected to be associated with isoprenylated CaaX proteins (see Table S1 in the supplemental material). It remains to be determined which among these presumptive CaaX proteins undergoes isoprenylation, let alone CaaX proteolysis. Among the T. brucei GTPases previously identified, only a few have CaaX motifs (17). These include the Ras-like GTPase T. brucei RLP (CTML), the Rho-like GTPase T. brucei RHP (CVIM), the Rab-like GTPase T. brucei Rab23 (CSVM), and the Rag-like GTPase T. brucei Rab28 (CAVM). All other identified trypanosomal GTPases, including those of the Arf and Ran families, do not contain CaaX motifs, while additional members of the Rab family possess canonical dicysteine geranylgeranylation motifs. Future studies may ascertain the role of T. brucei Rce1 and/or T. brucei Ste24 in the maturation of trypanosomal GTPases containing a CaaX motif and help elucidate the reported essential role of T. brucei Rce1 (20).
Our study cautions the use of a single reporter system to specifically determine which CaaX protease cleaves a particular motif. Nonetheless, such reporters still retain value for assessing whether a particular set of CaaX protease orthologs has conserved or dissimilar specificity. For example, the observation that yeast and human Rce1p better recognize certain motifs (Fig. 1D, CAMQ and CTAQ) than does T. brucei Rce1 in the context of the a-factor reporter implies that T. brucei Rce1 has an intrinsic specificity difference from its relatives. Similar arguments can be made for T. brucei Ste24 by comparing its specificity profile to that of its orthologs, although the observed differences are less dramatic (Fig. 1D, CASQ and CAMQ).
The inhibition of T. brucei Rce1 could hold therapeutic potential, since the loss of T. brucei Rce1 function appears to be correlated with the loss of parasite viability (20); the impact of Ste24 loss of function has not yet been addressed. Should trypanosomal and human Rce1 ultimately have overlapping substrate specificities, substrate-based inactivators of T. brucei Rce1 will likely target the human enzyme, thereby leading to unintended and undesirable side effects for patients. We have observed some differences, however, in the activities of trypanosomal and human Rce1 that may indicate that specific targeting of the parasitic enzyme may be possible. First, we determined that T. brucei Rce1 does not readily cleave CTAQ and CAMQ motifs in the context of the a-factor reporter, and thus does not have the exact specificity profile of its human and yeast counterparts (Fig. 1D). Second, we observed that T. brucei Rce1 has a distinct preference, by comparison to the yeast and human enzymes, for the optimal placement of a lysine dinitrophenol quenching group on a synthetic fluorogenic peptide reporter (Fig. 4) (37). Lastly, we identified two small molecule agents that inhibit T. brucei Rce1 preferentially over the human enzyme in vivo (Fig. 5). The specificity and inhibitor profiles of T. brucei Rce1 seem to reflect enzymatic differences between it and human Rce1, suggesting the exciting prospect that trypanosome-specific agents can be identified and developed. This conclusion is predicated on the observed specificity differences holding true independent of protein context.
In conclusion, our study continues to support the observation that eukaryotic systems generally possess two distinct CaaX proteolytic activities. The purpose for this redundancy is not immediately obvious and is likely tied to the need to accommodate the great variety of substrate CaaX proteins encoded in eukaryotic genomes. We argue, based on our results, that these substrates possess information both within their CaaX motifs and protein backbones that provides specificity for one or both proteolytic systems.
Supplementary Material
Acknowledgments
We are grateful to T. Ochsenreiter for providing genomic T. brucei DNA, M. P. Terns for access to a Zeiss Axioskop 2 Mot Plus microscope, T. M. Dore and members of the Schmidt laboratory (all of the University of Georgia) for critical discussions and technical assistance, and R. G. Mallon (Wyeth Research) for initial samples of fluorogenic K-Ras4B peptides.
This study was supported by an R01 grant (GM067092) from the National Institutes of Health (W.K.S.) and an associated research supplement (K.A.C.).
Footnotes
Published ahead of print on 9 October 2009.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Anderson, J. L., B. S. Henriksen, R. A. Gibbs, and C. A. Hrycyna. 2005. The isoprenoid substrate specificity of isoprenylcysteine carboxylmethyltransferase: development of novel inhibitors. J. Biol. Chem. 280:29454-29461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashby, M., D. King, and J. Rine. 1992. Endoproteolytic processing of a farnesylated peptide in vitro. Proc. Natl. Acad. Sci. USA 89:4613-4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyartchuk, V. L., M. N. Ashby, and J. Rine. 1997. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science 275:1796-1800. [DOI] [PubMed] [Google Scholar]
- 4.Bracha, K., M. Lavy, and S. Yalovsky. 2002. The Arabidopsis AtSTE24 is a CAAX protease with broad substrate specificity. J. Biol. Chem. 277:29856-29864. [DOI] [PubMed] [Google Scholar]
- 5.Cadiñanos, J., W. K. Schmidt, A. Fueyo, I. Varela, C. Lopez-Otin, and J. M. Freije. 2003. Identification, functional expression and enzymic analysis of two distinct CaaX proteases from Caenorhabditis elegans. Biochem. J. 370:1047-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cadiñanos, J., I. Varela, D. Mandel, W. K. Schmidt, A. Díaz-Perales, C. López-Otín, and F. JMP. 2003. AtFACE-2, a prenylated-protein protease from Arabidopsis thaliana related to Ras converting enzymes. J. Biol. Chem. 278:42091-42097. [DOI] [PubMed] [Google Scholar]
- 7.Caplan, A. J., J. Tsai, P. J. Casey, and M. G. Douglas. 1992. Farnesylation of Ydj1p is required for function at elevated growth temperatures in Saccharomyces cerevisiae. J. Biol. Chem. 267:18890-18895. [PubMed] [Google Scholar]
- 8.Caplin, B. E., L. A. Hettich, and M. S. Marshall. 1994. Substrate characterization of the Saccharomyces cerevisiae protein farnesyltransferase and type-I protein geranylgeranyltransferase. Biochim. Biophys. Acta 1205:39-48. [DOI] [PubMed] [Google Scholar]
- 9.Cox, A., and C. Der. 2002. Farnesyltransferase inhibitors: promises and realities. Curr. Opin. Pharmacol. 2:388-393. [DOI] [PubMed] [Google Scholar]
- 10.Dai, Q., E. Choy, V. Chiu, J. Romano, S. R. Slivka, S. A. Steitz, S. Michaelis, and M. R. Philips. 1998. Human prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J. Biol. Chem. 273:15030-15034. [DOI] [PubMed] [Google Scholar]
- 11.Dolence, E. K., J. M. Dolence, and C. D. Poulter. 2000. Solid-phase synthesis of a farnesylated CaaX peptide library: inhibitors of the Ras CaaX endoprotease. J. Comb. Chem. 2:522-536. [DOI] [PubMed] [Google Scholar]
- 12.Dolence, J. M., L. E. Steward, E. K. Dolence, D. H. Wong, and C. D. Poulter. 2000. Studies with recombinant Saccharomyces cerevisiae CaaX prenyl protease Rce1p. Biochemistry 39:4096-4104. [DOI] [PubMed] [Google Scholar]
- 13.Eastman, R. T., F. S. Buckner, K. Yokoyama, M. H. Gelb, and W. C. Van Voorhis. 2006. Thematic review series: lipid posttranslational modifications: fighting parasitic disease by blocking protein farnesylation. J. Lipid Res. 47:233-240. [DOI] [PubMed] [Google Scholar]
- 14.Elble, R. 1992. A simple and efficient procedure for transformation of yeasts. BioTechniques 13:18-20. [PubMed] [Google Scholar]
- 15.Esteva, M. I., K. Kettler, C. Maidana, L. Fichera, A. M. Ruiz, E. J. Bontempi, B. Andersson, H. M. Dahse, P. Haebel, R. Ortmann, G. Klebe, and M. Schlitzer. 2005. Benzophenone-based farnesyltransferase inhibitors with high activity against Trypanosoma cruzi. J. Med. Chem. 48:7186-7191. [DOI] [PubMed] [Google Scholar]
- 16.Farh, L., D. Mitchell, and R. Deschenes. 1995. Farnesylation and proteolysis are sequential, but distinct steps in the CaaX box modification pathway. Arch, Biochem, Biophys. 318:113-121. [DOI] [PubMed] [Google Scholar]
- 17.Field, M. C. 2005. Signalling the genome: the Ras-like small GTPase family of trypanosomatids. Trends Parasitol. 21:447-450. [DOI] [PubMed] [Google Scholar]
- 18.Fujimura-Kamada, K., F. J. Nouvet, and S. Michaelis. 1997. A novel membrane-associated metalloprotease, Ste24p, is required for the first step of NH2-terminal processing of the yeast a-factor precursor. J. Cell Biol. 136:271-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Veronneau, S. Dow, A. Lucau-Danila, K. Anderson, B. Andre, A. P. Arkin, A. Astromoff, M. El-Bakkoury, R. Bangham, R. Benito, S. Brachat, S. Campanaro, M. Curtiss, K. Davis, A. Deutschbauer, K. D. Entian, P. Flaherty, F. Foury, D. J. Garfinkel, M. Gerstein, D. Gotte, U. Guldener, J. H. Hegemann, S. Hempel, Z. Herman, D. F. Jaramillo, D. E. Kelly, S. L. Kelly, P. Kotter, D. LaBonte, D. C. Lamb, N. Lan, H. Liang, H. Liao, L. Liu, C. Luo, M. Lussier, R. Mao, P. Menard, S. L. Ooi, J. L. Revuelta, C. J. Roberts, M. Rose, P. Ross-Macdonald, B. Scherens, G. Schimmack, B. Shafer, D. D. Shoemaker, S. Sookhai-Mahadeo, R. K. Storms, J. N. Strathern, G. Valle, M. Voet, G. Volckaert, C. Y. Wang, T. R. Ward, J. Wilhelmy, E. A. Winzeler, Y. Yang, G. Yen, E. Youngman, K. Yu, H. Bussey, J. D. Boeke, M. Snyder, P. Philippsen, R. W. Davis, and M. Johnston. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387-391. [DOI] [PubMed] [Google Scholar]
- 20.Gillespie, J. R., K. Yokoyama, K. Lu, R. T. Eastman, J. G. Bollinger, W. C. Van Voorhis, M. H. Gelb, and F. S. Buckner. 2007. C-terminal proteolysis of prenylated proteins in trypanosomatids and RNA interference of enzymes required for the posttranslational processing pathway of farnesylated proteins. Mol. Biochem. Parasitol. 153:115-124. [DOI] [PubMed] [Google Scholar]
- 21.Hollander, I., E. Frommer, and R. Mallon. 2000. Human ras-converting enzyme (hRCE1) endoproteolytic activity on K-ras-derived peptides. Anal. Biochem. 286:129-137. [DOI] [PubMed] [Google Scholar]
- 22.Hrycyna, C. A., and S. Clarke. 1992. Maturation of isoprenylated proteins in Saccharomyces cerevisiae. J. Biol. Chem. 267:10457-10464. [PubMed] [Google Scholar]
- 23.Hrycyna, C. A., S. K. Sapperstein, S. Clarke, and S. Michaelis. 1991. The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. EMBO J. 10:1699-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang, G. F., K. Yokoyama, and M. H. Gelb. 1993. A prenylated protein-specific endoprotease in rat liver microsomes that produces a carboxyl-terminal tripeptide. Biochemistry 32:9500-9507. [DOI] [PubMed] [Google Scholar]
- 25.Kim, S., A. Lapham, C. Freedman, T. Reed, and W. Schmidt. 2005. Yeast as a tractable genetic system for functional studies of the insulin-degrading enzyme. J. Biol. Chem. 280:27481-27490. [DOI] [PubMed] [Google Scholar]
- 26.Lane, K. T., and L. S. Beese. 2006. Thematic review series: lipid posttranslational modifications: structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J. Lipid Res. 47:681-699. [DOI] [PubMed] [Google Scholar]
- 27.Leung, K. F., R. Baron, B. R. Ali, A. I. Magee, and M. C. Seabra. 2007. Rab GTPases containing a CAAX motif are processed post-geranylgeranylation by proteolysis and methylation. J. Biol. Chem. 282:1487-1497. [DOI] [PubMed] [Google Scholar]
- 28.Manandhar, S. P., E. R. Hildebrandt, and W. K. Schmidt. 2007. Small-molecule inhibitors of the Rce1p CaaX protease. J. Biomol. Screen. 12:983-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martzen, M. R., S. M. McCraith, S. L. Spinelli, F. M. Torres, S. Fields, E. J. Grayhack, and E. M. Phizicky. 1999. A biochemical genomics approach for identifying genes by the activity of their products. Science 286:1153-1155. [DOI] [PubMed] [Google Scholar]
- 30.Michaelis, S., and I. Herskowitz. 1988. The a-factor pheromone of Saccharomyces cerevisiae is essential for mating. Mol. Cell. Biol. 8:1309-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moores, S. L., M. D. Schaber, S. D. Mosser, E. Rands, M. B. O'Hara, V. M. Garsky, M. S. Marshall, D. L. Pompliano, and J. B. Gibbs. 1991. Sequence dependence of protein isoprenylation. J. Biol. Chem. 266:14603-14610. [PubMed] [Google Scholar]
- 32.Ohkanda, J., J. W. Lockman, K. Yokoyama, M. H. Gelb, S. L. Croft, H. Kendrick, M. I. Harrell, J. E. Feagin, M. A. Blaskovich, S. M. Sebti, and A. D. Hamilton. 2001. Peptidomimetic inhibitors of protein farnesyltransferase show potent antimalarial activity. Bioorg. Med. Chem. Lett. 11:761-764. [DOI] [PubMed] [Google Scholar]
- 33.Oldenburg, K. R., K. T. Vo, S. Michaelis, and C. Paddon. 1997. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 25:451-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otto, J. C., E. Kim, S. G. Young, and P. J. Casey. 1999. Cloning and characterization of a mammalian prenyl protein-specific protease. J. Biol. Chem. 274:8379-8382. [DOI] [PubMed] [Google Scholar]
- 35.Pei, J., and N. V. Grishin. 2001. Type II CAAX prenyl endopeptidases belong to a novel superfamily of putative membrane-bound metalloproteases. Trends Biochem. Sci. 26:275-277. [DOI] [PubMed] [Google Scholar]
- 36.Plummer, L. J., E. R. Hildebrandt, S. B. Porter, V. A. Rogers, J. McCracken, and W. K. Schmidt. 2006. Mutational analysis of the ras converting enzyme reveals a requirement for glutamate and histidine residues. J. Biol. Chem. 281:4596-4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porter, S. B., E. R. Hildebrandt, S. R. Breevoort, D. Z. Mokry, T. M. Dore, and W. K. Schmidt. 2007. Inhibition of the CaaX proteases Rce1p and Ste24p by peptidyl (acyloxy)methyl ketones. Biochim. Biophys. Acta 1773:853-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romano, J. D., W. K. Schmidt, and S. Michaelis. 1998. The Saccharomyces cerevisiae prenylcysteine carboxyl methltransferase Ste14p is in the endoplasmic reticulum membrane. Mol. Biol. Cell 9:2231-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlitzer, M., A. Winter-Vann, and P. J. Casey. 2001. Non-peptidic, non-prenylic inhibitors of the prenyl protein-specific protease Rce1. Bioorg. Med. Chem. Lett. 11:425-427. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt, W. K., A. Tam, K. Fujimura-Kamada, and S. Michaelis. 1998. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc. Natl. Acad. Sci. USA 95:11175-11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt, W. K., A. Tam, and S. Michaelis. 2000. Reconstitution of the Ste24p-dependent N-terminal proteolytic step in yeast a-factor biogenesis. J. Biol. Chem. 275:6227-6233. [DOI] [PubMed] [Google Scholar]
- 42.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siliciano, P., and K. Tatchell. 1984. Transcription and regulatory signals at the mating type locus in yeast. Cell 37:969-978. [DOI] [PubMed] [Google Scholar]
- 44.Reference deleted.
- 45.Tam, A., F. Nouvet, K. Fujimura-Kamada, H. Slunt, S. S. Sisodia, and S. Michaelis. 1998. Dual roles for Ste24p in yeast a-factor maturation: NH2-terminal proteolysis and COOH-terminal CAAX processing. J. Cell Biol. 142:635-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tam, A., W. K. Schmidt, and S. Michaelis. 2001. The multispanning membrane protein Ste24p catalyzes CAAX proteolysis and NH2-terminal processing of the yeast a-factor precursor. J. Biol. Chem. 276:46798-46806. [DOI] [PubMed] [Google Scholar]
- 47.Trueblood, C. E., V. L. Boyartchuk, E. A. Picologlou, D. Rozema, C. D. Poulter, and J. Rine. 2000. The CaaX proteases, Afc1p and Rce1p, have overlapping but distinct substrate specificities. Mol. Cell. Biol. 20:4381-4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winter-Vann, A. M., R. A. Baron, W. Wong, J. dela Cruz, J. D. York, D. M. Gooden, M. O. Bergo, S. G. Young, E. J. Toone, and P. J. Casey. 2005. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. USA 102:4336-4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winter-Vann, A. M., and P. J. Casey. 2005. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 5:405-412. [DOI] [PubMed] [Google Scholar]
- 50.Young, S. G., P. Ambroziak, E. Kim, and S. Clarke. 2001. Postisoprenylation protein processing: CXXX (CaaX) endoproteases and isoprenylcysteine carboxyl methyltransferase, p. 155-213. In F. Tamanoi and D. S. Sigman (ed.), The enzymes, vol. XXI. Academic Press, Inc., New York, NY. [Google Scholar]
- 51.Young, S. G., M. Meta, S. H. Yang, and L. G. Fong. 2006. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 281:39741-39745. [DOI] [PubMed] [Google Scholar]
- 52.Zhang, Y., G. Nijbroek, M. L. Sullivan, A. A. McCracken, S. C. Watkins, S. Michaelis, and J. L. Brodsky. 2001. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12:1303-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu, K., A. D. Hamilton, and S. M. Sebti. 2003. Farnesyltransferase inhibitors as anticancer agents: current status. Curr. Opin. Investig. Drugs 4:1428-1435. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.